
Good practices for reporting the photocatalytic evaluation of a visible-light active semiconductor: Bi2O3, a case study†
Agileo
Hernández-Gordillo‡
 a,
Monserrat
Bizarro
a,
Tanveer A.
Gadhi
b,
Ana
Martínez
a,
Alberto
Tagliaferro
cd and
Sandra E.
Rodil
*a
a,
Monserrat
Bizarro
a,
Tanveer A.
Gadhi
b,
Ana
Martínez
a,
Alberto
Tagliaferro
cd and
Sandra E.
Rodil
*a
aInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior SN, Ciudad Universitaria, CP 04510, Coyoacán, Cd. De México, Mexico. E-mail: srodil@unam.mx
bU.S. Pakistan Center for Advanced Studies in Water (USPCASW), Mehran, University of Engineering and Technology, Jamshoro 76062, Pakistan
cDepartment of Applied Science and Technology, Politecnico di Torino, Italy
dUOIT, Canada
First published on 15th February 2019
Abstract
In this paper, we discuss the importance of distinguishing the contributions of photolysis, adsorption, sensitization, degradation and mineralization processes to the photocatalytic activity of a visible-light active semiconductor: Bi2O3. Based on this case study, we propose a follow-up work plan to obtain the relevant information and achieve proper interpretation of reported data to adequately infer the photocatalytic activity of the Bi2O3 material. To do so, we compared the changes in the theoretical and experimental absorbance spectra of three different dyes during the photodegradation process: rhodamine-B (RhB), acid blue 113 (AB) and indigo carmine (IC). Photocatalytic degradation of these dyes using the same semiconductor material (Bi2O3) was performed using the standard spectrocolorimetric method while taking care to appropriately consider the competing processes mentioned above. Furthermore, the degree of mineralization achieved due to the photocatalytic degradation of the dyes was obtained using the total organic carbon (TOC) analysis. The commonly used evaluation of the results suggests that a certain degree of photodegradation and mineralization was achieved. However, careful analysis indicates that this was mainly due to a decrease in the relative concentration of the dye molecules in the solution because of their adsorption on the surface of the semiconductor and not to complete degradation.
Introduction
Heterogeneous visible-light photocatalysis is becoming an important alternative for the decomposition of recalcitrant organic pollutants in water, such as phenols, surfactants, dyes, and organic pesticides as well as emergent pharmaceutical and personal care products. Heterogeneous photocatalysis exploits light for the activation of a photocatalytic material that enables oxidation/reduction reactions through the generation of reactive oxygen species (ROS), leading to degradation or complete mineralization of organic pollutants.1,2 An important goal to achieve during heterogeneous photocatalysis is the mineralization, i.e., the degradation of the parent pollutant and its intermediate products to CO2 and H2O without any residual contaminants.3,4 When this mineralization process can be carried out using solar light, heterogeneous visible-light photocatalysis becomes a viable and sustainable alternative for water treatment. Hence, there is worldwide interest in the development of new or modified photocatalytic materials which are photoactive under visible light illumination to take full advantage of the solar spectrum.5–8Regarding dyes, it has been reported that approximately 1% to 15% of synthetic textile dyes are discharged in wastewater streams.9,10 The discharge of colored effluent obstructs light penetration, disrupting biological processes. Many dyes are harmful to organisms and may cause direct destruction of aquatic species.11 Therefore, the removal of dyes from industrial wastewater is a major environmental issue. It is hence compulsory to put effort toward the development of new technologies to achieve the elimination of dyes before wastewater discharge. By using UV-active photocatalytic materials, such as TiO2 and ZnO, it has been clearly demonstrated that different organic dyes can be converted into non-toxic and colorless products, achieving complete mineralization in some cases (CO2 and water byproducts).12–14 However, the quest for visible-light active photocatalytic materials capable of degrading organic dyes is not yet complete. One reason is that the ideal photocatalytic material should have an optimum band gap (Eg < 3 eV), but at the same time, it should fulfill the thermodynamic conditions that allow the fast generation of ROS.15 Moreover, the evaluation of the photocatalytic activity of the material is not as easy as it appears because there are several parameters that must be properly considered, such as solution pH, stirring rate, dissolved oxygen, and dye concentration. We consider that relevant data and discussion about processes that occur during photocatalysis are usually disregarded in many published papers. Identification of processes such as (i) photodiscoloration due to adsorption or photolysis, (ii) dye self-sensitization, (iii) actual photodegradation with the unavoidable formation of intermediate products, and (iv) mineralization of the intermediate products is significant for the correct interpretation of the photocatalytic activity of a semiconductor.
Currently, it is very common to evaluate the photocatalytic activity of “new” photocatalytic materials (nanostructures or thin films) using colored dye solutions as probe pollutants to degrade because the kinetics of the degradation process can be easily followed using a spectrophotometric technique,16 assuming that the absorbance is solely proportional to the relative dye concentration. However, the simplicity of this quantitative technique has led to false results or incorrect interpretations; this has raised valid criticism by the specialized community,3,15,17–19 who insist on the importance of understanding the correlation between photocatalysis and heterogeneous catalysis. One important point to consider is that the whole absorbance spectrum of the dye solution should be acquired from UV to visible wavelengths as a function of the irradiation time, rather than only determining the absorbance at a fixed wavelength. Moreover, for dyes, it has been shown that color removal (decoloration, discoloration or decolorization) is not equivalent to dye mineralization; indeed, total color removal can be achieved without mineralization.12,20 It has also been established that the results obtained from specific semiconductor-dye couples cannot be generalized to other organic molecules, not even to other dyes; this is due to possible synergistic reactions established between the particular dye molecule and the semiconductor, such as sensitization.14 However, we consider that sensitization and other common misinterpretations of results are not unique to the use of dye solutions but are inherent to the spectrophotometric evaluation of probe solutions using supernatants or aliquots.
In this paper, we propose a methodology consisting of a number of fundamental steps for the photocatalytic evaluation of visible-light active photocatalytic materials; this methodology is applicable to different organic molecules, including dyes. By following the proposed methodology, which includes steps for the determination of certain competing processes occurring during evaluation under dark and illumination conditions, we show that it is possible to reach more reliable conclusions about the photocatalytic activity of a material. Some of the proposed key experiments are already included in many papers; however, good interpretation of the results has not necessarily been provided in all cases, leading to false assessments. The follow-up methodology is supported by experiments using a candidate semiconductor (Bi2O3) and three different dyes. We also performed quantum chemical calculations of the absorbance spectra of the dyes, their intermediates or degradation products and their photobleaching processes. The procedure that we propose for the analysis of photocatalytic activity can be used for any semiconductor material and/or probe molecule.
