
Comparative study of the mobility of Pd species in SSZ-13 and ZSM-5, and its implication for their activity as passive NOx adsorbers (PNAs) after hydro-thermal aging†
Jaeha
Lee
a,
YoungSeok
Ryou
a,
Sungha
Hwang
 a,
Yongwoo
Kim
a,
Sung June
Cho
b,
Hyokyoung
Lee
c,
Chang Hwan
Kim
c and
Do Heui
Kim
*a
a,
Yongwoo
Kim
a,
Sung June
Cho
b,
Hyokyoung
Lee
c,
Chang Hwan
Kim
c and
Do Heui
Kim
*a
aSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul, 151-744, Rep. of Korea. E-mail: dohkim@snu.ac.kr
bClean Energy Technology Laboratory and Department of Applied Chemical Engineering, Chonnam National University, Gwangju 500-757, Rep. of Korea
cHyundai-Kia Motors R&D center, Hwaseong, 445-706, Rep. of Korea
First published on 22nd November 2018
Abstract
The comparative behavior of Pd species in ZSM-5 and SSZ-13 zeolite supports resulting from thermal treatment in the presence and absence of water was studied. Thermal treatment at 750 °C caused the atomic distribution of Pd species in ZSM-5, while clustered Pd species remained in SSZ-13. The addition of water in the feed during thermal treatment resulted in the Pd sintering in the Pd/ZSM-5 catalyst, although Pd dispersion and agglomeration were concurrently observed in the Pd/SSZ-13 catalyst, depending on the Pd loading. The prepared catalysts were vacuum-treated to induce the auto-reduction of the atomic Pd species, and to compare the mobility of the Pd species within the different zeolite frameworks. The isolated Pd species in ZSM-5 were completely reduced to form large Pd clusters after vacuum treatment, while most of the Pd species in SSZ-13 remained atomically dispersed, indicating that the Pd mobility is higher in the former than in the latter. Hence, the different behavior of Pd/ZSM-5 and Pd/SSZ-13 catalysts against the oxidative and hydrothermal treatments could be explained by the intrinsic difference in the Pd mobility within the different types of zeolite. It is likely that the smaller pore size of SSZ-13 than ZSM-5 contributed to the lesser Pd mobility in the former. The hydrothermal treatment deactivated the NO adsorption ability of the other prepared Pd/zeolite catalysts with larger pore openings (Beta and Modernite zeolites), which further supports the present proposal.
1. Introduction
Precious-group metal (PGM)-supported zeolite catalysts have gained much attention over the past several decades, because of their potential applicability to various kinds of reactions; a few examples include the selective catalytic reduction of NOx (SCR),1–5 methane oxidation,6–8 and hydrogenation.9–11 The use of zeolite as a supporting material of PGMs is beneficial in several respects. First, high PGM dispersion could be attained on zeolites relatively easily, because of the large pore volume and the high surface area of zeolites, as well as the strong ionic interaction between PGMs and Al species.12,13 In addition, the strong electrostatic interaction between PGMs and Al species in zeolite could benefit the catalytic activities of catalysts. For example, Tang et al. reported that the electronic interaction between Pd and Beta zeolite enhances the sulfur tolerance of the Pd species, which promoted the deep hydrogenation of aromatics.10 Therefore, the proper utilization of zeolite as a supporting material of PGMs could be an attractive strategy in preparing the catalysts with high catalytic performance.In the automotive industry, reducing the emission of NOx from the lean-burn engine is a great issue. NOx storage reduction (NSR) and selective catalytic reduction (SCR) catalysts require a temperature higher than 250 °C to eliminate the NOx species from exhaust gas.14–19 Therefore, the majority of NOx is reported to be emitted during the cold start period of the vehicle operation, i.e., before the NSR or SCR catalysts reach their operating temperature.20,21 One strategy to reduce the NOx emission is to store NO at low temperature, and to release it at high temperature, when the SCR and NSR catalysts can eliminate NOx. The catalysts with this concept are frequently referred to as cold-start catalysts (CSCs) or passive-NOx absorbers (PNAs).22,23 Several catalysts have been investigated as candidate materials to attain this purpose; among them, Pd/CeO2-based catalysts are reported to have good functionality.22 However, the CeO2-based catalysts are vulnerable to the exposure to SO2.24–26 The catalytic activities are completely lost when the catalysts are sulfur aged, and only a portion of the functionality could be restored after the regeneration process.27 Recently, Pd/zeolite catalysts have been reported to have good PNA functionality, while they can also endure the sulfur aging processes.28 The interaction between the Pd ions in zeolite and SO2 seems to be weak, rendering the catalysts with high sulfur tolerance, originating from the intimate interaction between Pd ions and Al species in the zeolite.10 Therefore, Pd/zeolite catalysts would be a strong candidate material as cold-start catalysts.28–34
Chen et al. reported that the ability to store and release NOx over Pd/zeolite catalysts changes significantly depending on the types of zeolite.28 This indicates that the interaction between Pd ions and zeolite is the critical factor in determining the catalytic functionalities.28 We recently reported that oxidative treatment at the high temperature of 750 °C is necessary to activate the catalytic activity of Pd/ZSM-5.31 On the other hand, for the case of Pd/SSZ-13, the addition of water at elevated temperature is essential for catalytic activation.30 In both cases, the atomically dispersed Pd species were found to be the active sites involved in adsorbing NO.30,31 Because catalysts are exposed to water at elevated temperature in real-world applications, understanding the behavior of Pd species in zeolite at high temperature in the presence of water is required. Therefore, comparing the behavior of Pd species in ZSM-5 and SSZ-13 under harsh hydrothermal aging conditions is meaningful.
The comparative behavior of the Pd species in ZSM-5 and SSZ-13 is studied in the present work. The catalysts were exposed to the elevated temperature of 750 °C, and the effect of the presence of water on the behavior of Pd species was extensively studied by applying various characterization methods, such as X-ray diffraction (XRD), N2 adsorption/desorption, Raman spectroscopy, extended X-ray absorption fine-structure spectroscopy (EXAFS), X-ray adsorption near-edge spectroscopy (XANES), CO-chemisorption, inductively coupled plasma-atomic emission spectroscopy (ICP-AES), high-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM), magic-angle spinning-nuclear magnetic resonance (MAS-NMR), and chemisorptive NO adsorption/desorption.
2. Experimental
Catalyst preparation
The Pd precursor was impregnated into the zeolite according to previously reported procedures but with some variations.31 Ammonium (NH4)-SSZ-13 (Zeolyst) with a Si to Al2 molar ratio (SARs) of 35 and ammonium (NH4)-ZSM-5 with SARs of 30 and 50 (Alfa Chemicals, U.K.) were ion-exchanged with palladium(II) nitrate hydrate, Pd(NO3)2(H2O)2 (Sigma-Aldrich, U.S.A.). If Pd(NO3)2(H2O)2 corresponding to 2 wt% Pd was dissolved in water and ion-exchanged in SSZ-13, it is denoted as “Pd(2)/SSZ-13”. Table S1 of the ESI† lists the actual Pd loading and the Pd-to-Al molar ratios of the prepared catalysts, based on the ICP-AES analysis. Most of the Pd precursors dissolved in water were successfully loaded in zeolite. The SAR of zeolite is denoted after the sample name; for example, if Pd was loaded on SSZ-13 with an SAR of 35, then it is named as “Pd/SSZ-13 (35)”. Afterward, the prepared catalysts were oxidized at 500 °C or 750 °C for 2 h under a 100 mL min−1 flow of 15% O2/N2. To indicate this, “500C” and “750C” were attached after the sample name and are also used to refer to the treatments. For example, if Pd(2)/SSZ-13 was oxidized at 750 °C, then it is named as “Pd(2)/SSZ-13 750C.” The catalysts were also oxidized at 750 °C for 25 h both in the presence and absence of water (10% H2O, and 15% O2). The former and the latter are designated as HTA and 750C(25 h) after the sample name, respectively. ‘HTA’ represents that the catalysts are ‘hydrothermally aged’ to simulate catalysts operated under the harsh condition. In order to compare the stability of Pd species in ZSM-5 and SSZ-13, vacuum treatment was also carried out at 300 °C for 4 h. Detailed discussion is provided in section 3.2.Catalyst evaluation
The PNA ability of the catalysts was evaluated according to previously reported procedures.31 The catalyst samples of 0.035 g and 0.1 g of the alpha-phase Al oxide (α-Al2O3) were blended. The catalysts were pretreated at 500 °C for 30 min with water (5%) and oxygen (10%), balanced with N2 at a flow rate of 200 mL min−1 (GHSV of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). After cooling the sample temperature to 120 °C, 100 ppm of NO was introduced to the lean-gas mixture for 100 s. Afterward, the temperature was raised to 500 °C at the ramping rate of 10 °C min−1, where it was maintained at 500 °C for 30 min to completely remove the stored NOx. The NOx concentration was measured using a 42i-HL NOx analyzer (Thermo Fisher Scientific, U.S.A.). Fig. S1 of the ESI† shows the NOx concentrations profiles on the Pd(2)/SSZ-13 (35) 750C HTA catalyst during the NO adsorption/desorption test. The total NOx amount estimated during the NO adsorption/desorption was similar to the amount of NO introduced into the reactor. Since the error was small (less than 2%), the amount of desorbed NOx during the temperature ramping could be regarded as the amount of adsorbed NOx on the catalyst at 120 °C.
