
Total synthesis of verucopeptin, an inhibitor of hypoxia-inducible factor 1 (HIF-1)†
Nobuaki
Takahashi
,
Hideaki
Hayashi
,
Viktors
Poznaks
and
Hideaki
Kakeya
 *
*
Department of System Chemotherapy and Molecular Sciences, Division of Bioinformatics and Chemical Genomics, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: scseigyo-hisyo@pharm.kyoto-u.ac.jp
First published on 18th September 2019
Abstract
Verucopeptin is an inhibitor of hypoxia-inducible factor 1 (HIF-1), which is a promising target for cancer chemotherapy. Here, we report the first total synthesis of verucopeptin via condensation of the depsipeptide core and the polyketide side chain unit including three branched methyl groups after the synthesis of each segment.
Tumor cells are usually exposed to hypoxic conditions or a starvation state. For survival and cell growth of tumor cells in these severe environments, hypoxia-inducible factor 1 (HIF-1) plays an important role as a transcriptional factor that regulates the expression of a number of genes involved in angiogenesis, gluconeogenesis, and metastasis.1,2 Therefore, HIF-1 is a promising target for cancer chemotherapy, and studies on HIF-1 by using a chemical and biological approach have been carried out in our group.3
For the purpose of identifying new HIF-1 inhibitors, we screened natural resources using a hypoxia-responsive luciferase reporter gene assay and we re-discovered verucopeptin (1) as a potent inhibitor of HIF-1 from a culture broth of Streptomyces sp. KUSC_A08.3b,4 Verucopeptin (1) consists of a cyclic depsipeptide core and a polyketide side chain including three branched methyl groups. Unlike the other derivatives such as azinothricin5a and dentigerumycin5b1 has a unique feature in that it exists in equilibrium between an open form and a closed form via its tetrahydropyran (THP) ring.
Recently, we determined the stereochemistry of 1 and revealed that 1 inhibits HIF-1 via the mTORC1 pathway.3b For elucidation of the mode of action of 1 in more detail, investigation of various derivatives of 1 is necessary. Although the synthesis of the peptide core has been reported by the Hale group,6 the total synthesis of 1 has not been achieved. To investigate its chemical and biological properties, we carried out the total synthesis of verucopeptin (1) and report it herein.
The summary of the total synthesis of 1 is shown in Scheme 1. Condensation of the depsipeptide core 2 and the side chain 3 followed by removal of the protective group for construction of the THP ring was carried out in the final stage. Carboxylic acid 3 was obtained by coupling alkyne 7 and aldehyde 6 following structural conversions. Aldehyde 6 was transformed from known alcohol 8. Alkyne 7 was obtained from alcohol 9, which was prepared from known monoacetate 10.7 Depsipeptide 2 was transformed from the linear peptide 4 by the same method as reported in Hale's work.6 Compound 4 was prepared by Fmoc-based solid phase peptide synthesis (SPPS) from Fmoc glycine loaded resin 5.
