
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The catalytic and radical mechanism for ethanol oxidation to acetic acid†
Sotiria
Mostrou
 a,
Andreas
Nagl
b,
Marco
Ranocchiari
c,
Karin
Föttinger
b and
Jeroen A.
van Bokhoven
*ac
a,
Andreas
Nagl
b,
Marco
Ranocchiari
c,
Karin
Föttinger
b and
Jeroen A.
van Bokhoven
*ac
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch; Tel: +41 44 632 55 42
bInstitute of Material Chemistry, Division Physical Chemistry, TU Wien, Getreidemarkt 9/165, 1060 Wien, Austria
cLaboratory for Catalysis and Sustainable Chemistry, Paul Scherrer Institute, WLGA 135, 5232 Villigen, Switzerland
First published on 2nd September 2019
Abstract
Au/TiO2 is a much-used catalyst for the conversion of ethanol to acetic acid. The proposed mechanism speaks of two essential reaction steps on the catalytic surface. The first is the ethanol to acetaldehyde and the second the acetaldehyde to acetic acid. When operating in the gas phase, acetic acid is usually absent. This work focuses on determining what triggers the second step by comparing the ethanol with acetaldehyde oxidation and the liquid with gas-phase reaction. We propose an updated reaction mechanism: acetaldehyde autoxidises non-catalytically to acetic acid, likely driven by radicals. The requirement for the autoxidation is the presence of oxygen and water in the liquid-phase. The understanding of the interplay between the catalytic ethanol to acetaldehyde and the following non-catalytic reaction step provides guiding principles for the design of new and more selective alcohol oxidation catalysts.
With the continuous increase in bioethanol production and the global need to decrease the use of fossil resources, ethanol oxidation ranks as one of the most promising alternatives for the production of bulk chemicals, such as acetaldehyde and acetic acid.1 Aqueous ethanol and oxygen convert to acetic acid already at 363 K with no additives over gold-based catalysts.2,3 The reaction can reach up to 88% selectivity to acetic acid in the batch, only with residence times above 20 h. The two key reaction steps are the oxidative dehydrogenation of adsorbed ethanol to acetaldehyde, which is the proposed rate-determining step, and its subsequent oxidation to acetic acid.3 Operation in a flow system shortens the residence time to a few minutes and results in similar conversion levels to those in batch, but the selectivity to acetic acid decreases to about 60%.4 Carbon dioxide is the second-most abundant product with selectivities up to 30%. Tan et al. described a possible pathway to the C–C cleavage.5 Ethanol adsorption and oxidation of the catalyst lead to acetate species, followed by the formation of oxalate (C2O42−) and finally to carbon dioxide.
Low selectivity to carbon dioxide and fast reaction are possible when operating in the gas-phase, but mainly acetaldehyde is produced in up to 70% yield, despite acetaldehyde oxidation (second step) being energetically less demanding.6 Hence, independent of temperature and the conversion level, the selectivity trend reverses when altering between gas and liquid phase oxidation. Mechanistic and theoretical studies on the role of water in the liquid-phase reaction have proposed that the acid is formed with the direct involvement of hydroxides in the presence of water.7–9 A gold surface covered with hydroxides also reduces the activation energy of ethanol adsorption to ethoxy species and dehydrogenation of the latter to acetaldehyde. These studies assume a high concentration of hydroxides and are valid only under such specific conditions. The addition of strong bases is undesirable; hence, most experimental research has been conducted in a neutral pH environment.2–4,10–12
A deep understanding of the selectivity driving factor is still missing, despite being vital for improving the overall process. This study focuses on understanding what allows the oxidation of acetaldehyde to acetic acid in the liquid-phase by investigating the two reaction steps independently. We further compared the gas and liquid-phase operation, using our recently established trickle-flow system, which enables direct assessment of the two phases.4 The isolated testing of aqueous acetaldehyde identified that under the typical ethanol oxidation reaction conditions acetaldehyde oxidises to acetic acid non-catalytically. The reaction is likely radical initiated, similarly to the well-established, non-catalytic oxidation of aliphatic aldehydes. The non-catalytic oxidation of acetaldehyde is feasible in the liquid phase and not in the gas phase.