000 h−1). After cooling the sample temperature to 120 °C, 100 ppm of NO was introduced to the lean-gas mixture for 100 s. Afterward, the temperature was raised to 500 °C at the ramping rate of 10 °C min−1, where it was maintained at 500 °C for 30 min to completely remove the stored NOx. The NOx concentration was measured using a 42i-HL NOx analyzer (Thermo Fisher Scientific, U.S.A.). Fig. S1 of the ESI† shows the NOx concentrations profiles on the Pd(2)/SSZ-13 (35) 750C HTA catalyst during the NO adsorption/desorption test. The total NOx amount estimated during the NO adsorption/desorption was similar to the amount of NO introduced into the reactor. Since the error was small (less than 2%), the amount of desorbed NOx during the temperature ramping could be regarded as the amount of adsorbed NOx on the catalyst at 120 °C.
X-ray diffraction (XRD)
XRD patterns were obtained at a voltage and current of 40 kV and 30 mA with Cu-Kα radiation (λ = 0.1542 nm) using a Mode 1 Smartlab diffractometer (Rigaku, Japan). A scanning-step size of 0.02° at a speed of 2.5° min−1 was used to collect the patterns in the 2θ range from 5° to 90°. The XRD scan rate was decreased to 0.2° min−1 to detect the XRD peaks from small Pd oxide (PdO).Extended X-ray absorption fine-structure spectroscopy (EXAFS)
The XAFS data of the catalyst over the Pd K-edge (24350 eV) were collected using the 7D-XAFS beamline of the Pohang Light Source (PLS-II) at the Pohang Accelerator Laboratory (South Korea). A Si(111) crystal monochromator was used, and the beam energy and the ring current were 2.5 GeV and 300 mA, respectively. Pd foil was used for the energy calibration (E0 = 24350 eV). The EXAFS data were analyzed by the Athena and Artemis (Demeter, U.S.A.).35 Detailed analytical procedures can be found in the previous publication.31
X-ray adsorption near-edge spectroscopy (XANES)
The linear combination fitting (LCF) was performed on XANES data after the background subtraction and normalization, by using the Athena software package to measure the oxidative states of the Pd species. Pd foil and PdO were used as standard materials. The mean square sum of the misfit at each data point was reduced, to minimize the discrepancy between the raw data and the fitted model.Raman spectroscopy
Raman-spectroscopy analysis was conducted using a T64000 Raman spectrometer (Horiba Jobin Yvon, Japan) with an argon (Ar)-ion laser (λ = 514.5 nm) at the National Center for Inter-University Research Facilities (South Korea).Inductively coupled plasma-atomic emission spectroscopy (ICP-AES)
The ICP-AES analysis was performed using an Optima-4300 DV spectrometer (PerkinElmer, U.S.A.).Scanning electron microscopy (SEM)
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) investigations were carried out with a JSM-6360 (JEOL) instrument with an accelerating voltage of 20 kV. Prior to analysis, Pt metal was coated on the samples with MSC-101 (JEOL). Fig. S2 of the ESI† shows the results. It was observed that the average size of the zeolite agglomerates was in the order: ZSM-5 (50) < SSZ-13 (35) < ZSM-5 (30).High-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM)
HAADF-STEM and energy-dispersive spectroscopy (EDS) analysis were performed at an accelerating voltage of 200 kV in a JEM-2100F (JEOL) electron microscope equipped with a field emission gun.Magic-angle spinning-nuclear magnetic resonance (MAS-NMR)
A WB Avance II 500 MHz (Bruker, Germany) system with a 4 mm MAS probe was used for 27Al and 29Si MAS-NMR experiments. A spinning frequency of 10 kHz, a spectral window of 65 kHz, complex points of 6572, and a pulse delay of 0.1 s were used to obtain 512 time-averaged scans. The Si to Al2 molar ratio was calculated from the 29Si MAS-NMR spectra. In addition, the Al-NMR and Si-NMR spectra in Fig. S3 and S4,† respectively, of the ESI† indicate that the hydrothermal treatment caused the de-alumination of the zeolite structures. The Al-NMR spectra in Fig. S3† shows that after the hydrothermal treatment, the peak intensity from the tetrahedral Al species in the framework at −60 ppm decreased, while the peak intensity from the octahedral Al species in the extra-framework at −0 ppm increased. The Si-NMR spectra in Fig. S4† also show that the peak intensity at −105 ppm from the Si species coordinated to the framework Al species decreased, while the peak intensity from the Si species at −113 ppm not coordinated to the framework Al species increased. A similar extent of de-alumination appeared to occur in the Pd/ZSM-5 and Pd/SSZ-13 catalysts after the hydrothermal treatment.N2 adsorption/desorption
N2 adsorption/desorption isotherms were measured at liquid-N2 temperature (−196 °C) using ASAP 2010 (Micromeritics Instruments, U.S.A.) apparatus. Before analysis, the catalysts were vacuum-treated at 300 °C for 12 h. The BJH and BET methods were used to measure the mesopore volume and the specific surface area, respectively. The single point adsorption at P/P0 = 0.97 was used to measure the pore volume with a pore diameter smaller than ∼70 nm. The micropore volume was calculated by subtracting the mesopore volume from the single point pore volume. Table 1 lists the results.| Sample | BET surface area (mb g−1) | Micro-pore volumea (cmc g−1) | Total pore volumeb (cmc g−1) | Si/Al2 | |