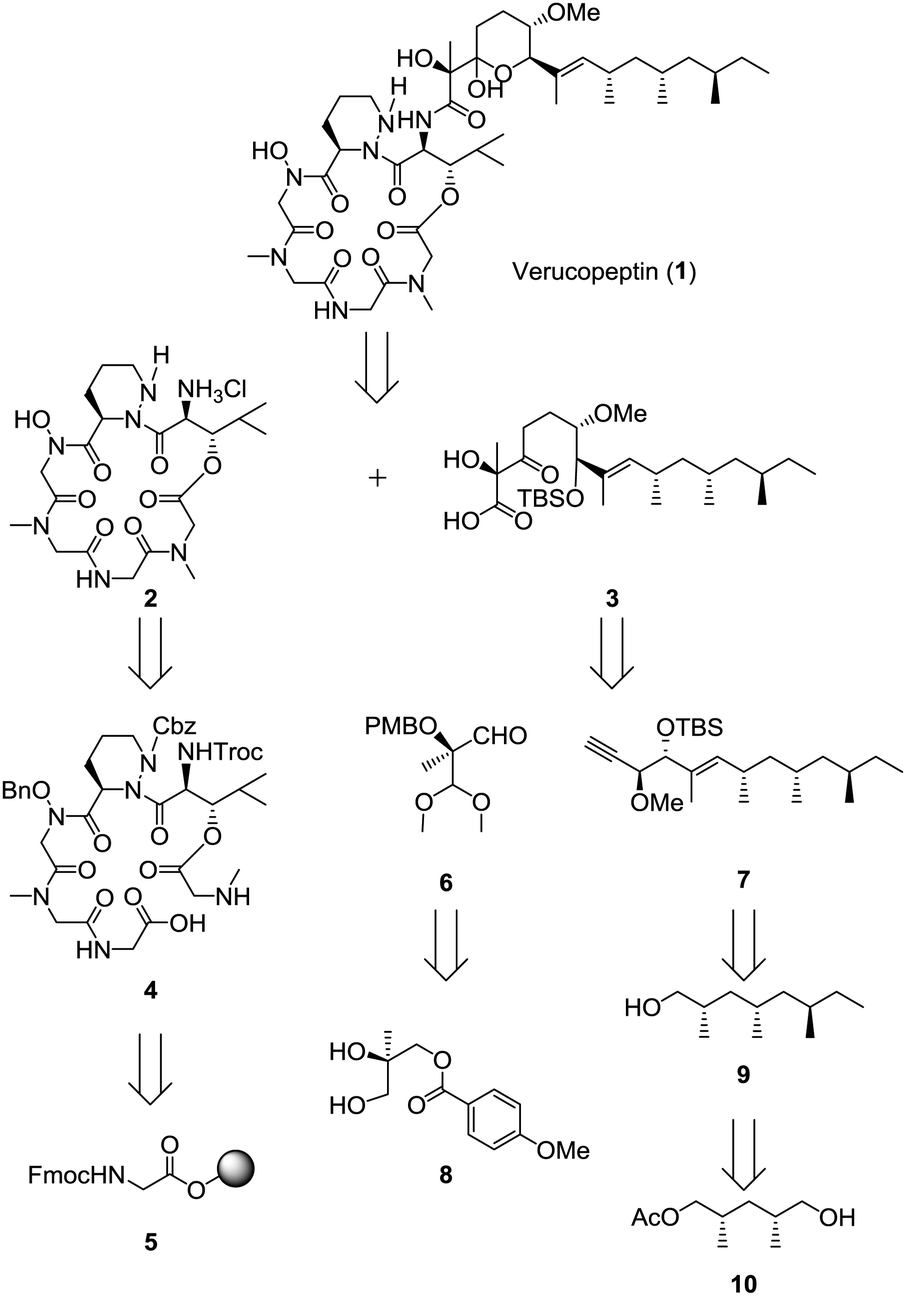 | ||
| Scheme 1 Retrosynthesis of verucopeptin (1). | ||
Alcohol 9 with three branched methyl groups was prepared from known monoacetate 107 in 6 steps (Scheme 2). After oxidation of 10 with 2-iodoxybenzoic acid (IBX), the resulting aldehyde was converted to propargyl alcohol 11 by Pd-catalyzed propargylation8 using chiral (S)-mesylate 12.9 To determine the absolute configuration, alcohol 11 was transformed to diol 13 whose enantiomer is a known compound10 and the spectral data, including the sign of optical rotation, were compared. After hydrogenation of the alkyne, the unnecessary hydroxy group was removed by radical deoxygenation in two steps. Next, hydrolysis of the acetyl group was conducted to afford alcohol 9 in 96% yield.