We tested both the ethanol (EtOH) and acetaldehyde (MeCHO) oxidation over a silicon carbide bed and a 1% Au/TiO2 catalyst, under typical liquid-phase ethanol oxidation conditions, using a trickle flow reactor at 423 K and 15 bar.4Fig. 1 shows the conversion of ethanol and acetaldehyde (top) and the product selectivity (bottom). In the oxidation of ethanol, the non-catalytic silicon carbide (solid bars) did not exhibit any conversion. The catalytic 1% Au/TiO2 (patterned bars) showed about 20% conversion under the tested conditions. The main product was acetic acid (AcOH, orange) with about 60% selectivity, while acetaldehyde (blue) and ethyl acetate (EtOAc, green) were formed, each with less than 10% selectivity. The remaining 25% selectivity was carbon dioxide, the only observable gas product.4
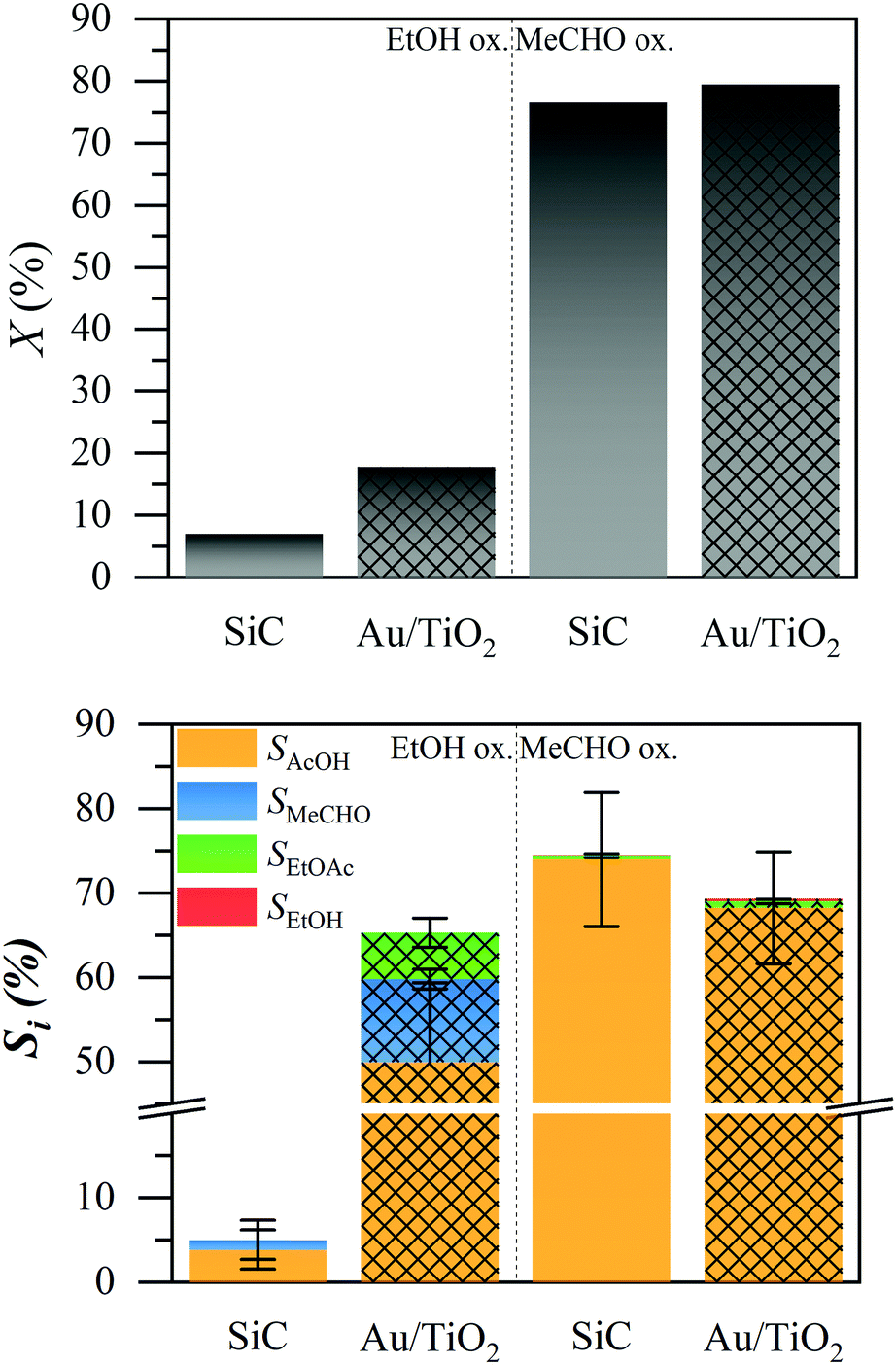 | ||
| Fig. 1 Top: Ethanol and acetaldehyde conversion over silicon carbide (solid) and 1% Au/TiO2 (patterned). Bottom: Selectivity to the liquid products. Conditions in flow: 423 K, 15 bar, 5 ml min−1 O2, 0.3 ml min−1 5% solution, 0.15 mg catalyst and 0.15 g SiC (catalytic), 0.30 g SiC (blank). | ||
In the oxidation of acetaldehyde, the conversion (top graph) was approximately the same (ca. 80%) in the presence and absence of the catalyst. We observed trace amounts of ethanol and ethyl acetate, each below 1% selectivity. In both cases, the major liquid product was acetic acid, with 73% and 68% selectivity in the absence and presence of the catalyst, respectively. The presence of the catalyst slightly decreased the selectivity to acetic acid. We observed no other liquid products (ESI†). Combustion products are not expected in the absence of the catalyst. The difference from 100% selectivity is attributed to the evaporation of acetaldehyde; we estimated that about 30% of acetaldehyde was evaporated, due to the high volatility of the reactant mixture (ESI†). To identify whether any contamination was responsible for the conversion, we also tested the reaction in the batch system, as well as with a different water source, all resulting in very similar production of acetic acid (ESI†).
We further studied the seemingly non-catalytic oxidation of acetaldehyde by altering the reaction parameters, such as the solvent and the physical state (Table 1). Entry 1.1 shows the acetic acid yield when operating under ethanol oxidation conditions: liquid–gas mixture, 423 K and 15 bar. Under these conditions, acetic acid was produced with 49% yield in flow and 76 in batch. When operating either in the gas phase (entry 1.2) or under oxygen-free (entry 1.3) or