Experimental and theoretical procedures
The follow-up methodology includes spectrophotometric evaluation of the dye concentration (Cn) measured for each process, namely photolysis (Cphoto), adsorption (Cads), sensitization (Csen), photobleaching (Cblea), and photodegradation (Cdeg); finally, we propose that a non-spectrophotometric technique should be used to determine the degree of mineralization (% Mine) of the intermediate products in solution. In this case, we present data on mineralization obtained using measurements of the change in the total organic carbon content (TOC).a) Dye solutions
Three dyes were selected: the cationic dye rhodamine-B (RhB), with a concentration of 5 mg L−1, and the anionic dyes indigo carmine (IC) and acid blue 113 (AB), with concentrations of 10 mg L−1 at pH 6.6 ± 0.2, close to the neutral value. For the AB dye, an additional solution was adjusted to an alkaline pH close to 9.5 using drops of NaOH. For each experiment, 10 mg of the α/β-Bi2O3 powder sample (see the ESI,† for synthesis and characterization details) were added to 15 mL of the dye solution, leading to a photocatalyst load of 0.67 g L−1. The stirring rate was kept constant at 1200 rpm while exposing the solution to air to allow oxygen diffusion. The experiments were performed under irradiation of a 9 W UV lamp (irradiating in the wavelength range of 350 to 400 nm) with an irradiance of 27 W m−2 and a 9 W white lamp (irradiating in the wavelength range of 420 to 640 nm) with an irradiance of 33 W m−2.21b) Spectrophotometric experiments
The whole absorbance spectra of the dye solutions in the dark and during illumination were measured using a Shimadzu 1800 UV-vis spectrophotometer in the wavelength range from 200 to 800 nm at intervals of time (t) by extracting a 3 mL aliquot from the suspension and separating the solid by centrifugation. Typical spectrophotometric experiments rely on measuring the maximum absorbance, Amax; from quantum chemistry, we know that the characteristic absorbance corresponds to an allowed quantized electronic transition. Moreover, classical studies have demonstrated that the absorbance, Amax, is directly proportional to the concentration of the probe-molecule in solution as described by the Beer–Lambert law (A = ε l C), where ε is the absorption coefficient (also called absorptivity, L g−1 cm), l is the path length of the light through the cell in cm and C is the concentration in g L−1. Therefore, a concentration calibration curve of the probe-molecule in solution can be easily obtained by measuring the Amax value of dye solutions with different concentrations. As a first step, a correlation curve was obtained between the concentration of dye in solution (g L−1) and the maximum absorbance value (Amax) for each dye measured at their corresponding absorption wavelengths (λmax), i.e. at 554 nm for RhB (λ554nm), 610 nm for IC (λ610nm) and 564 nm for AB (λ564nm).c) Kinetics
The time evolution of the dye concentration in solution during the experiments was monitored using the equivalence between absorbance and concentration (Amax = C), however, we measure the absorbance at λmax looking at the whole absorbance spectrum. Concentration data were considered to be reliable as long as the position of each Amax peak was not shifted or the shape of the spectrum peak did not change during illumination. The variation of the concentration or relative concentration (Cn/C0) of a probe-molecule in solution as a function of irradiation time (t) is estimated using eqn (1): | (1) |
 | (1a) |
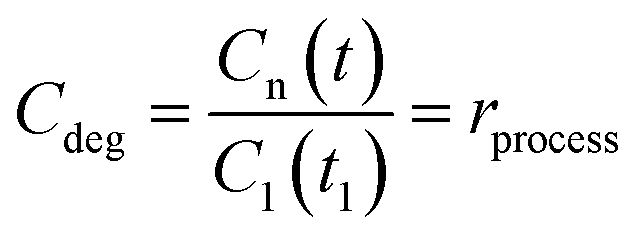 | (2) |
d) Computational calculations of electronic spectra
To understand the different processes that can lead to discoloration of the solution, quantum chemical calculations of the absorbance spectra of the three dyes and their corresponding discoloration/degradation intermediate products were performed. All the electronic calculations were performed with the Gaussian 09 package of programs.22 Fully optimized structures and harmonic frequencies were obtained at the M05/6-311+G(2d,p) level of theory23–27 in conjunction with the continuum SMD model using water to mimic a polar environment.28 Local minima were identified by the absence of imaginary frequencies. The absorption spectra were computed with TD-DFT using the optimized geometries at the same level of theory.29,30 This TD-DFT investigation was performed in the so-called vertical approximation, i.e. the transition energies toward the lowest excited state were computed considering a frozen geometry (the optimal ground state structure obtained from the optimization). In this approximation, the vibrational degrees of freedom and hence the coupling between nuclear and electronic degrees of freedom (vibronic effects) are neglected. This approach allowed us to determine the nature of different excited states. The accuracy of the relative position of the maximum value and the relative intensities of the peaks depends on the treated case.31 To achieve accuracy, it would have been necessary to perform TD-DFT in its adiabatic kernel approximation. However, for the purpose of this investigation, TD-DFT vertical approximation is effective because we are not interested in the exact value but rather in whether the shape of the spectrum is comparable with the experimental spectrum.32Results and discussion
The “photocatalytic activity” of a semiconductor is typically quantified by monitoring the variation of the Cn/C0 value of the dye/molecule in solution (eqn (1)) with irradiation time assuming simple Langmuir–Hinshelwood kinetics,13 in which an adsorbed monolayer of the probe-molecule is completely in equilibrium. The apparent kinetic rate constant (Kapp) of the photocatalytic process (discoloration or degradation) is estimated rather than the rate constant (KROS) of the formation of ROS, which is assumed as a constant33–36 independently of the dye concentration. However, some authors consider that a first order pseudo steady-state approach must be used for the kinetics equation to fully describe the photocatalysis.18,37–39 Following this procedure means that the photocatalytic activity is not directly measured but is indirectly estimated from measurements of the absorbance spectra, Amax, of the probe-molecule in solution or, in some cases, by measurements of the degradation products (intermediates) or total organic carbon (TOC) content in the solution. Therefore, any photocatalytic experiment using dye molecules should take into account all the possible phenomena that could simultaneously induce a decrease in Cn/C0 of the dye molecule in solution, such as photolysis (Cphoto), adsorption (Cads), or the formation of new intermediate products. These intermediates may cause variations in the original absorbance spectrum, Amax, which invalidate the use of eqn (1), and this is common during photobleaching or photodegradation (Cblea, Cdegra). In the following, we describe the procedures, and the information obtained from them, to accomplish correct evaluation of the photocatalytic activity of a semiconductor material using visible light and dyes as the organic pollutants. The processes described are photolysis, adsorption, photocatalytic degradation and mineralization and the results are summarized in Tables 1–3, respectively.Photolysis