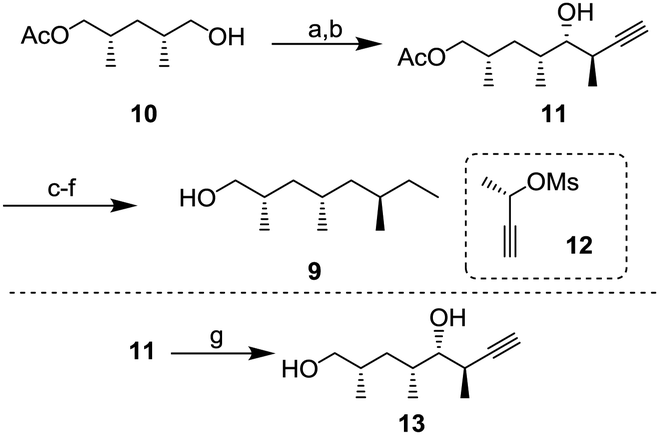 | ||
| Scheme 2 Synthesis of alcohol 9; (a) IBX, DMSO, r.t., 3 h, 85%; (b) 12, Pd(OAc)2, PPh3, ZnEt2, THF, −78 °C to −20 °C, 6 h, 70%; (c) Pd/C, H2, MeOH, r.t., 18 h, 85%; (d) phenyl chlorothionoformate, DMAP, pyridine, CH2CI2, r.t., 3.5 h, 74%; (e) nBu3SnH, AIBN, toluene, 100 °C, 16 h; (f) K2CO3, MeOH, r.t., 2 h, 96% (2 steps); and (g) K2CO3, MeOH, r.t., 3 h, 85%. | ||
Next, we focused on the construction of the two adjacent chiral centers of the methoxy group and allylic hydroxy group (Scheme 3). After oxidation of 9 with 85% yield, the resulting aldehyde was transformed to α,β-unsaturated ester 15 by the Wittig reaction using 14. Conversion of 15 into α,β-unsaturated aldehyde 16 was conducted by diisobutylaluminium hydride (DIBAL-H) reduction and followed by oxidation with 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) and PhI(OAc)2 with 75% yield (2 steps). Next, the aldehyde 16 was subject to the Evans aldol reaction with oxazolidinone 17. Under the typical conditions with boron enolate, the diastereomerically pure aldol product 18 was obtained in 96% yield. Next, reductive removal of the chiral auxiliary of 18 gave diol 1911 in 55% yield.
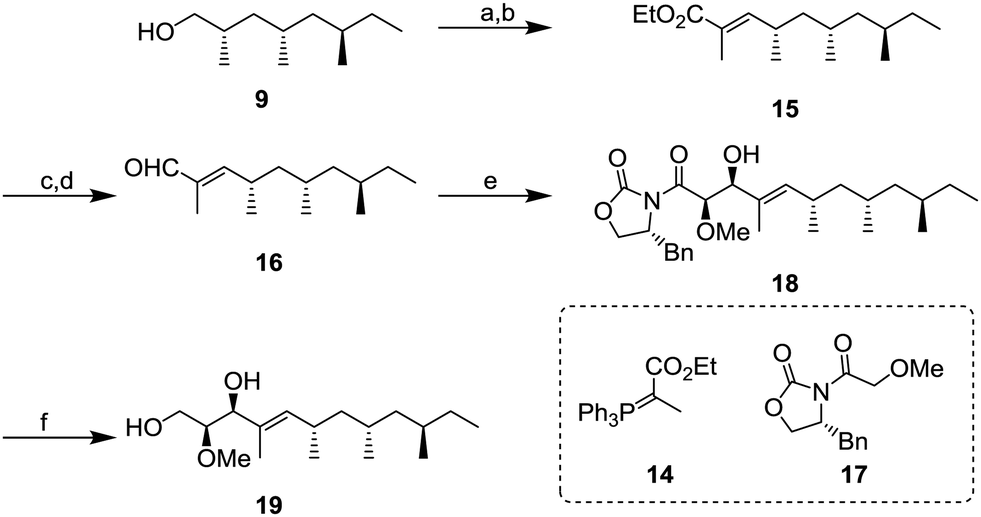 | ||
| Scheme 3 Synthesis of diol 6; (a) TEMPO, Phl(OAc)2, CH2Cl2, r.t., 3 h, 88%; (b) 14, CH3CN, 80 °C, 12 h, 75%; (c) DIBAL-H, toluene, −78 °C, 4 h; (d) DMP, CH2Cl2, r.t., 2 h, 75% (2 steps); and (e) 17, nBu2BOTf, NEt3, CH2Cl2, −78 °C to 0 °C, 1 h, 96%; (f) LiCl, NaBH4, EtOH, THF, r.t., 2 h, 55%. | ||
Next, the diol 19 was transformed to alkyne 7 for a coupling reaction with aldehyde 6 (Scheme 4). After protection of the primary alcohol with a tert-butyldimethylsilyl (TBS) group to obtain the intermediate in 81% yield,12 the Mitsunobu reaction was conducted to obtain benzoate 20 whose stereochemistry was the same as that of verucopeptin (1). The benzoate group was removed by methanolysis with K2CO3 and the resulting alcohol was protected by a TBS group to obtain a di-silylated compound 21. The compound 21 was converted to alkyne 7 by selective deprotection of the primary alcohol, Dess–Martin periodinane (DMP) oxidation, and alkyne formation using the Ohira–Bestmann reagent.13