water-free (entry 1.4) conditions, the acetaldehyde to acetic acid conversion was much lower, yielding less than 3% acetic acid. The presence of water, 10% in ethanol (entry 1.5), produced acetic acid in 34% yield. We also observed a noticeable amount of ethyl acetate (40%). The formation of ethyl acetate reduces the apparent acetic acid selectivity, as it derives from the side-reaction of acetic acid with ethanol.3 The addition of the radical scavenger BHT yielded 5.9% acetic acid (entry 1.6).13 The above test was conducted under the same conditions as entry 1.5, i.e. 5% MeCHO/10% H2O/EtOH, to increase the solubility of BHT. We also observed the apparent conversion of about 60%, mainly due to evaporation during sampling, and small peaks in the chromatograph (ESI†), which are related to BHT but not identified here.
| Experiment | Conditions | Yield in flow (%) | Yield in batch (%) | |
|---|---|---|---|---|
| 1.1 | Ethanol oxidation conditions | O2/423 K/15 bar | 48.9 | 76.0 |
| 1.2 | Role of the MeCHO phase | Liquid → gas | 2.5 | — |
| 1.3 | Role of oxygen | O2 → N2 | — | 1.8 |
| 1.4 | Role of water | H2O → EtOH | 2.8 | — |
| 1.5 | Role of water | H2O → 10% H2O–EtOH | — | 34.3 |
| 1.6 | Radical trap | + BHT | — | 5.9 |
Finally, we studied the impact of the non-catalytic oxidation of acetaldehyde on the overall reaction mechanism of ethanol oxidation by altering the conditions, such that the non-catalytic oxidation of acetaldehyde cannot proceed. Fig. 2 presents the selectivity to acetaldehyde and acetic acid during the catalytic ethanol oxidation while altering the system pressure, moving from the gas to the liquid regime. The reaction phase, gas, liquid, or partial evaporation, was determined by comparison of the temperature on the catalyst bed (green) and the theoretical boiling point of the mixture at the respective pressure (black), while the oven temperature was constant at 473 K. At low pressure and in the gas-phase reaction regime, acetaldehyde (blue squares) was the main product with up to 60% selectivity. Acetic acid (orange triangles) was below 10%. When liquid reactants were present, even in a gas–liquid mixture, the selectivity to acetaldehyde decreased almost linearly, down to 25% in the liquid regime. At the same time, the selectivity to acetic acid increased from 10 up to 50% in the liquid. We observed the same behaviour independent of the conversion levels or water content (ESI†). Adding radical scavengers into the reaction mixture led to leaching of the gold from the support. Therefore the role of radicals in the ethanol oxidation could not be determined.