Photolysis refers to the chemical decomposition of molecules by light (photo-decomposition or photochemical reaction), which is defined as catalyzed photolysis according to Serpone.40 Some chemical compounds are degraded into smaller units by the direct absorption of photons or by reaction with oxidative species generated from the water solution by UV lamps.41 This is a well-known phenomenon in dyes and pharmaceuticals. The absorption of light by the dye molecule results in photochemically induced homolytic cleavage of the chromophore group, such as the double bond in the indigoid group (NH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH)42 or the C–OC group in RhB dye.43 This leads to a loss of color and a decrease in the Amax. Therefore, photolysis of the probe-molecule must be evaluated under the defined experimental conditions (λ of lamp, dye concentration, pH, aerated conditions, scavenger substances, etc.), without adding the photocatalyst material. The degree of photo-decomposition [% Cphoto = (1 − Cphoto) × 100] of a dye due to light must be known before that dye can be used to evaluate the photocatalytic activity of a material. This effect is critical, especially for high power lamps; however, it is not always reported.44,45
C–NH)42 or the C–OC group in RhB dye.43 This leads to a loss of color and a decrease in the Amax. Therefore, photolysis of the probe-molecule must be evaluated under the defined experimental conditions (λ of lamp, dye concentration, pH, aerated conditions, scavenger substances, etc.), without adding the photocatalyst material. The degree of photo-decomposition [% Cphoto = (1 − Cphoto) × 100] of a dye due to light must be known before that dye can be used to evaluate the photocatalytic activity of a material. This effect is critical, especially for high power lamps; however, it is not always reported.44,45
In our example, the % Cphoto values of the three dyes were investigated by irradiating the dye solutions with UV and visible light at the corresponding working dye concentrations and maintaining the solution under magnetic stirring for at least 180 minutes. Each experiment was performed without adding the photocatalytic material, and the profile of Cphoto(Cn/C0) was monitored every 30 minutes using eqn (1) through the corresponding Amax of each dye. Fig. 1 shows the decrease of Cn/C0 of all the dyes vs. time during the photolysis evaluation process. The three dyes suffered small decreases in absorbance, suggesting slight decomposition under irradiation, except for RhB, which showed greater stability under visible light. From Table 1, we know that about 10% to 12% of dye was degraded (% Cphoto) by the action of light; therefore, this 10% to 12% decrease in the absorbance cannot be attributed to photocatalytic action of the photocatalyst because it is due to photolysis.
 | ||
| Fig. 1 Variations of the relative concentrations Cphoto of the three dyes due to the photolysis process when the solution is irradiated with visible light (open symbols) and UV light (filled symbols). | ||
| Dye/irradiation | Dye sol. (ppm) | C photo = Cn/C0 | % Cphoto = [(1 − Cphoto) × 100] | Dyedegraded (ppm) |
|---|---|---|---|---|
| C 0 = initial dye concentration in solution (equal to unity). Cn = dye concentration in solution at 180 min of irradiation. Cphoto = relative dye concentration after photolysis. % Cphoto = degree of photolysis of the dye by irradiation. | ||||
| RhB/UV | 5 | 0.92 | 8 | 0.4 |
| RhB/visible | 5 | 0.98 | 2 | 0.1 |
| IC/UV | 10 | 0.88 | 12 | 1.2 |
| IC/visible | 10 | 0.90 | 10 | 1.0 |
| AB/UV | 10 | 0.88 | 12 | 1.2 |
| AB/visible | 10 | 0.88 | 12 | 1.2 |
The next experiment was the evaluation of the fraction of dye adsorbed on the semiconductor surface.
Adsorption
Adsorption of the probe-molecule on the surface of a photocatalyst is a relevant process in photocatalysis that can be estimated by monitoring the concentration (Cads = Cn/C0) of the probe-molecule in solution as a function of immersion time with the specific load of the photocatalytic material in dark and stirred conditions. Adsorption plays an important role in photocatalysis because it is a critical step to achieve the oxidation–reduction of species by the photogenerated electron-hole pairs. Herein, we show that to correctly evaluate the photocatalytic activity of a material, it is essential to measure the fraction of dye molecules adsorbed on its surface once equilibrium is attained, i.e. when a monolayer of adsorbed molecules is formed. For this, we use the following equation:| θads = (1 − Cads) × 100 | (3) |
The adsorption will influence the effective C0 of the dye, i.e., the real concentration of the probe-molecule in solution when illumination starts (C1); therefore, it will influence the estimated photodegradation percentage (% Cdeg or conversion) and mineralization (% Mine). It should be remembered that adsorption is also a classical method to decrease the concentration of a probe-molecule in solution; this can be tailored depending on the pH of the solution, the catalyst load and the C0 of the probe-molecule. The θads should be measured prior to the irradiation experiments under the experimental conditions (pH, catalyst load, dye concentration, stirring conditions) at which the photocatalytic assays will be performed,46 but in dark conditions. When dye molecules are adsorbed on the surface of a photocatalyst, the maximum absorbance Amax of the dye in solution is decreased in comparison to its initial value (C0). Hence, Amax should be measured at periodic time intervals until no more changes are observed in the spectra, indicating that the adsorption–desorption equilibrium has been reached. The decrease in Amax is proportional to the fraction of dye molecules adsorbed: θads.
To investigate the profiles of Cads of the three different dyes onto α/β-Bi2O3 powders, 15 mL of each dye solution at neutral pH were stirred in the dark. The Amax of the dye in solution was acquired every 30 minutes until no further changes were observed. In the case of AB, where a high adsorption was observed at neutral pH, additional experiments were conducted at alkaline pH (9.5) in order to decrease the dye adsorption.
Fig. 2A–C show the absorbance spectra of the three dyes (RhB, IC and AB) at t = 0 (continuous line spectra) and after adsorption–desorption equilibrium was reached under the dark-stirred conditions (dashed line spectra). It can be seen that the conditions to apply eqn (1) are fulfilled (no peak shifts); Fig. 2D shows the decreases of Cn/C0 of all the dyes vs. time, indicating that adsorption–desorption equilibrium was attained after 30 minutes for the three dyes. It is important to use the whole spectrum to measure Amax or to take care to use proper Amax values according to the pH because some dyes (such as methyl orange, which is not included in this investigation) are pH sensitive and their λmax changes with pH.