 | ||
| Scheme 4 Synthesis of alkyne 7; (a) TBSCI, imidazole, CH2Cl2, r.t., 2 h, 81%; (b) PhCOOH, DEAD, PPh3, THF, r.t., 15 h, 95%; (c) K2CO3, MeOH, r.t., 12 h; (d) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 2 h, 98% (2 steps); (e) PPTS, EtOH, r.t., 16 h, 80%; (f) DMP, CH2Cl2, r.t., 2 h; and (g) dimethyl(1-diazo-2-oxopropyl)phosphonate, K2CO3, MeOH, r.t., 18 h, 58% (2 steps). | ||
Aldehyde 6 was prepared from known diol 814 as shown in Scheme 5. Protection of the diol by p-methoxybenzylidene acetal gave compound 22 in quantitative yields.15 After removal of the p-methoxybenzyl (PMB) ester group, TBS protection of the hydroxy group gave O-silylated compound 23. Next, reduction of the p-methoxybenzylidene acetal with DIBAL-H was conducted to afford 24 whose tertiary alcohol was protected by the PMB group. After oxidation of 24, the resulting aldehyde was transformed to a dimethyl acetal to generate 25. Finally, removal of the TBS group followed by oxidation gave the aldehyde 6.
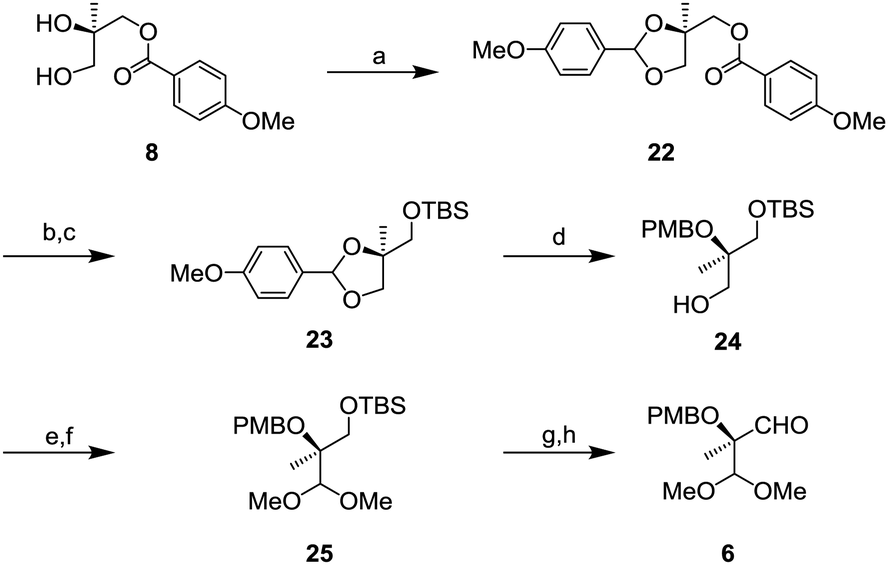 | ||
| Scheme 5 Synthesis of aldehyde 6; (a) p-methoxybenzaldehyde dimethyl acetal, PPTS, CH2Cl2, r.t., 18 h, 98%; (b) NaOMe, MeOH, r.t., 18 h; (c) TBSCI, imidazole, CH2Cl2, r.t., 16 h, 92% (2 steps); (d) DIBAL-H, CH2Cl2, 0 °C, 4 h, 36%; (e) SO3-pyridine, NEt3, DMSO, r.t., 2 h, 82%; (f) PPTS, trimethyl orthoformate, CH2Cl2, r.t., 16 h; (g) TBAF, THF, r.t., 16 h, 91% (2 steps); and (h) SO3-pyridine, NEt3, DMSO, r.t., 4 h, 69%. | ||
Completion of the side chain unit is described in Scheme 6. The addition of alkyne 7 to aldehyde 6 using lithium hexamethyldisilazide (LHMDS) gave 26 in 95% yield as a mixture of diastereomers. Oxidation of the resulting secondary alcohol with DMP and the subsequent hydrogenation of the triple bond were performed to obtain ketone 27. After deprotection of the dimethyl acetal in the presence of trimethylsilyl triflate (TMSOTf) and 2,4,6-collidine,16 the resulting aldehyde was oxidized to carboxylic acid 3 by the Pinnick oxidation.