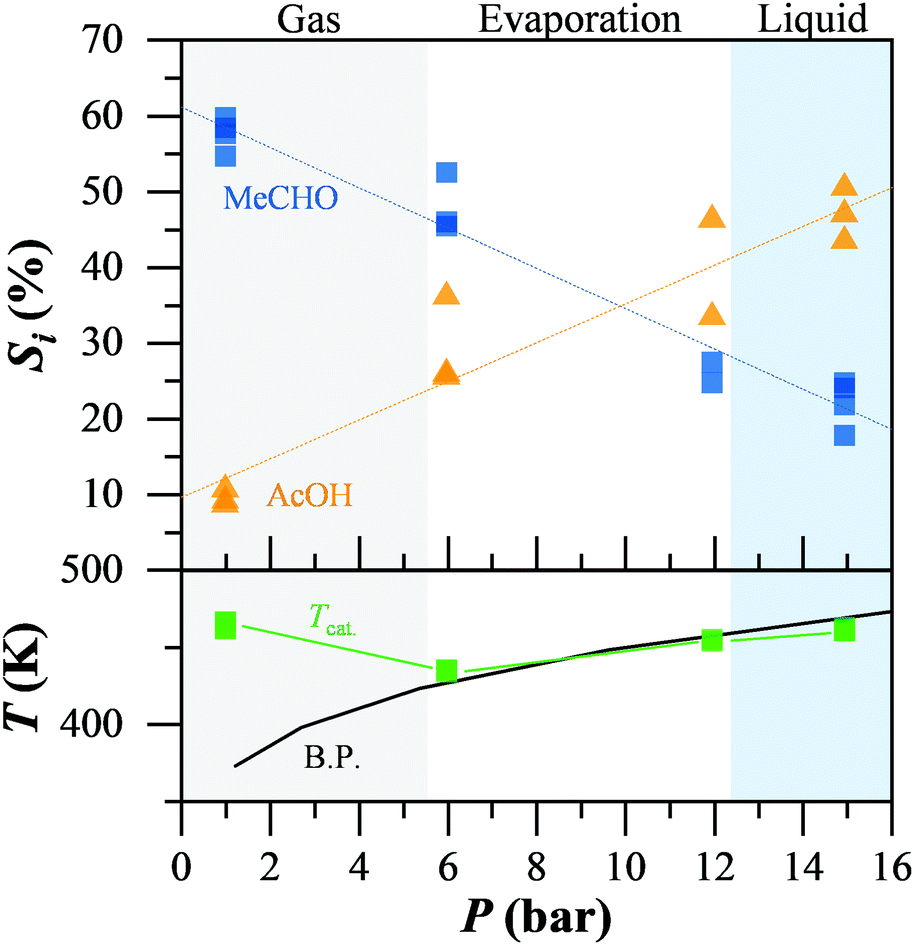 | ||
Fig. 2 Selectivity of acetic acid and acetaldehyde dependence on the phase of matter of the ethanol solution. The physical state of the ethanol solution (5% EtOHaq.) was identified by comparison of the temperature of the catalyst bed and the theoretical boiling point of the mixture at the operating pressure. Conditions in flow: 423 K, 15.0 bar, 5 ml min−1 O2, 0.3 ml min−1 5% EtOH/H2O, 0.3 mg fixed bed, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 catalyst:SiC. :1 catalyst:SiC. | ||
The results of ethanol and acetaldehyde oxidation revealed that, under the conditions required for catalytic ethanol oxidation, acetaldehyde oxidation proceeds non-catalytically (Fig. 1). There are three requirements for the non-catalytic oxidation of acetaldehyde: (a) the presence of oxygen, (b) the presence of water, and (c) operation in the liquid phase (Table 1). The non-catalytic acetaldehyde oxidation utilises liquid acetaldehyde at moderate oxygen pressure and relatively low temperature (353 K) for the production of acetic acid.14
The well-established literature indicates that a formyl group readily oxidises to the corresponding carboxyl group in oxygen/air or other oxidants, in an autoxidation cycle initiated by a radical initiator.15–20 Benzaldehyde is one of the most common examples and more extensively studied. The autoxidation cycle is initiated by a radical initiator, which breaks the relatively weak aldehyde C–H bond (86 kcal).17 The formed benzoyl radical reacts with oxygen forming benzoylperoxy radicals, which results in the formation of benzoic acid and one new benzyl radical. The formed acid is very resistant to direct reduction back to the aldehyde. Sankar et al. identified the radicals involved in the benzaldehyde oxidation, by adduct trapping and detection with electron paramagnetic resonance.20