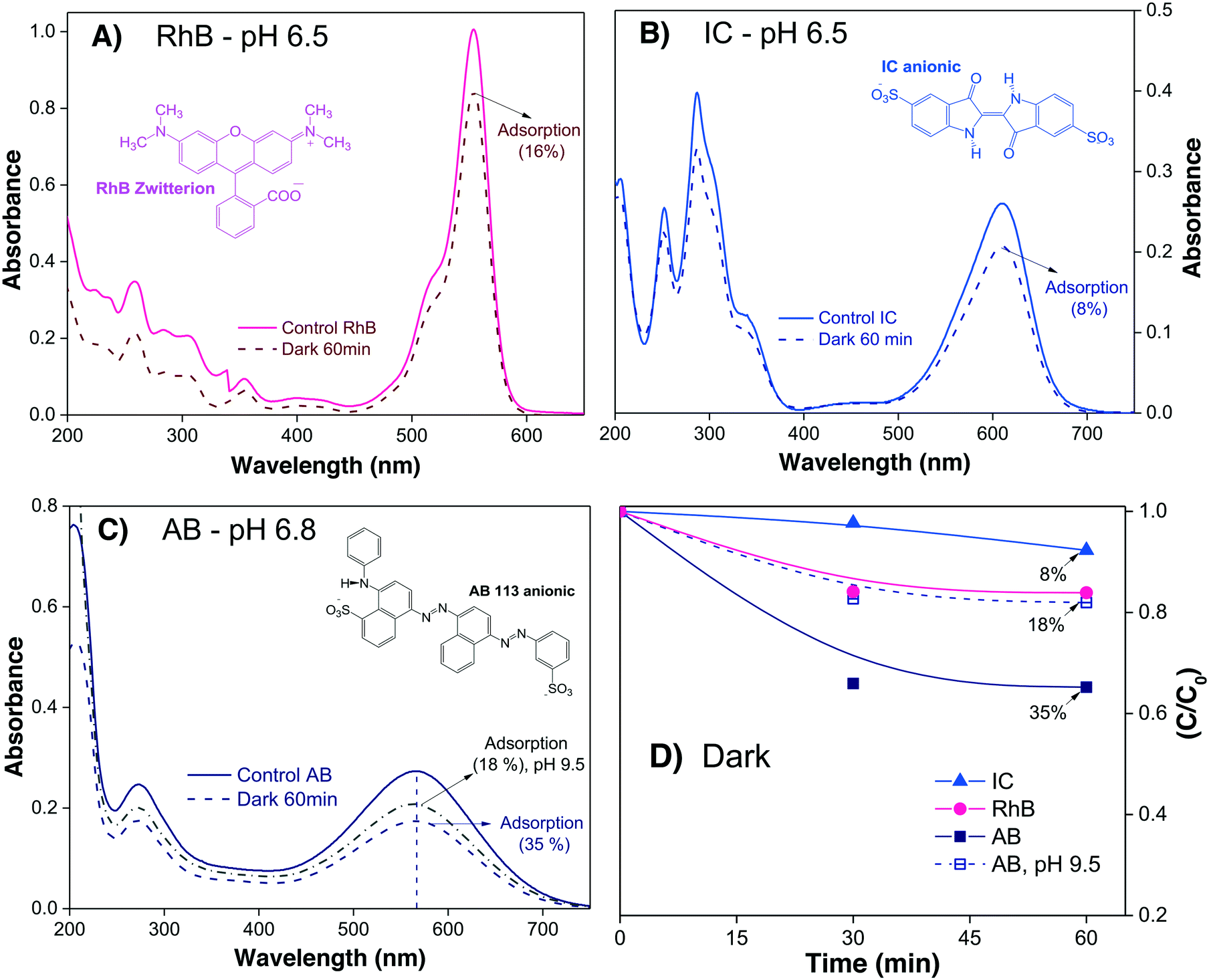 | ||
| Fig. 2 (A–C) Absorbance spectra of three dye solutions and their evolution after 60 minutes in the dark in the presence of α/β-Bi2O3 powder. D) Variations of Cn/C0 due to adsorption of the dye molecules on the semiconductor surface in dark conditions; lines are drawn as a visual aid. | ||
From Table 2, it is then possible to estimate the maximum θads by taking the saturated Cn value at 60 minutes; θads changes according to the dye molecule and pH. The adsorption of the dye molecules (and, later, of their degradation products) onto the surface of the photocatalyst is a complex phenomenon that depends strongly on the ionic properties and the pKa of the molecule, the point of zero charge (PZC) of the photocatalyst, and coulombic and hydrophobic/hydrophilic interactions.
| Dye | Dye sol. (ppm) | C ads = Cn/C0 | θ ads = [(1 − Cads)] | Dyeadsorbed (ppm) | C 1 (ppm) |
|---|---|---|---|---|---|
| C 0 = initial dye concentration in solution (equal to unity). Cn = dye concentration in solution at 60 minutes in dark conditions. Cads = relative concentration of dye in solution after the adsorption process. θads = fraction of molecules of the dye adsorbed. | |||||
| RhB | 5 | 0.84 | 0.16 | 0.8 | 4.2 |
| IC | 10 | 0.92 | 0.08 | 0.8 | 9.2 |
| AB (pH 6.8) | 10 | 0.65 | 0.35 | 3.5 | 6.5 |
| AB (pH 9.5) | 10 | 0.82 | 0.18 | 1.8 | 8.2 |
The different percentages of adsorbed molecules observed in these experiments can be readily correlated to the physicochemical properties of the photocatalyst surface and the dyes. For the RhB cationic dye, the θads was 0.16 (Table 2), in agreement with our previous report.21 At pH 6.5, the Bi2O3 surface is partially protonated (H+), because the measured PZC of the α/β-Bi2O3 sample was 6.8.21 The pKa of the RhB dye is 3.5; thus, at pH 6.5, the –COOH groups of the cationic RhB+ are deprotonated to –COO−, yielding the zwitterion RhB± (inset Fig. 2A), which can be adsorbed in the amphoteric sites of Bi2O3 through oxygen bonds.47 For the two anionic dyes, IC and AB, the θads values were 0.08 and 0.35, respectively. In both dyes, the anionic behavior is related to the two sulfonated (–SO3−) groups (inset of Fig. 2B and C), which can be adsorbed onto the photocatalyst surface by forming hydrogen bonds with the protonated surface at pH < 6.8.48 However, the larger adsorption of the AB dye in comparison to IC suggests a contribution of coulombic interactions. At neutral pH, AB is negatively charged (AB2−) due to its two negatively charged sulfonated groups (pKa = 0.5);49 thus, it can be electrostatically attracted to the protonated α/β-Bi2O3 composite. Similar electrostatic attractions have been reported for azo dyes on TiO2 and bismuth(III) nitrates.9,50–52 Thus, the θads of the AB dye can be controlled by modifying the solution pH to alkaline, as can be observed in Fig. 2C and D (the dotted line in Fig. 2C refers to pH 9.5). The θads of the AB dye decreased to 0.18 at pH 9.5 because at this pH, the Bi2O3 surface is rich in −OH species that repel the AB2− species. However, it is also important to consider that in addition to electrostatic interactions, adsorption can occur via hydrophobic–hydrophobic interactions or hydrogen and oxygen bonding. A summary of the different adsorption mechanisms observed for the three dyes on the Bi2O3 surface is schematically shown in Fig. 3.
 | ||
| Fig. 3 Schematic of the adsorption processes of the three dyes on Bi2O3 at different pH values. | ||
Formally, θads should be quantified at different initial dye concentrations to determine the critical value after which no more adsorption is observed; this value will depend on the surface properties of the semiconductor, the probe molecule and the pH value of the solution. However, it is more common to use a fixed initial concentration, even when different semiconductors are compared.