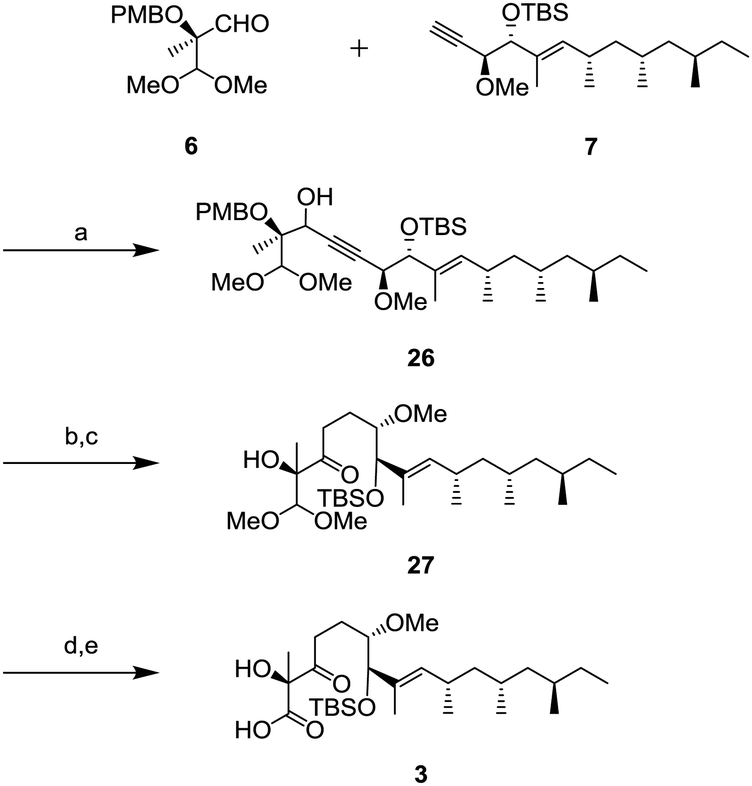 | ||
| Scheme 6 Synthesis of carboxylic acid 3; (a) LHMDS, −78 °C to 0 °C, 1 h, 95%; (b) DMP, CH2Cl2, r.t., 1 h, 96%; (c) Pd/C, AcOEt, r.t., 3 h; (d) TMSOTf, 2,4,6-collidine, CH2Cl2, 0 °C, 5 h; and (e) NaClO2, NaH2PO4 monohydrate, 2-methyl-2-butene, H2O, tBuOH, r.t., 3 h, 69% (3 steps). | ||
We started the synthesis of the depsipeptide core as shown below. The building blocks for SPPS are described in Scheme 7. Although resin 5 and Fmoc sarcosine 28 were commercially available, N-hydroxy glycine 29, piperazic acid 30 and dipeptide 31 needed to be synthesized. Among them, 3017 and 316 were synthesized by previously reported methods. However, N-hydroxy glycine 29 was an unknown compound, and thus it needed to be synthesized. From the tert-butyl ester 32, protection of the amino group with the Fmoc group and subsequent removal of the tert-butyl group under acidic conditions gave the amino acid 29.