|---|---|---|---|---|---|
| a Total pore volume – meso-pore volume. b Single point adsorption total pore volume of pores less than 69.6 nm diameter at P/P0 = 0.97. c Measured from SEM-EDX, averaged over 10 images. d Si/Al2f, framework Al (Alf) measured from Si-MAS-NMR. | |||||
| ZSM-5 (30) | 396 | 0.16 | 0.22 | 30.4 ± 2.1c (36.3)d | |
| Pd(2)/ZSM-5 (30) | 355 | 0.13 | 0.23 | ||
| 750C HTA | |||||
| ZSM-5 (50) | 393 | 0.17 | 0.24 | 50.7 ± 3.3c (58.2)d | |
| Pd(2)/ZSM-5 (50) | 374 | 0.17 | 0.24 | ||
| 750C HTA | |||||
| SSZ-13 (35) | 686 | 0.26 | 0.27 | 35.3 ± 3.1c (44.2)d | |
| Pd(2)/SSZ-13 (35) | 723 | 0.27 | 0.30 | ||
| 750C HTA | |||||
CO chemisorption
Static CO chemisorption was measured on a Micromeritics ASAP 2010 following the previously reported procedures.36,37 A sample of 0.12 g was placed in a U-shaped quartz tube. Samples were reduced with H2 gas at 350 °C followed by the evacuation at 350 °C. The temperature was cooled to 35 °C and CO chemisorption was performed. After obtaining the first CO isotherm, the sample was evacuated at 35 °C for 4 h to remove the physisorbed CO. The second CO isotherm estimates the amount of physisorbed CO. The amount of chemisorbed CO is represented by the difference between the first and the second isotherms.3. Results and discussion
3.1 Influence of the thermal treatment at 750 °C in the presence and absence of water
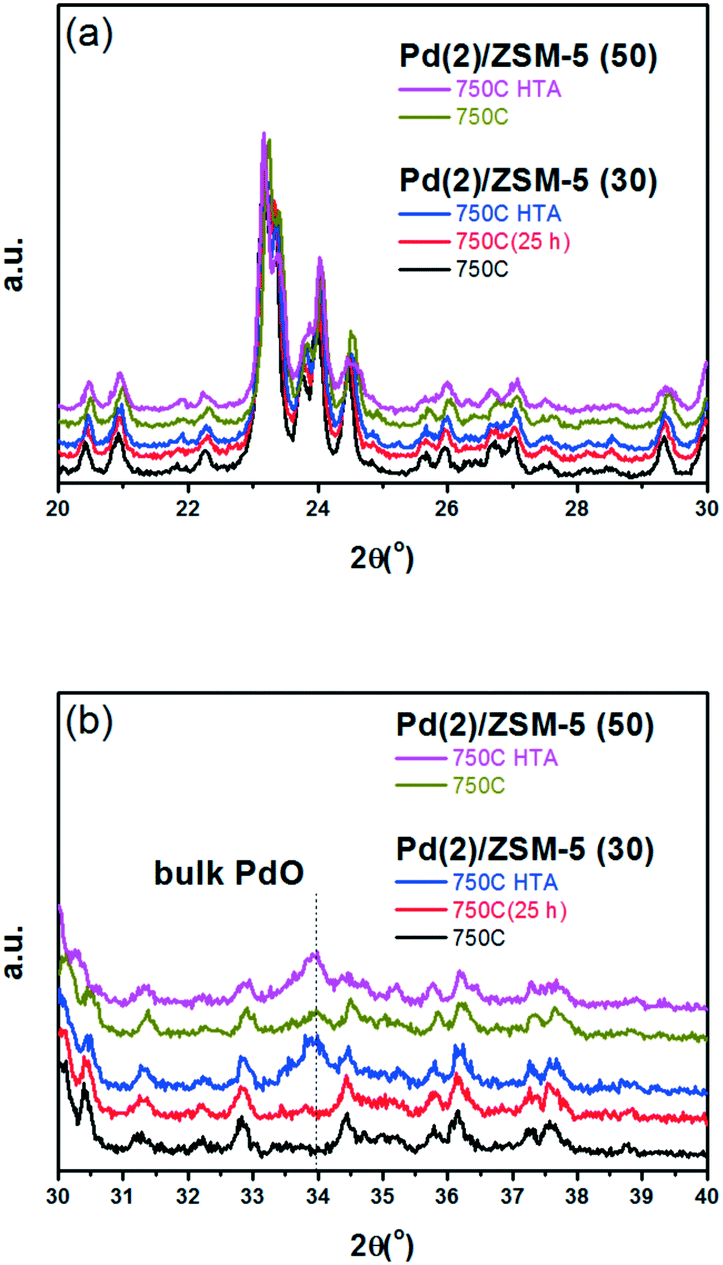 | ||
| Fig. 1 XRD patterns of Pd(2)/ZSM-5 (30 and 50) catalysts, after the oxidative at 750 °C, or after the HTA treatment. (a) The 2θ region between 20° and 30° was magnified to observe the change in XRD pattern from ZSM-5 support after different thermal treatments. (b) The 2θ region between 30° and 40° was magnified to observe the change in XRD pattern arising from the bulk PdO phase after different thermal treatments. | ||
A similar set of thermal treatments was performed on the Pd/SSZ-13 catalysts, and their XRD patterns are displayed in Fig. 2. Fig. 2a shows that the SSZ-13 structure was preserved after the thermal treatments at 750 °C, both in the presence and absence of water. The XRD peak arising from the PdO phase could be observed at 34.0° after the hydrothermal treatment, while it was not observable after the thermal treatment without water (Fig. 2b). However, the XRD patterns from the bulk PdO and the SSZ-13 structure overlapped with each other at 34.0°, which made it difficult to distinguish the XRD peak from the bulk PdO. Therefore, the Raman analysis was performed on the Pd/SSZ-13 catalysts to observe the evolution of the Raman shift at 650 cm−1 assigned to the bulk PdO.42 In this manner, the formation of the bulk PdO was expected to be more clearly observable without the interference from the SSZ-13 structure. Indeed, the Raman spectra in Fig. S6† show that the formation of the bulk PdO was accelerated only after the hydrothermal treatment.
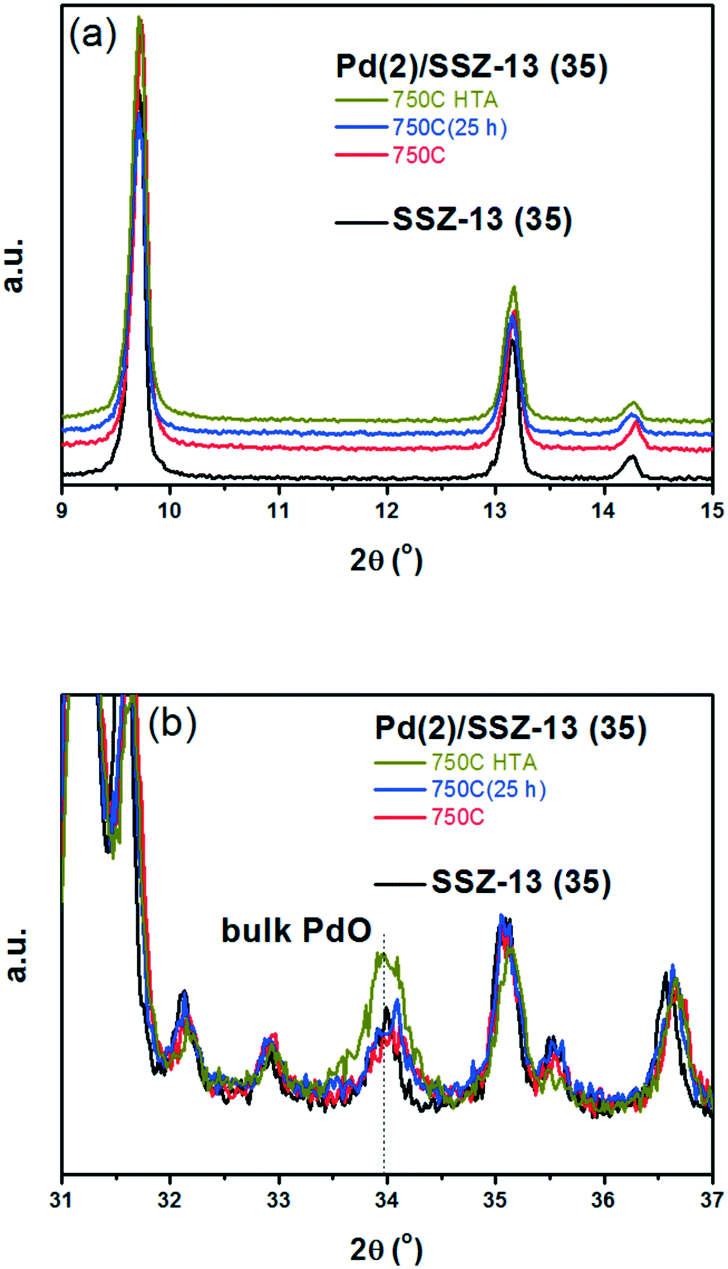 | ||
| Fig. 2 XRD patterns of Pd(2)/SSZ-13 (35) after the oxidative at 750 °C, or after the HTA treatment. (a) The 2θ region between 9° and 15° was magnified to observe the change in XRD pattern from SSZ-13 support after different thermal treatments. (b) The 2θ region between 31° and 37° was magnified to observe a change in the XRD pattern arising from the bulk PdO phase after different thermal treatments. | ||
Fig. 3 displays the EXAFS spectra of the Pd(2)/SSZ-13 and Pd(2)/ZSM-5 catalysts and their analyses. EXAFS oscillations are displayed in Fig. 3a, and their Fourier transforms in R-space are displayed in Fig. 3b and c for the Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts, respectively. Table S2 of the ESI† also provides the fitting results of the EXAFS spectra. Fig. 3b shows that after the 750C treatment, Pd(2)/ZSM-5 with SARs of 30 only displayed the first shell derived from the Pd–O bond, whereas those with SARs of 50 revealed the second shell from the Pd–O–Pd bond. The second shell should originate from the formation of the bulk PdO, while the first shell would originate from the bulk PdO, the isolated PdO species, or the interaction of the isolated Pd2+ ions with the framework oxygen species.13 This represents that, after the thermal treatment without water, the Pd species were well dispersed in ZSM-5 with a relatively high Al content (SARs of 30), while the bulk PdO was formed in ZSM-5 with a relatively low Al content (SARs of 50), consistent with the XRD results in Fig. 1. After the hydrothermal treatment, the second shell intensity from the Pd–O–Pd bonds increased in the Pd(2)/ZSM-5 catalysts, regardless of their SARs. This indicates that water promoted the Pd sintering. Even after the hydrothermal treatment, the intensity of the second shell was lower in Pd(2)/ZSM-5 with the higher Al content, implying its important role in the Pd–Al interaction. In the case of Pd(2)/SSZ-13, after the 750 °C treatment, only the first shell was observed, while after the hydrothermal treatment, the intensity of the second shell increased. All the XRD (Fig. 1b), Raman (Fig. S6 of the ESI†), and EXAFS (Fig. 3c) results unambiguously confirmed that the hydrothermal treatment formed PdO particulates in SSZ-13.