The correlation between acetaldehyde and benzaldehyde requires caution due to the aromatic ring. Non-aromatic aldehydes are not as extensively studied, but their non-catalytic oxidation is precedent, e.g. octanal.21 The involvement of radicals is suggested because (a) the GC and NMR analysis detected the Criegee intermediate, (b) the addition of a radical trap suppressed the reaction, and (c) the acyl adduct was identified by electron paramagnetic resonance.21 An indication of radical involvement in our reaction is the suppression of the reaction under BHT (Table 1, entry 1.6), a radical scavenger that is used to suppress aldehyde autoxidation.13 The experimental identification of the intermediate species mentioned requires an extensive investigation in due course. Nonetheless, peracetic acid is a stable compound, industrially produced by liquid acetaldehyde and air; the acetic acid yield reaches up to 97% with acetic acid as solvent (Hoechst process).19 We exclude the possibility of acid–base catalysis, because the reaction is performed under neutral solutions.22 From this study, the nature of the active oxygen, such as singlet oxygen, oxygen radicals, and hydrogen peroxide, cannot be determined.23,24
Hence, the ethanol oxidation in the liquid-phase involves two consecutive steps, shown in Scheme 1: (1) the oxidative dehydrogenation of ethanol to acetaldehyde on the catalytic surface (reaction (1)) and (2) the non-catalytic, likely radical-involved, autoxidation of acetaldehyde to acetic acid in the liquid phase (reaction (2)). A catalytic pathway of acetaldehyde oxidation is possible depending on the catalyst and conditions. The oxidation of acetaldehyde over manganese or cobalt acetate is a well-established industrial route.1 Recent studies on the gaseous acetaldehyde oxidation over Pd–Au surfaces have shown high selectivity to acetic acid at low temperatures, but they observed the formation of by-products, such as CO2, above 375 K.25 In our system, we expect that the adsorption of acetaldehyde