From the above results referring to the same photocatalytic material but different dyes, we can see that the degree of adsorption can be significantly different. Therefore, if the adsorption is not properly considered and eqn (1) is directly used to estimate the photocatalytic degradation achieved after illumination (Cdeg), i.e., the initial concentration C0 is used, the degradation will be overestimated. The reason for this overestimation is that the absorbance (Amax) is decreased by two different processes, namely adsorption and degradation, while being attributed only to degradation. It was explained earlier that adsorption is necessary for photocatalytic reactions to occur; we cannot assume that all adsorbed molecules are or will be degraded, but it is factual that adsorbed molecules lead to a decrease in the absorbance. Overestimation of photocatalytic degradation due to neglect of the adsorption is a very common error found in the literature;53–55 this is critical when different materials are compared and the data are used to select the best photocatalyst. Moreover, the θads will also have an impact on the estimation of the % Mine using any technique based on analysis of the solution because the concentration of total organic carbon (% CTOCn) in the solution will also decrease.46
Once the θads is determined, it is possible to evaluate the photocatalytic activity of a photocatalyst knowing the effective concentration of probe-molecules in solution (C1); proper evaluation should use eqn (2). However, a few competing processes which can occur during the illumination should also be considered.
Competing photocatalytic processes
The evaluation of the “photocatalytic activity” of a photocatalyst material is performed by measuring the color removal (also called discoloration/decoloration) as a function of the irradiation time, preferably under stirring-aerated conditions in order to guarantee that the reaction is not limited by oxygen diffusion. This color removal, % Cdisc, measured as the decrease in the absorbance Amax, is usually interpreted as the photodegradation (% Cdeg = (1 − Cdeg) × 100) of the dye molecules due to the action of semiconductor-light interactions. However, there are two competing processes which are not directly due to the photocatalytic activity of the material that can also provoke a decrease in the absorbance Amax of the dye in solution under irradiation: photobleaching and sensitization. These require further explanation prior to the analysis of the photodegradation results.We propose that by comparing the whole experimental absorption spectrum and the theoretical spectra of the two leuco-forms of a molecule under study, it is possible to discriminate between photobleaching and photodegradation. For some dyes, such as IC, the spectrum of the leuco-form has already been studied;57,58 therefore, it is known that the formation of the leuco-indigo form can be observed as a strong peak around 390 to 400 nm instead of the peak around 550 to 600 nm of IC. For less well-known dyes, theoretical calculations give us useful information.
Fig. 4A shows the calculated spectra for both IC forms; it can be observed that leuco-IC presents a strong absorption at lower wavelengths, in agreement with experimental observations.57,58 Thus, during a photocatalytic experiment using IC, the formation of leuco-IC due to the electron transfer process (reduction of IC) can be detected as a shift of the Amax to lower wavelengths and/or through the presence of two peaks and two isosbestic points, indicating that the two-chemical species (IC and leuco-IC) are in equilibrium.
 | ||
| Fig. 4 Calculated absorbance spectra of A) IC and B) RhB and their corresponding leuco forms. | ||
Meanwhile, Fig. 4B shows the calculated spectra for RhB± and its corresponding leuco-form, indicating that leuco-RhB does not show a strong absorbance peak in the visible region. Therefore, when using RhB, it is more difficult to discriminate between photobleaching and photodegradation processes because the Amax situated around 550 nm for RhB± will decrease without other spectral changes that can indicate the formation of leuco-RhB. Although Ma et al.43 suggested that leuco-RhB presents a slightly yellow color with a weak absorbance peak at 420 nm, this was not confirmed by calculations or a literature review.
It is important to mention that the formation of the leuco forms, i.e. leuco-IC58,59 and leuco-RhB dyes,43 typically occurs under conditions where the oxygen diffusion rate is limited, such as in N2 atmosphere, low stirring conditions or in the presence of hydrogenating/reducing agents (NaHB4, NH2NH2), also called sacrificial electron donors.
In the present work, the photocatalytic degradation of the three dyes was performed at a high stirring rate, i.e. 1200 rpm, generating oxidizing conditions where leuco forms are not favored. The absorption spectra of the RhB and the IC dye solutions after irradiation (both UV and visible) in the presence of the test α/β-Bi2O3 semiconductor are shown in Fig. 5 and 6, respectively. The figures show that on the one hand, similarities exist between the experimental and calculated spectra of both dyes. On the other hand, for the IC dye (Fig. 6), there is no signal for either the leuco-IC absorption or isosbestic points, indicating that the photobleaching process of the dianionic IC2− dye is not relevant to our experiments. For RhB, nothing certain can be stated because only a decrease in the maximum (Amax) is observed; this decrease could be due to either photodegradation or photobleaching. Thus, the above analysis suggests that the use of RhB as a probe-molecule to evaluate the photocatalytic activity of a material is dubious, even though it has been widely employed.60–65
 | ||
| Fig. 5 Absorbance spectra of zwitterion RhB solution during photodegradation under (A) visible light and (B) UV light. C) Relative concentrations of dye with and without correction due to the change in the initial concentration by adsorption and D) linear fits of the ln(C0/C) plots of the degradation under visible and UV light to obtain the apparent reaction rate. | ||
 | ||
| Fig. 6 Absorbance spectra of the IC solution during photodegradation under (A) visible light and (B) UV light. C) Relative concentrations of dye with and without correction due to the change in the initial concentration by adsorption and D) linear fits of the ln(C0/C) plots of the degradation under visible and UV light to obtain the apparent reaction rate. | ||
The other mechanism that can lead to a decrease in the Cn/C1 ratio but does not reflect the catalytic activity of the photocatalyst is sensitization; it is usually associated with dyes but can occur in any organic molecule with absorption bands in the UV or visible region of the spectrum, as long as the incident light is resonant with the absorption.
One proposed method to detect sensitization is based on the measurement of an action spectrum, as suggested by Lee et al.71 The action spectrum is a plot of the photonic efficiency versus the wavelength; this is not easily obtained for all materials, but it enables clear discrimination between sensitization and photocatalytic activity.72 When the action spectra cannot be measured, an indirect method to recognize if the sensitization process has occurred is to compare the degrees of photodiscoloration obtained using visible light and using another wavelength range which is not resonant with the absorbance spectrum of the dye, such as UV. If the visible-light reaction rate (r) is greater than or similar to that obtained using UV light, sensitization may be playing an important role in the final response.3,73
In order to determine the possible contribution of the sensitization process for the Bi2O3 material, photodiscoloration experiments were performed using both UV and visible light. The solution volumes and photocatalyst loads were the same as previously mentioned. The Amax values of the dyes are centered at λ554nm (RhB), λ610nm (IC) and λ565nm (AB), and the white lamp used emits in the whole range between 420 and 640 nm. Therefore, it is possible to excite all three dye molecules by resonant absorption.