 | ||
| Fig. 3 (a) k3 weighted Pd K-edge EXAFS oscillations and their Fourier transforms for (b) Pd(2)/ZSM-5 (30 and 50) catalysts, and (c) Pd(2)/SSZ-13 (35) catalysts. EXAFS oscillation and its Fourier transform for the bulk PdO are also plotted in the figure as a reference. | ||
 | ||
| Fig. 4 NOx desorption curves of (a) Pd(2)/ZSM-5 (30, 50), and (b) Pd(2)/SSZ-13 (35) catalysts, after various thermal treatments. (c) Comparison of the PNA ability of Pd(2)/ZSM-5 (30) and Pd(2)/SSZ-13 (35) catalysts, after various thermal treatments. | ||
Fig. 4b shows the NOx adsorption/desorption profiles of the Pd/SSZ-13 catalysts with SARs of 35. After the 500C treatment, the NO adsorption ability of the catalyst was not activated. In contrast to Pd/ZSM-5, the 750C treatment did not activate the NO adsorption ability of the Pd/SSZ-13 catalysts (Fig. 4b and c). Only after the hydrothermal treatment could Pd/SSZ-13 adsorb a significant amount of NO; the NO storage ability of Pd(2)/SSZ-13 increased significantly from 3.3 to 25.4 μmol g−1. The presence of water in the feed played the crucial role in activating the NO adsorption ability, since the oxidative treatment without water for the same period of time (25 h) did not activate the NO adsorption ability (Table 2). The NOx adsorption/desorption profiles displayed in Fig. 4 appear to be inconsistent with the characterization results displayed in the previous section, where the PdO bulk was formed in both Pd(2)/ZSM-5 and Pd(2)/SSZ-13, which is inactive in adsorbing NO (Fig. 1 and 2).31
| Catalysts | NOx desorbed (μmol gcatal−1) (750C catalysts) | ||
|---|---|---|---|
| Below 300 °C | Above 300 °C | Total amount | |
| Pd(2)/ZSM-5 (30) 500C | 0.3 | 4.3 | 4.6 |
| Pd(2)/ZSM-5 (30) 750C | 8.9 | 15.6 | 24.5 |
| Pd(2)/ZSM-5 (50) 750C | 0.0 | 4.6 | 4.6 |
| Pd(2)/ZSM-5 (30) 750C(25 h) | 5.7 | 17.7 | 23.4 |
| Pd(2)/ZSM-5 (30) 750C HTA | 2.4 | 9.5 | 11.9 |
| Pd(2)/ZSM-5 (50) 750C HTA | 1.0 | 6.5 | 7.5 |
| Pd(2)/SSZ-13 (35) 500C | 0.0 | 0.2 | 0.2 |
| Pd(1)/SSZ-13 (35) 750C | 0.0 | 1.7 | 1.7 |
| Pd(2)/SSZ-13 (35) 750C | 0.0 | 3.3 | 3.3 |
| Pd(2)/SSZ-13 (35) 750C(25 h) | 1.9 | 5.6 | 7.5 |
| Pd(1)/SSZ-13 (35) 750C HTA | 11.1 | 6.9 | 18.0 |
| Pd(2)/SSZ-13 (35) 750C HTA | 10.3 | 15.1 | 25.4 |
To reduce the effect of Pd loading on the sintering behavior of Pd and to more vividly observe the effect of hydrothermal aging on the PNA ability, the Pd loading was reduced to 1 wt% in Pd/SSZ-13. Fig. 5 shows the EXAFS spectra of the Pd(1)/SSZ-13 catalysts before and after the hydrothermal treatment. Unlike Pd(2)/SSZ-13, the hydrothermal treatment did not induce the formation of bulk PdO in Pd(1)/SSZ-13, according to the Fourier transforms of EXAFS oscillations in Fig. 5b. In addition, HAADF-STEM images of Pd(1)/SSZ-13 in Fig. 6 and S7 of the ESI† clearly show that the bulk PdO disappeared after the hydrothermal treatment through the re-dispersion of Pd species, in agreement with our previous reports.30 Even though the bulk PdO species were observed in TEM images in Fig. 6a before the hydrothermal treatment, the Pd–O–Pd shell was not observed in the R-space of EXAFS oscillations in Fig. 5b. Since the X-ray adsorption is a bulk technique, one possibility is that the majority of the Pd species were atomically distributed, while some Pd species were locally aggregated into PdO bulk; the X-ray interference would not occur sufficiently on the small-sized PdO species formed in a small amount. The HTA treatment activated the NO adsorption ability of Pd/SSZ-13 catalysts with the Pd loading of 1 and 2 wt% (Fig. 7b). In addition, the same catalytic activity was observed when the prepared catalyst was directly hydrothermally treated without the thermal treatment, indicating that the thermal treatment was not a critical step for the adsorption activity (Fig. S8 of the ESI†). At the Pd loading of 1 wt%, the NOx desorption peak at the lower temperature (∼250 °C) was more dominant than the one at the higher temperature (∼400 °C). On the other hand, in the Pd(2)/SSZ-13 catalyst, the amount of NOx desorbed at the high temperature was more dominant than the one at the lower temperature (Fig. 7b). This indicates that both the amount of NOx stored, and the adsorption strength of the stored NOx in the catalyst, were influenced by the amount of isolated Pd species. When comparing Pd/SSZ-13 catalysts with the Pd loading of 1 and 2 wt%, the maximum capacity of SSZ-13 to maintain the Pd species in the isolated state seemed to lie in between 1 and 2 wt%. Below that value, all the Pd species loaded on SSZ-13 would become atomically distributed after the hydrothermal treatment, while above that value, the formation of bulk PdO would be inevitable.