 | ||
| Scheme 7 Building blocks of the depsipeptide core 3; (a) FmocCl, sat. NaHCO3 aq., 1,4-dioxane, r.t., 1 h, 83% and (b) TFA, CH2Cl2, r.t., 1 h, 90%. | ||
The SPPS was started from the Fmoc-glycine loaded Wang resin 5 (Scheme 8). Two cycles of removal of the Fmoc group using 20% piperidine/DMF and introduction of the amino acid (Fmoc-sarcosine 28 or hydroxy glycine 29) in the presence of a condensing agent gave the tripeptide 33. Then, synthesis of the tetrapeptide 34 was examined. After deprotection of the Fmoc group, the resulting free amine was condensed with piperazic acid 30 using O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), 1-hydroxy-7-azabenzotriazole (HOAt), and N,N-diisopropylethylamine (DIEA). Because the reactivity of the Nα-amine in the piperazic acid of 34 was remarkably low,18 the protection of the Nα-amine in 30 was not necessary under these conditions. In the final step of the SPPS, introduction of dipeptide 31 was investigated to generate the hexapeptide 36. After conversion of 31 to the acid chloride 35,6,18 a condensation reaction with 34 in toluene at 60 °C in the presence of AgCN gave 36. Next, removal from the resin and deprotection of the tert-butoxycarbonyl (Boc) group was conducted. Treatment with 98% trifluoroacetic acid (TFA) and purification by HPLC gave a pure linear peptide 4 in 36% yield from resin 5. Synthesis of the peptide core 2 from 4 was carried out by Hale's method.6 Macrolactamization, replacement of the 2,2,2-trichloroethoxycarbonyl (Troc) group to the benzyloxycarbonyl (Cbz) group, and removal of a benzyl group and two Cbz groups gave 2, whose spectral data were consistent with those in Hale's report.
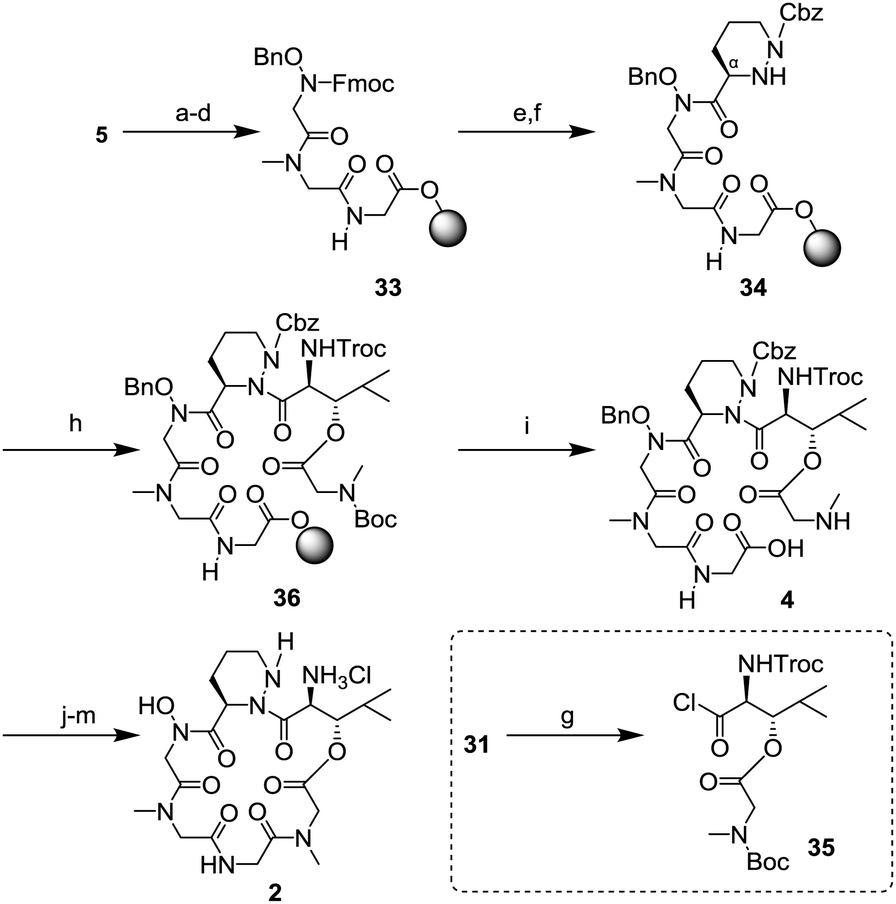 | ||