on the catalyst as acetate species can contribute towards the formation of carbon dioxide.5 As the ethanol-to-acetaldehyde is the only catalytic step, future catalytic designs for the liquid-phase ethanol oxidation should target the improvement of the oxidative dehydrogenation step and the inhibition of the acetaldehyde oxidation on the catalyst surface. Our discovery provides a reaction mechanism relevant for the oxidation of ethanol in the absence of a strong base and challenges the hitherto accepted reaction mechanism of two in-series reaction steps on the catalyst surface.7–9
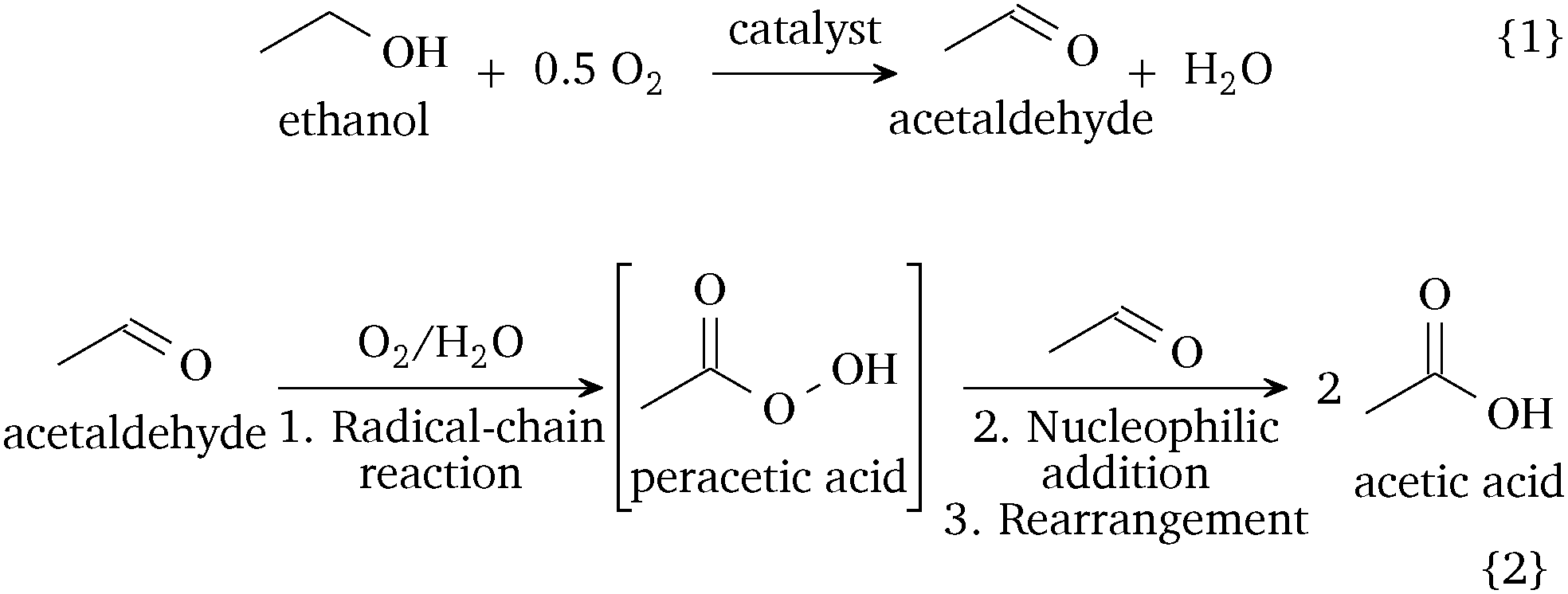 | ||
| Scheme 1 Proposed reaction mechanism of the catalytic liquid-phase oxidation of ethanol in a neutral environment. | ||
This study focuses on the reactivity of acetaldehyde, the intermediate of ethanol-to-acetic acid, under typical ethanol oxidation operating conditions. When operating in the liquid regime, acetaldehyde oxidises to acetic acid in the absence of a distinct heterogeneous catalyst, provided that oxygen and water are present. Based on the available mechanism of the non-catalytic oxidation of other aldehydes and radical scavenger experiments, the most plausible reaction mechanism is the radical initiated autoxidation cycle, which proceeds only in the liquid phase and not in the gas phase.
We evaluated the extent of the oxidation mechanism mentioned above in the formation of acetic acid during the catalytic ethanol oxidation by an immediate comparison of operation in the liquid and, respectively, the gas phase. We proposed that the liquid-phase oxidation of acetaldehyde is the main path to acetic acid in the ethanol oxidation. This work is a milestone in the field of heterogeneous catalysis of primary alcohol oxidation, which has until now considered that all reaction steps were occurring on the catalytic surface, leading to misguided reaction mechanisms and misdirected catalyst designs.