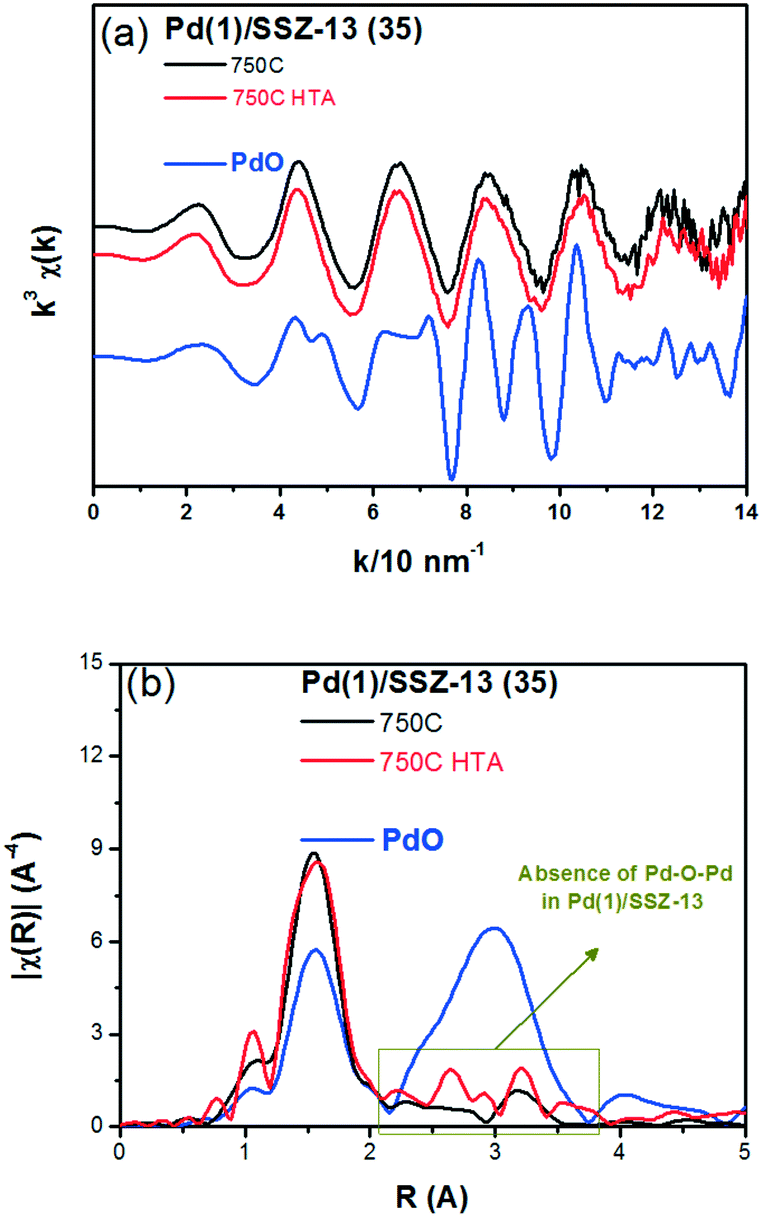 | ||
| Fig. 5 (a) k3 weighted Pd K-edge EXAFS oscillations, and (b) Fourier transforms for the Pd(1)/SSZ-13 (35) catalysts and the bulk PdO (as reference). | ||
 | ||
| Fig. 6 HAADF-STEM images of (a) Pd(1)/SSZ-13 (35) 750C, and (b) Pd(1)/SSZ-13 (35) 750C HTA catalysts. | ||
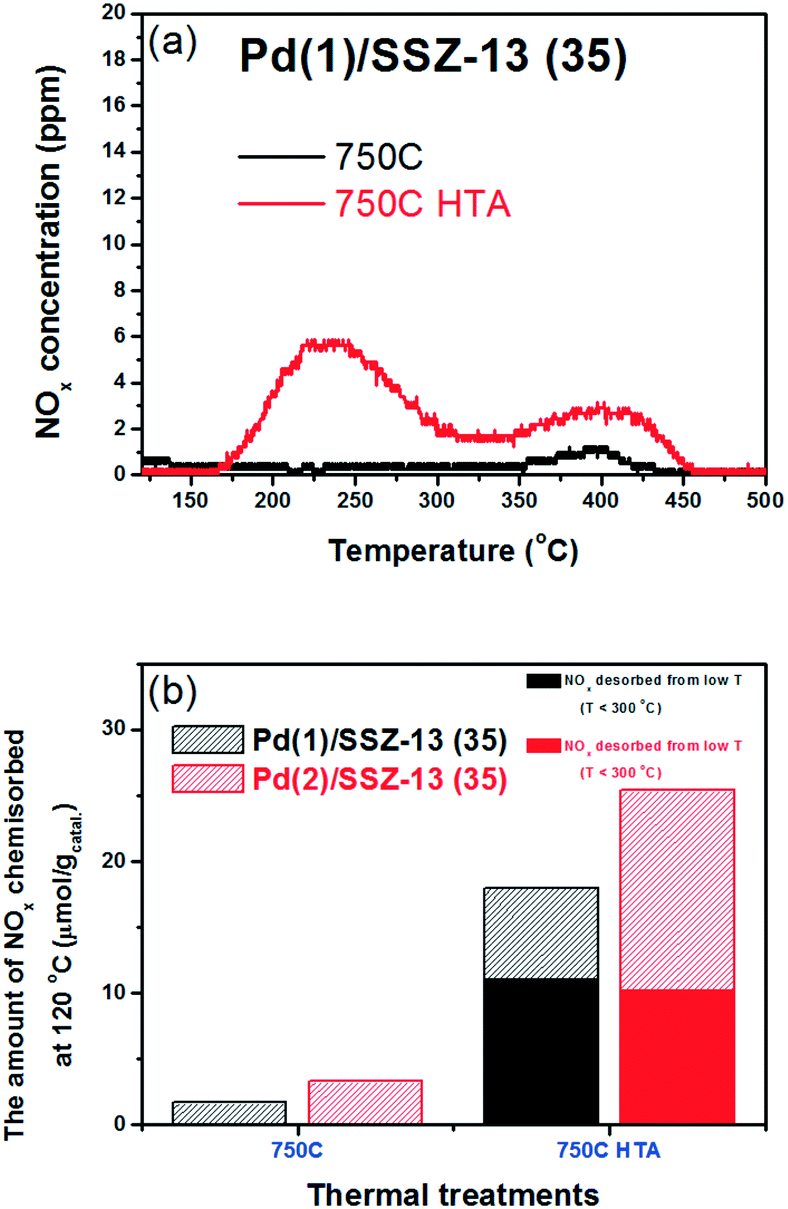 | ||
| Fig. 7 (a) NOx desorption curves of Pd(1)/SSZ-13 (35) catalysts. (b) Comparison of the PNA ability of Pd/SSZ-13 (35) catalysts with the Pd loading of (1 and 2) wt%. | ||
The NO adsorption capacity of Pd/SSZ-13 catalysts after 1 h adsorption of NO (fully adsorbed) was also evaluated. The maximum Pd to NO molar ratios were 0.58 and 0.41 for Pd(1)/SSZ-13 and Pd(2)/SSZ-13 catalysts, respectively (Table 3). The higher portion of Pd species in the Pd(1)/SSZ-13 catalyst appears to be able to participate in the PNA capability more than in the Pd(2)/SSZ-13 catalyst. This can be understood by the fact that clustered PdO species that do not chemisorb NO under the conditions used in this study are present in Pd(2)/SSZ-13 but not in the Pd(1)/SSZ-13 catalyst. Also, despite the absence of clustered PdO species in Pd(1)/SSZ-13, the maximum NO to Pd molar ratio was less than 1. This indicates that some Pd species in SSZ-13 cannot chemically adsorb NO even in the cationic state. A recent DFT study by Mei et al. also highlighted the various chemical states that the Pd species in BEA zeolite can have, and the different reactivities of the Pd species to the adsorption of NO.43 Last, in Table 3, it could be observed that only 33% of the available Pd species in SSZ-13 could be exploited when exposed to 100 ppm of NO for 100 s. This may indicate that the reactivity of Pd species in SSZ-13 should be improved to chemisorb NO more swiftly to achieve a better PNA ability.
| Catalysts (750C HTA) | (α) NO adsorbed (100 s) (μmol g−1) | (β) NO adsorbed (1 h) (μmol g−1) | NO to Pd molar ratio (100 s) | NO to Pd molar ratio (1 h) | α/β |
|---|---|---|---|---|---|
| Pd(1)/SSZ-13 (35) | 18.0 | 54.2 | 0.19 | 0.58 | 0.33 |
| Pd(2)/SSZ-13 (35) | 25.4 | 77.0 | 0.14 | 0.41 | 0.33 |
Before discussing the relationship between the Pd–zeolite interaction and the PNA ability, the origin of each NOx desorption peak of Pd/ZSM-5 and Pd/SSZ-13 catalysts should be discussed. Both catalysts showed two NOx desorption peaks in a similar temperature range: one below 300 °C and the other above 300 °C. Atomically dispersed cationic Pd species (Pd2+) would be the NO adsorption sites in the Pd/SSZ-13 catalysts,30,44 but atomically dispersed PdO and Pd2+ would contribute to the low and high temperature NOx desorption peaks of Pd/ZSM-5 catalysts, respectively, because a considerable amount of NO2 desorption was observed at the low temperature desorption peak of Pd/ZSM-5 (Fig. S9†).31
3.2 Investigation of the Pd–zeolite interactions by the auto-reduction technique
The behavior of the Pd species in SSZ-13 seems surprising, considering that the presence of water facilitated the aggregation of Pd species into PdO bulk in the Pd/ZSM-5 catalyst, as observed in the XRD patterns in Fig. 1 and the EXAFS spectra in Fig. 3. These opposite behaviors of the Pd species in ZSM-5 and SSZ-13 may be caused by the differences in the interaction between the Pd species and different kinds of zeolites. Such differences in the strength of interaction between Pd species and zeolites could be expected to cause differences in the mobility of Pd species in different kinds of zeolite. It has been reported that the vacuum treatment of the metal-exchanged zeolite catalysts leads to the auto-reduction of the cationic metals by the loss of the interaction with the framework Al species.45,46 Such treatment is known to reduce the cationic species, leading to metal sintering.47 Therefore, the strength of the interaction between the Pd species and different kinds of zeolites was investigated by performing vacuum treatment, and by observing the resultant Pd distributions. The Pd(2)/ZSM-5 (30) 750C and Pd(2)/SSZ-13 (35) 750C catalysts were thus vacuum-treated at 350 °C to compare the behavior of the Pd species in both zeolites.After the vacuum treatment, the pore structure of Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts were preserved. The surface area and the pore volume of the catalysts were also maintained from the vacuum treatment according to the N2 adsorption/desorption analysis. The XRD patterns also showed that the structures of the catalysts were not changed by the vacuum treatment (data not shown). The XRD patterns in the range 30° to 44° were fine-scanned, as shown in Fig. S10 of the ESI,† to observe the evolution of the metallic Pd clusters. The broad peak from the Pd metal was observed in Pd(2)/ZSM-5 after the vacuum treatment. However, the XRD pattern from the SSZ-13 structure overlapped with that from the Pd metal clusters. Therefore, the Pd K-edge XAFS spectra of the two catalysts were obtained to compare the distribution and chemical states of Pd species after the vacuum treatment.