| Scheme 8 Synthesis of cyclic depsipeptide 2; (a) 20% piperidine/DMF, r.t., 1 h; (b) 28, DIC, HOBt, DIEA, DMF, r.t., 2 h; (c) 20% piperidine/DMF, r.t., 1 h; (d) 29, HATU, HOAt, DIEA, DMF, r.t., 1 h; (e) 20% piperidine/DMF, r.t., 1 h; (f) 30, HATU, HOAt, DIEA, DMF, r.t., 12 h; (g) (COCl)2 benzene, r.t., 2.5 h; (h) 35, AgCN, toluene, 60 °C, 20 min; (i) 98% TFA, r.t., 1 h, 36% (overall yield of Fmoc-SPPS); (j) HATU, N-ethyl morpholine, CH2Cl2 (0.0004 M), 0 °C to r.t., 48 h, 54%; (k) Zn, AcOH/H2O, r.t., 2 h; (l) CbzCl, 10% Na2CO3 aq., THF, r.t., 2 h, 79% (2 steps); and (m) Pd/C (degussa type), H2, AcCl, MeOH, r.t., 24 h, 92%. | ||
The final stage of the synthesis is shown in Scheme 9. The depsipeptide core 2 and the side chain unit 3 were combined in the presence of 1H-benzotriazol-1-yloxy-tri(pyrrolidino)phosphonium hexafluorophosphate (PyBop) and trimethylamine (NEt3) to afford compound 37. In the last step, the TBS group of 37 was removed to generate verucopeptin (1) in 23% (2 steps). The spectral data of synthetic 1 were consisted with those of the natural compound.
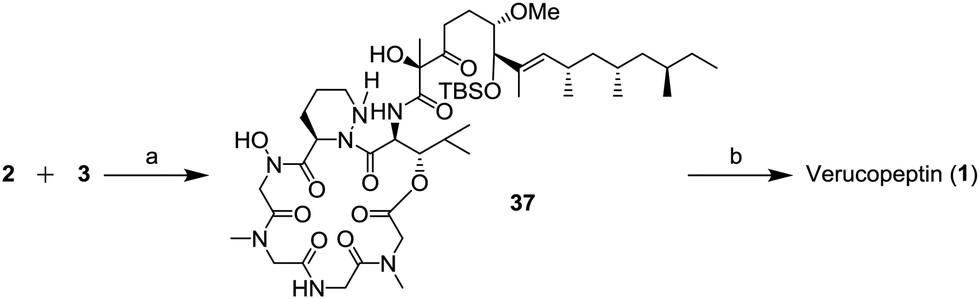 | ||
| Scheme 9 Completion of the total synthesis of verucopeptin (1); (a) PyBop, NEt3, CH2Cl2, −78 °C to r.t., 3 h and (b) 1 N HCl aq., THF, r.t., 8 h, 23% (2 steps). | ||
In conclusion, verucopeptin (1) was successfully synthesized. First, the side chain unit 3 was created via construction of six chiral centers, whereas the Fmoc-SPPS unit was subjected to macrolactamization to obtain the depsipeptide core 2. In the final stage, 2 and 3 were coupled to complete the first total synthesis of 1. Taking advantage of the convergent properties in our present synthetic scheme, derivatization of the synthetic product by changing the partial structure in the units is underway for a structure–activity relationship (SAR) study and elucidation of its mode of action.
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (17H06401 and 19H02840) and a research grant (JP19fk0310112 and JP19am0101092) from the Japan Agency of Medical Research and Development (AMED), Japan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. L. Semenza, Annu. Rev. Pathol.: Mech. Dis., 2014, 9, 47 CrossRef CAS PubMed.