We want to express our gratitude to Dr Marc Newton, and Mr Petr Šot for fruitful discussions on the organic chemistry of aldehydes. We are grateful to Stefania Geladari for experimental support. We acknowledge the experimental support and ideas given by Dr Maximilian Moser. We thank Ms Janne Soetbeer and the Electron Paramagnetic Resonance group of ETH for discussions. Finally, we are genuinely thankful to Mr Joel Jenni, Mr Fredy Mettler, and the mechanical workshop of chemical engineering in ETH for their critical input and technical support on the experimental set-up. This research was funded by the Swiss National Science Foundation (SNF 200021E-158188) and by the Austrian Science Fund (FWF I2158-N28).
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Yoneda, S. Kusano, M. Yasui, P. Pujado and S. Wilcher, Appl. Catal., A, 2001, 221, 253–265 CrossRef CAS.
- C. H. Christensen, B. Jørgensen, J. Rass-Hansen, K. Egeblad, R. Madsen, S. K. Klitgaard, S. M. Hansen, M. R. Hansen, H. C. Andersen and A. Riisager, Angew. Chem., Int. Ed., 2006, 45, 4648–4651 CrossRef CAS.
- B. Jørgensen, S. Egholm Christiansen, M. L. Dahl Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–337 CrossRef.
- S. Mostrou, T. Sipcz, A. Nagl, B. Fdi, F. Darvas, K. Föttinger and J. A. Van Bokhoven, React. Chem. Eng., 2018, 3, 781–789 RSC.
- T. H. Tan, J. Scott, Y. H. Ng, R. A. Taylor, K. F. Aguey-Zinsou and R. Amal, ACS Catal., 2016, 6, 8021–8029 CrossRef CAS.
- V. I. Sobolev, K. Y. Koltunov, O. A. Simakova, A.-R. Leino and D. Y. Murzin, Appl. Catal., A, 2012, 433–434, 88–95 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- Q. Meng, Y. Shen, J. Xu, X. Ma and J. Gong, Surf. Sci., 2012, 606, 1608–1617 CrossRef CAS.
- Q. Gu, P. Sautet and C. Michel, ACS Catal., 2018, 8, 11716–11721 CrossRef CAS.
- K. Q. Sun, S. W. Luo, N. Xu and B. Q. Xu, Catal. Lett., 2008, 124, 238–242 CrossRef CAS.
- D. Heeskens, P. Aghaei, S. Kaluza, J. Strunk and M. Muhler, Phys. Status Solidi, 2013, 250, 1107–1118 CrossRef CAS.
- P. Aghaei and R. J. Berger, Appl. Catal., B, 2013, 133, 195–203 CrossRef.
- G. H. Foster, Process of making acetic acid, US Pat., US2009838A, 1930.
- G. H. Foster, Process of making acetic acid, 1930, https://patents.google.com/patent/US2009838#citedBy.
- E. J. Corey, N. W. Gilman and B. E. Ganem, J. Am. Chem. Soc., 1968, 90, 5616–5617 CrossRef CAS.
- C. F. Hendriks, H. C. A. Van Beek, P. M. Heertjes, C. F. Hendriks and H. C. A. Van Beek, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 271–275 Search PubMed.
- J. D. Roberts and M. C. Caserio, Basic Principles of Organic Chemistry, W.A. Benjamin, 1977, ch. CARBONYL C, p. 1596 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidation of Organic Compounds, Elsevier, 1981, ch. Oxidations, pp. 17–32 Search PubMed.
- K. Weissermel and H. Arpe, Industrial Organic Chemistry, Wiley, 2003, ch. Secondary, p. 491 Search PubMed.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332 CrossRef.
- L. Vanoye, A. Aloui, M. Pablos, R. Philippe, A. Percheron, A. Favre-Réguillon and C. de Bellefon, Org. Lett., 2013, 15, 5978–5981 CrossRef CAS.
- R. N. Hayes, R. P. Grese and M. L. Gross, J. Am. Chem. Soc., 1989, 111, 8336–8341 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- G. Li, Q. Yan, X. Gong, X. Dou and D. Yang, ACS Sustainable Chem. Eng., 2019, 7(16), 14009–14015 CrossRef CAS.
- S. Han, K. Shin, G. Henkelman and C. B. Mullins, ACS Catal., 2019, 9, 4360–4368 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05813c |
| This journal is © The Royal Society of Chemistry 2019 |