Fig. 8 shows the EXAFS spectra of Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts. In Fig. 8b, the evolution of the Pd–Pd shell from the metallic Pd clusters in the R-space of the EXAFS oscillation at ∼2.4 Å was obvious in both the Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts after the vacuum treatment. The intensity of the peak was higher on the former than on the latter. The fitting results in Table S2 of the ESI† indicate that the coordination numbers of the Pd–Pd shell were 6.90 and 2.94 for the Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts, respectively, implying that the size of the Pd clusters formed in the former seems to be much larger than those in the latter.48 In addition, in Fig. 8b, the first shell from the Pd–O bond completely disappeared after the vacuum treatment on Pd/ZSM-5, while the shell was still visible on Pd/SSZ-13. Since the shell from the Pd–O–Pd bond was not observed on both Pd/ZSM-5 and Pd/SSZ-13 after the vacuum treatment in Fig. 8b, the possibility of having bulk PdO as the dominant species could be excluded. Therefore, the presence of Pd–O bonds indicates that after the vacuum treatment, most of the Pd species were atomically dispersed on Pd/SSZ-13. The XANES spectra in Fig. 9 compare the oxidation states of the Pd species in the Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts after the vacuum treatment; the XANES data was subjected to linear combination fitting (LCF) to estimate the oxidative states of the Pd species, and the results were inserted in Fig. 9 (see the Experimental section for detail). Fig. 9a shows that the chemical states of the Pd species became completely metallic by the vacuum treatment in Pd(2)/ZSM-5. On the other hand, Fig. 9b shows that only 34% of the Pd species became metallic after the vacuum treatment in Pd(2)/SSZ-13; 66% of the Pd species remained oxidized, even after the vacuum treatment. Therefore, the XANES spectra in Fig. 9 shows that the interaction between the Pd species and zeolite was more robust against the vacuum treatment in Pd/SSZ-13 than in Pd/ZSM-5. Consistent with the observations made from the EXAFS spectra, the CO chemisorption results in Fig. S11† also indicate that after the reductive treatment at 350 °C followed by the vacuum treatment at 350 °C, a larger amount of CO was chemisorbed on the Pd(2)/SSZ-13 catalyst than on the Pd(2)/ZSM-5 catalyst, indicating that the Pd dispersion was higher in the former after the vacuum treatment.
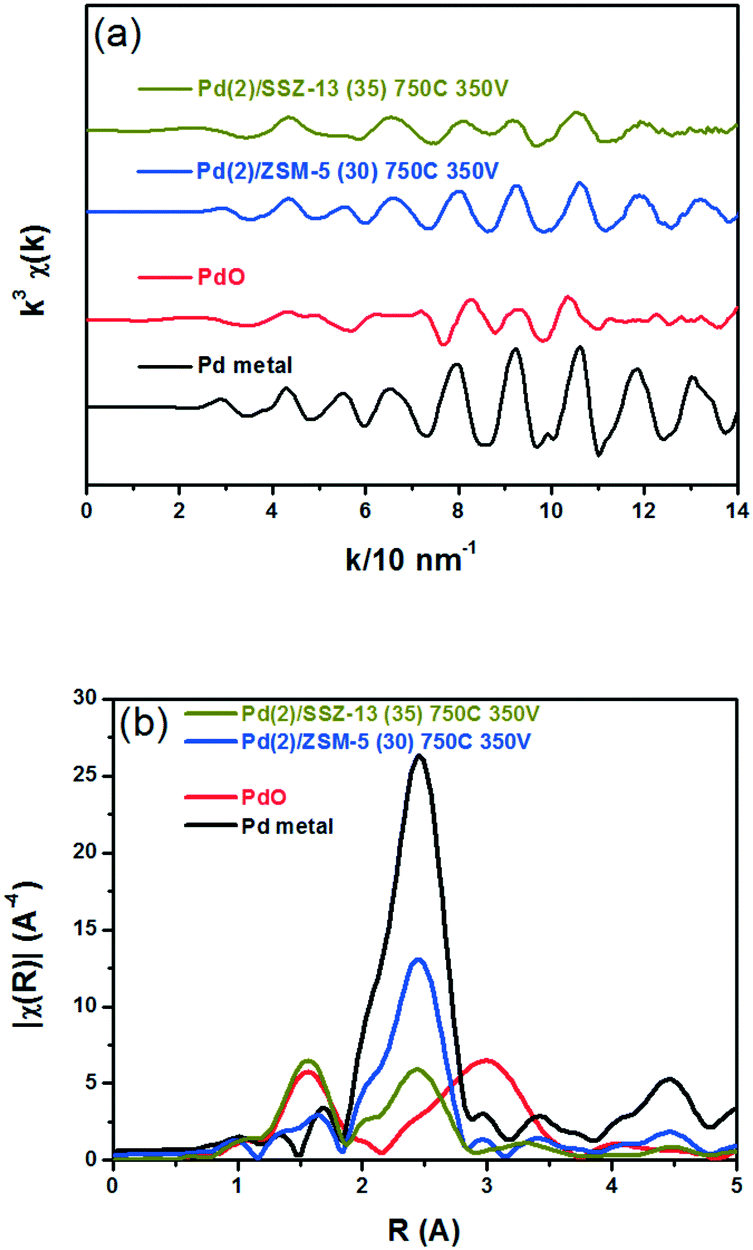 | ||
| Fig. 8 (a) k3 weighted Pd K-edge EXAFS oscillations, and (b) their Fourier transforms for Pd(2)/ZSM-5 (30) 750C 350 V and Pd(2)/SSZ-13 (35) 750C 350 V samples. Spectra from the bulk PdO and the metallic Pd are also included as references. | ||
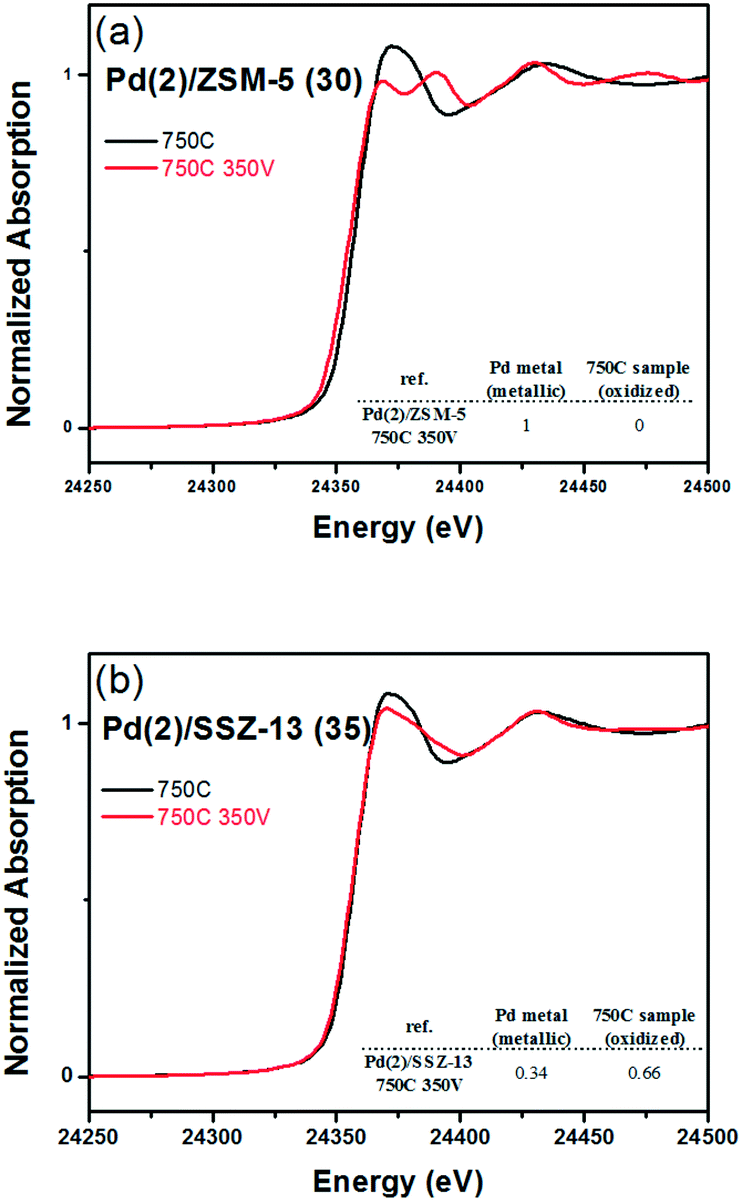 | ||
| Fig. 9 XANE spectra of (a) Pd(2)/ZSM-5 (30) 750C & Pd(2)/ZSM-5 (30) 750C 350 V, and (b) Pd(2)/SSZ-13 (35) 750C & Pd(2)/SSZ-13 (35) 750C 350 V samples. Metallic Pd and 750 °C treated samples were used as the reference materials for the linear fitting analysis. | ||
Fig. 10 shows the HAADF-STEM images of the Pd(2)/ZSM-5 and Pd(2)/SSZ-13 catalysts after the vacuum treatment. In Pd(2)/ZSM-5, the vacuum treatment led to the formation of the large Pd clusters. Interestingly, the Pd species in ZSM-5 aggregated linearly, the length of which extended to almost 50 nm. The size of the Pd clusters was much larger than the size of the pore diameter of ZSM-5. Therefore, these Pd clusters may be present at the external surface of the zeolite, or at the boundary of the zeolite particles. The EDX analysis of the Pd(2)/ZSM-5 catalyst after the vacuum treatment in Fig. S12 of the ESI† shows that the Pd particles were distinctly formed. Some of the representative images of the Pd(2)/ZSM-5 catalyst after the vacuum treatment are displayed in Fig. S13 of the ESI.† Very different behaviors were observed for the Pd species in SSZ-13 after the vacuum treatment. Fig. 10b shows that the Pd clusters in SSZ-13 were relatively small in size, which were dispersed like speckles. Some Pd clusters with the size up to ∼10 nm were frequently observed as well, but most of the Pd clusters were very small in size, and their size distribution was difficult to estimate. The EDX analysis of the Pd(2)/SSZ-13 catalyst after the vacuum treatment in Fig. S14 of the ESI† shows that the boundaries of the Pd particles were blurred by their small size. Some of the representative images of the Pd(2)/SSZ-13 catalysts after the vacuum treatment are displayed in Fig. S15 of the ESI.† Since 66% of the Pd species remained oxidized after the vacuum treatment according to the XANES spectra in Fig. 9, it is likely that the isolated Pd species and the small-sized Pd clusters coexisted in SSZ-13.
 | ||
| Fig. 10 HAADF-STEM image of (a) Pd(2)/ZSM-5 (30) 750C 350 V, and (b) Pd(2)/SSZ-13 (35) 750C 350 V catalysts. | ||