- S. Koyasu, M. Kobayashi, Y. Goto, M. Hiraoka and H. Harada, Cancer Sci., 2018, 109, 560 CrossRef CAS PubMed.
- (a) Y. Yasuda, T. Arakawa, Y. Nawata, S. Shimada, S. Oishi, N. Fujii, S. Nishimura, A. Hattori and H. Kakeya, Bioorg. Med. Chem., 2015, 23, 1776 CrossRef CAS PubMed; (b) A. Yoshimura, S. Nishimura, S. Otsuka, A. Hattori and H. Kakeya, Org. Lett., 2015, 17, 5364 CrossRef CAS PubMed; (c) H. Kakeya, Nat. Prod. Rep., 2016, 33, 648 RSC.
- A. Yoshimura, S. Kishimoto, S. Nishimura, S. Otsuka, Y. Sakai, A. Hattori and H. Kakeya, J. Org. Chem., 2014, 79, 6858 CrossRef CAS PubMed.
- (a) H. Maehr, C. M. Liu, N. J. Palleroni, J. Smallheer, L. Todaro, T. H. Williams and J. F. Blount, J. Antibiot., 1986, 39, 17 CrossRef CAS PubMed; (b) D. C. Oh, M. Poulsen, C. R. Currie and J. Clardy, Nat. Chem. Biol., 2009, 5, 391 CrossRef CAS PubMed.
- K. J. Hale, L. Lazarides and J. Cai, Org. Lett., 2001, 3, 2927 CrossRef CAS PubMed.
- J. C. Anderson, S. V. Ley and S. P. Marsden, Tetrahedron Lett., 1994, 35, 2087 CrossRef CAS.
- (a) J. A. Marshall and N. D. Adams, J. Org. Chem., 1998, 63, 3813 Search PubMed; (b) J. A. Marshall and N. D. Adams, J. Org. Chem., 1999, 64, 5201 CrossRef CAS; (c) Y. Tamaru, S. Goto, A. Tanaka, M. Shimizu and M. Kimura, Angew. Chem., Int. Ed. Engl., 1996, 35, 878 CrossRef CAS.
- J. R. Frost, C. M. Pearson, T. N. Snaddon, R. A. Booth, R. M. Turner, J. Gold, D. M. Shaw, M. J. Gaunt and S. V. Ley, Chem. – Eur. J., 2015, 21, 13261 CrossRef CAS PubMed.
- M. Jiménez, W. Zhu, A. Vogt, B. W. Day and D. P. Curran, Beilstein J. Org. Chem., 2011, 7, 1372 CrossRef PubMed.
- To confirm whether we obtained the syn aldol product, diol 19 was converted to an acetonide-protected compound and an NOE experiment was conducted (for details see the ESI†).
- The absolute configuration of the allylic position generated by the Evans aldol reaction was determined by a modified Mosher method (for details see the ESI†).
- (a) S. Ohira, Synth. Commun., 1989, 19, 561 CrossRef CAS; (b) S. Müller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 13 Search PubMed.
- T. Nagamitsu, D. Takano, M. Seki, S. Arima, M. Ohtawa, K. Shiomi, Y. Harigaya and S. Omura, Tetrahedron, 2008, 64, 8117 CrossRef CAS.
- The conversion of 8 to 22 was carried out using the same method with the case of the enantiomer; J. Xie and D. A. Horne, Tetrahedron Lett., 2009, 50, 4485 CrossRef CAS.
- A. Ahlers, T. de Haro, B. Gabor and A. Fîrstner, Angew. Chem., Int. Ed., 2016, 55, 1406 CrossRef CAS PubMed.
- Y. Chen, Y. Lu, Q. Zou, H. Chen and D. Ma, Org. Process Res. Dev., 2013, 17, 1209 CrossRef CAS.
- A. J. Oelke, D. J. France, T. Hofmann, G. Wuitschik and S. V. Ley, Nat. Prod. Rep., 2011, 28, 1445 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, procedures for the syntheses of verucopeptin (1) and copies of 1H- and 13C-NMR spectra. See DOI: 10.1039/c9cc06169j |
| This journal is © The Royal Society of Chemistry 2019 |