The sensitization process was tested for the three dyes by analyzing the whole absorbance spectrum of the dye solution and comparing the time to achieve similar decrements in the Amax value under UV and visible light.
The absorbance spectra of the zwitterion RhB± dye solution (pH 6.5) during the irradiation process under visible and UV light are presented in Fig. 5A and B, respectively. The RhB± in solution exhibited a characteristic Amax at λ554nm that slowly decreased over 180 min of visible irradiation. Under UV light irradiation (350 to 400 nm, Fig. 5B), the spectrum attained a similar decrease in Amax in only 90 minutes. For both irradiations, no shifts of Amax were observed, suggesting negligible contribution of the N-deethylation sensitization process described earlier; this was also expected from the quantum chemical calculations, as shown later.
The absorbance spectra of the IC dye solution (pH 6.5) during the irradiation process (visible and UV light) are presented in Fig. 6A and B, respectively. The IC solution exhibited the characteristic Amax at λ610nm, which decreased almost to zero over 30 min under UV irradiation; meanwhile, under visible light, the Amax of IC decreased but did not reach zero after 60 min of irradiation, suggesting that the sensitization process when using IC dye is negligible.
The evaluation of the anionic AB dye was performed at pH 9.5 to avoid the strong adsorption observed at neutral pH. The absorbance spectra of the AB dye during visible and UV irradiation are shown in Fig. 7A and B, respectively; it can be observed that the decreases in Amax are very similar for both lights, attaining similar values after 90 min. This effect can be taken as an indication that sensitization is playing an important role.
 | ||
| Fig. 7 Absorbance spectra of the AB solution at pH 9.5 during photodegradation under (A) visible light and (B) UV light. | ||
Finally, after estimating the contributions of different processes (photolysis (% Cphoto), adsorption (θads), photobleaching and sensitization) to the variation in the absorbance Amax of the dye solution, it is possible to proceed with evaluation of the photodegradation (% Cdeg) of the dye molecule.
Photodegradation
Photodegradation, also called photoconversion, is the molecular cleavage of a probe molecule into diverse intermediate products; it can be initiated by oxidative processes through the interaction of adsorbed dye molecules with either the photogenerated holes or the reactive oxygen species (superoxide or hydroxyl radicals) generated on the surface of the photocatalyst by the photocarriers.As described previously, the evolution of the dye concentration (Cdeg = Cn/C1) with respect to the irradiation time can be estimated from the variations in the Amax, assuming the same approach (Amax = C). However, when the dye molecule is degraded into secondary or intermediate products that have different ε values but absorb at similar λmax values to the original dye, the straightforward correlation between Amax and the concentration of the dye in solution (see eqn (1)) is no longer valid.20,74 This is usually the first source of error for the estimation of the photodegradation activity (% Cdeg) of a photocatalyst. Therefore, we propose that instead of reporting only the Cn/C1 ratio, a careful analysis of the spectral changes in the whole energy range should be presented. By analyzing the whole spectra, it is possible to detect the presence of intermediates or degradation products, which indicate that photodegradation of the probe-molecule is actually occurring; moreover, depending on the spectral changes, the validity of eqn (1) can be determined. The expected modifications in the whole absorbance spectrum include a) broadening of the main Amax due to intermediates that absorb close to the characteristic absorbance (λmax) of the selected molecule, but with different absorptivities; b) appearance of new absorbance peaks ( ) related to intermediates with characteristic
) related to intermediates with characteristic 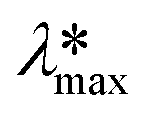 values well below those of the original probe-molecule, so that they do not overlap; and c) the presence of isosbestic points, which can indicate the presence of two or more molecules in equilibrium. Ideally, one could experimentally determine the contribution of the intermediates to the spectral changes. However, this requires the lengthy procedure of obtaining pure substances of all the intermediates and drawing calibration curves to compare their absorptivities to that of the test molecule.
values well below those of the original probe-molecule, so that they do not overlap; and c) the presence of isosbestic points, which can indicate the presence of two or more molecules in equilibrium. Ideally, one could experimentally determine the contribution of the intermediates to the spectral changes. However, this requires the lengthy procedure of obtaining pure substances of all the intermediates and drawing calibration curves to compare their absorptivities to that of the test molecule.
One feasible approach, which we propose in this paper, is the use of quantum chemical calculations to obtain prior knowledge about the processes expected during the degradation of a particular organic molecule.
Once the qualitative photodegradation and the validity of eqn (1) are confirmed, the percentage of degradation (% Cdeg = 1 − Cdeg × 100) or the profile Cn/C1 are estimated to evaluate the kinetic parameters. From the analysis, the apparent kinetic rate constant (Kapp) can be used to compare the activity of the photocatalyst to that of other materials. However, in general, the validity of eqn (1) is assumed but not demonstrated because the whole spectra are not analyzed. Before proceeding with the analysis for each of the dyes, we will first review the estimation of the kinetic parameters.
 | (4) |
 | (5) |
 | (6) |
This type of unimolecular surface reaction shows two limiting rate laws, corresponding to the two extreme behaviors of the Langmuir isotherm:
a) At high initial dye concentrations: For C1 values that lead to steady state surface coverage close to unity (θads ≈ 1), a zeroth order reaction is observed (r = kr). This means that the surface reaction rate depends neither on the dye concentration nor on the concentration of intermediate products in solution.
b) At low initial dye concentrations: For C1 values not high enough to saturate the adsorption sites, the steady state surface coverage is much smaller than unity (θads ≪ 1); thus, a first order reaction is obtained. This means that the surface reaction rate depends on the initial dye concentration and on the concentration of intermediate products in the solution.
In this case, eqn (5) can be approximated as
| r = krθads ≈ krKsC1(t) = KappC1(t) | (7) |
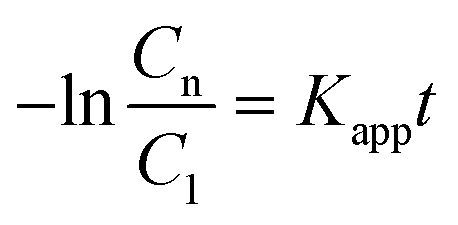 | (8) |
Therefore, by plotting the −ln(Cn/C1) data obtained from the analysis of the spectra using eqn (1)versus the irradiation time, t, Kapp can be estimated if a good linear correlation is observed.