Combining the analysis results show that the Pd species loaded on the SSZ-13 were less mobile than those loaded on the ZSM-5, probably because of the stronger interaction between the Pd species and the framework Al species in the former catalyst. After the 750C treatment, the Pd species in ZSM-5 became mobile, and could move to the sites where they could adsorb NO. However, the Pd sintering did not occur under the oxidative atmosphere, because the mobility of the Pd species in ZSM-5 would be spatially limited. On the other hand, the Pd species in SSZ-13 were not mobile under the oxidative atmosphere at 750 °C; neither the Pd sintering nor the Pd dispersion was observed in Pd/SSZ-13. When water was added to the feed, the PdO bulk was formed in Pd/ZSM-5. Previous reports suggested that the mobile Pd(OH)x species may be formed in the presence of water.38–41 The radius of the space that the Pd species in ZSM-5 could move would be widened in the presence of water, which would increase the chance for the Pd species to collide with each other, to cause Pd sintering. However, the addition of water induces an atomic distribution of Pd species in Pd/SSZ-13. Since the Pd species were strongly bound in SSZ-13, they might require water to break the strong bonding with the framework Al species to move to the sites where they could adsorb NO. However, above the maximum capacity of the SSZ-13 support to maintain the atomically dispersed Pd species, which lay between 1 and 2 wt% of Pd, Pd sintering would be inevitable.
Such intrinsic difference in the behavior of the Pd species in ZSM-5 and SSZ-13 may originate from the difference in the size of the pore opening of the zeolite. The ionic diameter of the Pd2+ ion was about 0.20 nm. The pore sizes of the 6- and 8-membered rings of SSZ-13 were around 0.22 and 0.39 nm, respectively, while the pore size of the 10 membered ring of ZSM-5 was around 0.53 nm.49 Therefore, the Pd species might encounter a higher energy barrier to penetrate the SSZ-13 structure with a smaller pore diameter.50 The behavior of the Pd species in the other types of zeolites with the larger pore openings also indicates that the Pd penetration through the structure was facile, where the hydrothermal treatment induced the catalytic deactivation (Table S3;† beta zeolite, and modernite were tested). Therefore, it can be concluded that the zeolite structure played a crucial role in determining the behavior of the Pd species against the exposure to the various thermal environments.
The suppressed Pd mobility in small pore zeolite SSZ-13 has significant implications for the use of the catalysts as a PNA material in real-world applications. Once the catalysts are loaded in the vehicle, it must remain functional for as long as possible. In addition, exhaust treatment catalysts loaded in diesel vehicles are exposed to high temperature (∼750 °C) during particulate filter regeneration.51,52 Therefore, the catalytic activity should be examined after HTA treatment at high temperature for a prolonged time, to test the practical applicability of the catalysts.53 The PNA activities of Pd/zeolite catalysts with medium and large pore openings (beta zeolite, modernite, and ZSM-5) were substantially reduced after HTA treatment. However, after HTA treatment, the PNA ability of Pd/SSZ-13 catalyst with the small pore opening was activated. Accordingly, when it comes to long-term durability for practical applications, the Pd/SSZ-13 catalyst would be a better choice.
Conclusions
Pd species were ion-exchanged into ZSM-5 and SSZ-13 zeolite supports, and then thermally treated in the absence and presence of water. It turned out that the oxidation at 750 °C distributed the PdO bulk in ZSM-5 into the atomic Pd species, but the treatment did not activate the mobility of the Pd species in SSZ-13. When the water was added to the feed, the Pd species in ZSM-5 agglomerated into the PdO bulk, leading to the loss of most of the NO adsorption ability. The Pd species in SSZ-13 responded very differently to the water treatment. For the Pd loading of 1 wt%, the Pd species became atomically distributed after the hydrothermal treatment, but for the Pd loading of 2 wt%, the Pd aggregation and the Pd distribution occurred concurrently. Therefore, the maximum capacity of SSZ-13 to hold the atomically dispersed Pd species might lie in between 1 and 2 wt%. In order to compare the interaction between the Pd species and the framework Al species, the catalysts were vacuum treated to induce Pd reduction and Pd sintering. It turned out that all the Pd species in ZSM-5 transformed into metallic Pd clusters. However, most of the Pd species in SSZ-13 remain oxidized after the vacuum treatment, which would suggest that the interaction between the Pd species and the framework Al species was stronger in SSZ-13 than in ZSM-5. The intrinsic difference in the mobility of the Pd species in the different types of zeolite might originate from the smaller pore size of SSZ-13 than that of ZSM-5. Such a proposal is supported by the NOx storage abilities of the other prepared catalysts with larger pore diameter (modernite and beta zeolites) where hydrothermal treatment led to the loss of catalytic activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2016R1A5A1009592). The experiments at PLS-II were supported in part by the Ministry of Science, ICT, and Future Planning (MSIP), and the Pohang University of Science and Technology (POSTECH).Notes and references
- C. J. Loughran and D. E. Resasco, Appl. Catal., B, 1995, 5, 351–365 CrossRef CAS
.