Despite the critical revisions made by many authors18,38,75,76 regarding the use of the Langmuir–Hinshelwood (L–H) approximation to describe the influence of the initial dye concentration (C1) on the reaction rate (kr), this approach is currently a “standard method” to compare the activities of different photocatalysts. It is beyond our objectives to analyze this controversy more deeply; however, the L–H is certainly a rough approximation which should be taken carefully. There is also the controversial point of choosing the zeroth or first order approximation. The common practice is that when there is a deviation from the first-order approximation, the logarithmic “Cn/C1vs. t” plot does not show a linear trend, and the authors must choose a decreased number of experimental points to fit the linear logarithmic plot. However, this can lead to significant errors in the Kapp values but also to a wrong interpretation of the results. In a recent paper, Asenjo et al.37 suggested performing direct fitting of the “Cn(t) vs. t” plot using non-linear minimization algorithms instead of arbitrarily assuming the order of the reaction.
In the following, we will examine the photodegradation of the three dyes, and when the conditions mentioned above are fulfilled, the % Cdeg and Kapp values will be estimated.
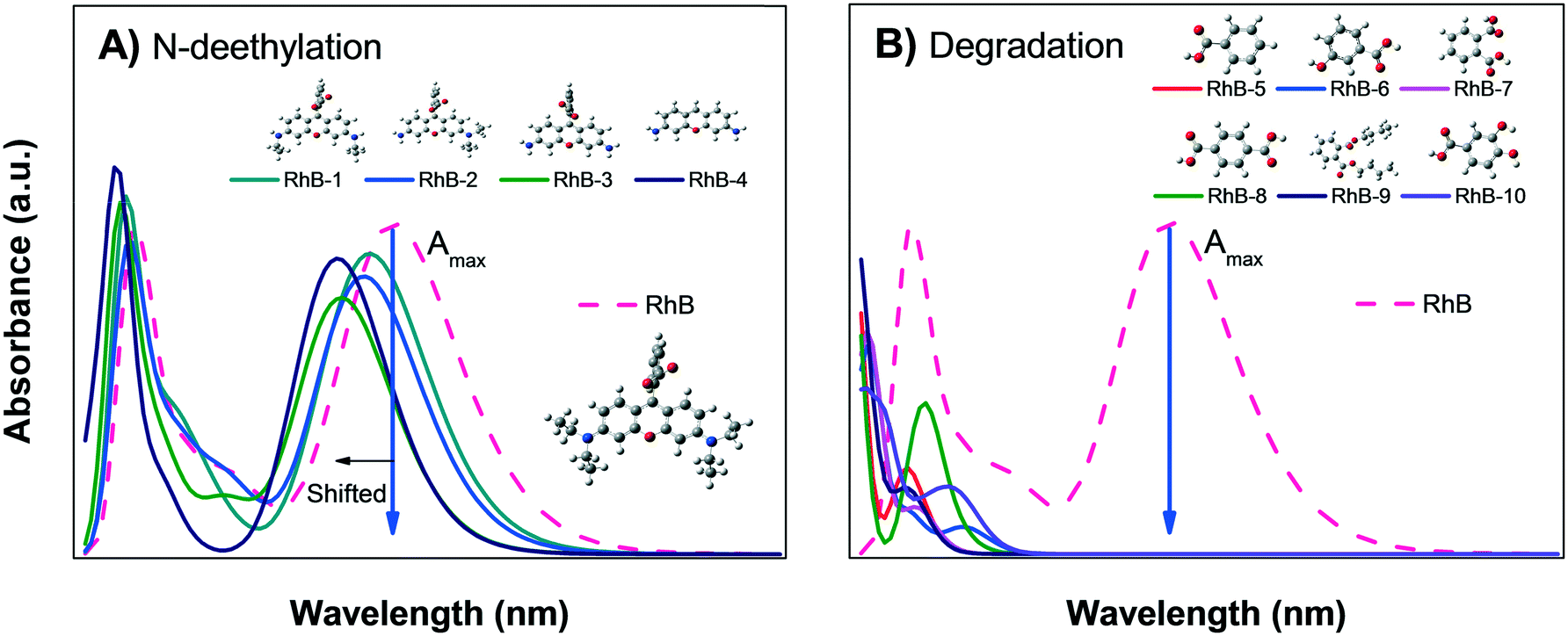 | ||
| Fig. 8 Calculated absorbance spectra of zwitterionic RhB; A) N-deethylation sensitization and B) degradation into small organic molecules. | ||
Fig. 8B shows the calculated absorbance spectra for the main degradation products (RhB-5 to RhB-10) of RhB± that might be formed during photodegradation; this shows that the degradation products have no absorbance peaks in the visible region. Therefore, when photodegradation occurs, the Amax of the RhB± solution will decrease without showing spectral changes except for a possible increment in the absorption at very low wavelengths UV region.
Comparison of the experimental data shown in Fig. 5A and B with Fig. 8A and B suggest that the decrease in the Amax of zwitterion RhB± dye solution as a function of irradiation time is due to photodegradation of the molecule; since no evidence of the N-deethylation process was observed. However, a similar decrease in Amax could also be due to the photobleaching of RhB, as explained in Fig. 4B. Hence, the only method to distinguish between these two processes (photodegradation and photobleaching of RhB) is to leave the solution in the dark for a certain time (overnight) and see if the RhB color returns; this is because photobleaching is reversible, while degradation is not.
After doing this, we can be certain that eqn (1) is valid, and it is possible to estimate the photodegradation rate (% Cdeg) by monitoring the decrease in Cn/C1 as a function of time, as shown in Fig. 5C.
Using the Cn/C1 data (Fig. 5C), Kapp (Table 3) was estimated from the linear fit of eqn (8). The results show that Kapp is two times larger under UV (1.1 × 10−2 min−1) light illumination than under visible light (0.7 × 10−2 min−1), supporting that sensitization and N-deethylation indeed did not take place for RhB.
| Dye/irradiation | C 1 (ppm) | % CTOC1 | C deg = Cn/C1 | % Cdeg = [(1 − Cdeg) × 100] | K app (×10−2 min−1) | Dyedeg (ppm) |
|---|---|---|---|---|---|---|
| C 1 = effective dye concentration. % CTOC1 = total organic carbon of effective dye solution calculated from C1. For AB, the photodegradation reaction was carried out at pH 9.5. Cdeg = relative concentration of dye in solution measured at 90 min of irradiation. | ||||||
| RhB/UV | 4.2 | 84 | 0.30 | 70 | 1.1 | 2.9 |
| RhB/visible | 0.37 | 63 | 0.7 | 2.6 | ||
| IC/UV | 9.2 | 92 | 0.03 (30 min) | 97 | 10.5 | 8.9 |
| IC/visible | 0.12 (60 min) | 86 | 3.4 | 7.9 | ||
| AB/UV | 8.2 | 82 | 0.27 | Invalid | Invalid | 5.9 |
| AB/visible | 0.23 | Invalid | Invalid | 6.3 | ||
The previous analysis confirmed that the color removal observed in Fig. 5A and B for RhB can be attributed to photodegradation of the dye molecules through the formation of intermediate products (Fig. 8B) and due to the catalytic activity of the α/β-Bi2O3 composite.