- C. J. Loughran and D. E. Resasco, Appl. Catal., B, 1995, 7, 113–126 CrossRef CAS
- M. Ogura, M. Hayashi, S. Kage, M. Matsukata and E. Kikuchi, Appl. Catal., B, 1999, 23, 247–257 CrossRef CAS
- M. Ogura, S. Kage, M. Hayashi, M. Matsukata and E. Kikuchi, Appl. Catal., B, 2000, 27, L213–L216 CrossRef CAS
- H. Ohtsuka and T. Tabata, Appl. Catal., B, 2000, 26, 275–284 CrossRef CAS
- Y. Lou, J. Ma, W. Hu, Q. Dai, L. Wang, W. Zhan, Y. Guo, X.-M. Cao, Y. Guo and P. Hu, ACS Catal., 2016, 6, 8127–8139 CrossRef CAS
- K. Okumura, S. Matsumoto, N. Nishiaki and M. Niwa, Appl. Catal., B, 2003, 40, 151–159 CrossRef CAS
- Q. Dai, Q. Zhu, Y. Lou and X. Wang, J. Catal., 2018, 357, 29–40 CrossRef
- T. Tang, C. Yin, L. Wang, Y. Ji and F.-S. Xiao, J. Catal., 2007, 249, 111–115 CrossRef CAS
- T. Tang, C. Yin, L. Wang, Y. Ji and F.-S. Xiao, J. Catal., 2008, 257, 125–133 CrossRef CAS
- J. Zhang, L. Wang, Y. Shao, Y. Wang, B. Gates and F.-S. Xiao, Angew. Chem., 2017, 129, 9879–9883 CrossRef
- A. W. Aylor, L. J. Lobree, J. A. Reimer and A. T. Bell, J. Catal., 1997, 172, 453–462 CrossRef CAS
- K. Okumura, R. Yoshimoto, T. Uruga, H. Tanida, K. Kato, S. Yokota and M. Niwa, J. Phys. Chem. B, 2004, 108, 6250–6255 CrossRef CAS PubMed
- D. H. Kim, Y.-H. Chin, G. G. Muntean, A. Yezeretz, N. W. Currier, W. S. Epling, H.-Y. Chen, H. Hess and C. H. Peden, Ind. Eng. Chem. Res., 2006, 45, 8815–8821 CrossRef CAS
- T. Szailer, J. H. Kwak, D. H. Kim, J. C. Hanson, C. H. Peden and J. Szanyi, J. Catal., 2006, 239, 51–64 CrossRef CAS
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets and J. T. Miller, Science, 2017, 357, 898–903 CrossRef CAS PubMed
- A. Wang, Y. Wang, E. D. Walter, R. K. Kukkadapu, Y. Guo, G. Lu, R. S. Weber, Y. Wang, C. H. Peden and F. Gao, J. Catal., 2018, 358, 199–210 CrossRef CAS
- F. Gao, D. Mei, Y. Wang, J. N. Szanyi and C. H. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed
- N. Akter, X. Chen, J. Parise, J. A. Boscoboinik and T. Kim, Korean J. Chem. Eng., 2018, 35, 89–98 CrossRef CAS
- T. V. Johnson, SAE Int. J. Engines, 2012, 5, 216–234 CrossRef
-
R. M. Heck, R. J. Farrauto and S. T. Gulati, Catalytic air pollution control: commercial technology, John Wiley & Sons, 2009 Search PubMed
- H.-Y. Chen, S. Mulla, E. Weigert, K. Camm, T. Ballinger, J. Cox and P. Blakeman, SAE Int. J. Fuels Lubr., 2013, 6, 372–381 CrossRef CAS
- T. Selleri, F. Gramigni, I. Nova, E. Tronconi, S. Dieterich, M. Weibel and V. Schmeisser, Catal. Sci. Technol., 2018, 8, 2467–2476 RSC
- P. Bazin, O. Saur, J. Lavalley, G. Blanchard, V. Visciglio and O. Touret, Appl. Catal., B, 1997, 13, 265–274 CrossRef CAS
- Y. Ji, T. J. Toops and M. Crocker, Catal. Lett., 2009, 127, 55–62 CrossRef CAS
- M. Waqif, P. Bazin, O. Saur, J. Lavalley, G. Blanchard and O. Touret, Appl. Catal., B, 1997, 11, 193–205 CrossRef CAS
- Y. Ryou, J. Lee, H. Lee, C. H. Kim and D. H. Kim, Catal. Today, 2017, 297, 53–59 CrossRef CAS
- H.-Y. Chen, J. E. Collier, D. Liu, L. Mantarosie, D. Durán-Martín, V. Novák, R. R. Rajaram and D. Thompsett, Catal. Lett., 2016, 146, 1706–1711 CrossRef CAS
- Y. Zheng, L. Kovarik, M. H. Engelhard, Y. Wang, Y. Wang, F. Gao and J. Szanyi, J. Phys. Chem. C, 2017, 121, 15793–15803 CrossRef CAS
- Y. Ryou, J. Lee, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Appl. Catal., B, 2017, 212, 140–149 CrossRef CAS
- J. Lee, Y. Ryou, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Appl. Catal., B, 2018, 226, 71–82 CrossRef CAS
- A. Vu, J. Luo, J. Li and W. S. Epling, Catal. Lett., 2017, 147, 745–750 CrossRef CAS
-
R. R. Rajaram, F.-M. Mckenna, H.-Y. Chen and D. Liu, US Pat., 14/563382, 2015 Search PubMed
- K. Khivantsev, F. Gao, L. Kovarik, Y. Wang and J. Szanyi, J. Phys. Chem. C, 2018, 122, 10820–10827 CrossRef CAS
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CrossRef CAS
- S. Tauster, S. Fung and R. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS
- H. Ohtsuka and T. Tabata, Appl. Catal., B, 1999, 21, 133–139 CrossRef CAS
- B. Pommier and P. Gelin, Phys. Chem. Chem. Phys., 2001, 3, 1138–1143 RSC
- B. Pommier and P. Gélin, Phys. Chem. Chem. Phys., 1999, 1, 1665–1672 RSC
- M. Suzuki, J. Amano and M. Miwa, Microporous Mesoporous Mater., 1998, 21, 541–547 CrossRef CAS
- W. Weber, R. Baird and G. Graham, J. Raman Spectrosc., 1988, 19, 239–244 CrossRef CAS
- D. Mei, F. Gao, J. Szanyi and Y. Wang, Appl. Catal., A, 2019, 569, 181–189 CrossRef CAS
- K. Khivantsev, N. R. Jaegers, L. Kovarik, J. C. Hanson, F. Tao, Y. Tang, X. Zhang, I. Z. Koleva, H. A. Aleksandrov and G. N. Vayssilov, Angew. Chem., Int. Ed., 2018 DOI:10.1002/ange.201809343
- S. Homeyer and W. Sachtler, J. Catal., 1989, 117, 91–101 CrossRef CAS
- K. Moller, D. C. Koningsberger and T. Bein, J. Phys. Chem., 1989, 93, 6116–6120 CrossRef CAS
- W. Vogel, H. Knözinger, B. Carvill, W. Sachtler and Z. Zhang, J. Phys. Chem. B, 1998, 102, 1750–1758 CrossRef CAS
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC
- D. Ohayon, R. Le Van Mao, D. Ciaravino, H. Hazel, A. Cochennec and N. Rolland, Appl. Catal., A, 2001, 217, 241–251 CrossRef CAS
- M. Choi, D. H. Lee, K. Na, B. W. Yu and R. Ryoo, Angew. Chem., 2009, 121, 3727–3730 CrossRef
- G. C. Koltsakis and A. M. Stamatelos, Ind. Eng. Chem. Res., 1997, 36, 4155–4165 CrossRef CAS
- B. R'mili, A. Boreave, A. Meme, P. Vernoux, M. Leblanc, L. Noel, S. Raux and B. D'Anna, Environ. Sci. Technol., 2018, 52, 3312–3319 CrossRef PubMed
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. Peden, J. Catal., 2012, 287, 203–209 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data. See DOI: 10.1039/c8cy02088d |
| This journal is © The Royal Society of Chemistry 2019 |