CNH) of the IC structure breaks via an oxidative process into isatin sulfonic or 2-amino-5-sulfobenzoic acid, both of which exhibit absorbance in the UV spectral region.82–84 This was also confirmed by the quantum chemical calculations of the spectra of the dianion IC2− and its corresponding intermediate products, as shown in Fig. 9.
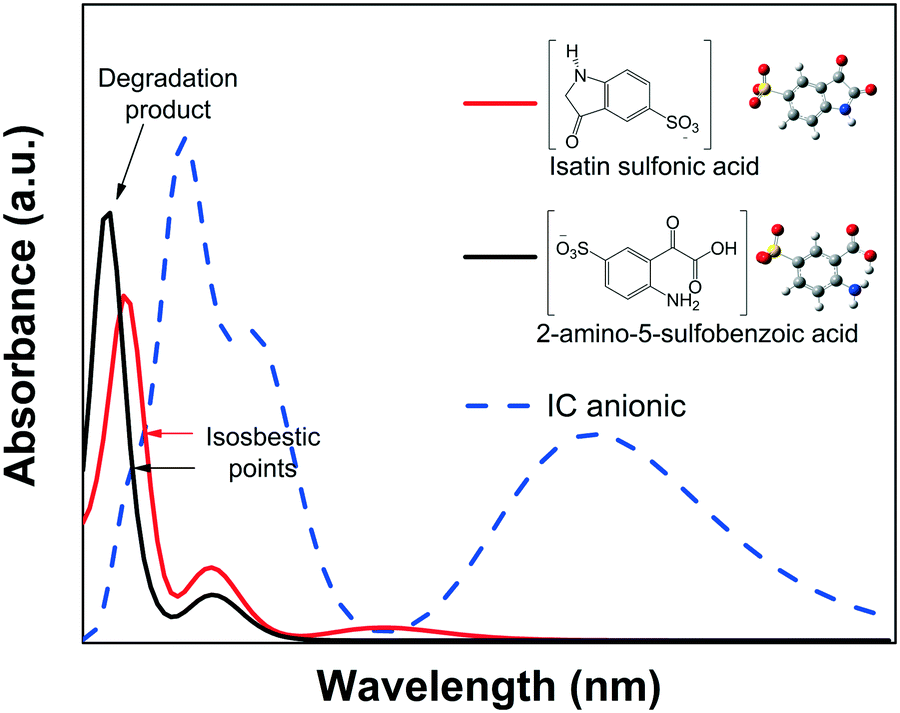 | ||
| Fig. 9 Calculated absorbance spectra of IC and its two intermediate products. | ||
None of the degradation products showed absorbance peaks that overlap the absorbance of the IC solution in the visible region; therefore, during photodegradation, the Amax of the IC dye should continuously decrease proportionally to the concentration. Thus, Cn/C1 can be properly estimated from the decrease of the Amax, and the photodegradation rate (% Cdeg) can be correctly determined. Moreover, when analyzing the whole calculated absorbance spectra, two interesting features are expected: a) the intensity of the low wavelength (λ < 285 nm) peaks might initially increase, but eventually, for a complete degradation, they should decrease following the main absorbance peak, and b) isosbestic points might be observed when the degradation products are in equilibrium with the IC dye in solution. These isosbestic points are expected to appear at lower wavelengths than that of the leuco form (see Fig. 4A); therefore, for IC dye, analysis of the isosbestic points allows us to differentiate between degradation and photobleaching processes.
The evolution of the spectra during the irradiation of the IC solution shown in Fig. 6A and B clearly demonstrates the photodegradation (% Cdeg) of the dye. Both signals due to IC (λ285nm and λ610nm) decrease with irradiation time, while the two UV peaks at λ219nm and λ245nm increase in intensity. These absorbace peaks due to the degradation products have been observed by other authors84,85 and the theoretical spectra are shown in Fig. 9; meanwhile, the corresponding isosbestic point is observed around λ251nm. The changes in all the spectral peaks are associated with the destruction of the indigoid group (NHCCNH) of the IC dye structure due to the photodegradation process, with the subsequent formation of the typical intermediate products. Moreover, from the spectral analysis, it is possible to conclude that the photodegradation process of the secondary products was not complete under UV light (up to 30 min) or under visible light (up to 60 min) irradiation because both UV peaks and the isosbestic point persist.
However, the evidence of the degradation of the IC chromophore group allows us to correctly estimate the decrease in the Cn/C1 value (Fig. 6C). Note that the correction due to the adsorption is less significant than for RhB. Fig. 6D shows the photodegradation kinetic plot, where the Kapp can be readily determined. It can be seen from Table 3 that Kapp is lower under visible light (3.4 × 10−2 min−1) than under UV light (10.5 × 10−2 min−1), confirming the negligible impact of the sensitization process.
 | ||
| Fig. 10 Calculated absorbance spectra of AB and multiple intermediates of the degradation process: A) intermediates that do not interfere with the AB absorbance and B) intermediates AB-1, AB-3 and AB-7, which clearly interfere with the Amax value. | ||
The results of the AB degradation shown in Fig. 7A and B also reflect the complexity of this dye in terms of its degradation pathway. First, the anionic AB dye is highly adsorbed onto the photocatalyst surface, and the color removal rate is similar under both lights; this suggests a contribution of the sensitization process. Second, the UV-light induced spectral evolution is different from the visible-light induced spectral evolution, suggesting that the intermediates arising from the destruction of the AB dye structure are peculiar to the illumination type. During visible illumination, it is possible to observe a shift in the Amax of the AB solution from λ565nm to λ550nm, and there is an isosbestic point at λ460nm after 40 min; this suggests the presence of two chemical species in equilibrium. Finally, after 90 min of irradiation, the spectrum displayed an increase in the absorbance around λ450nm, indicating the formation of another intermediate product. Meanwhile, under UV light irradiation (Fig. 7B), a continuous decrease in Amax is observed, without peak shifts or isosbestic points. However, the expected interferences deduced from the theoretical calculations indicate that the correlation between Amax and Cn of the AB dye is not valid; consequently, the % Cdeg value of the AB dye cannot be determined (Table 3). This suggests that AB dye is not a good probe molecule.
Mineralization
The degradation of the original organic probe-molecule to CO2 and H2O is the final goal of the photocatalytic process of organic pollutants, where the total organic carbon content (% CTOCn) at a given irradiation time is reduced to zero.1,3,4 The organic probe-molecule breaks down into intermediate products, which are finally mineralized through the oxidative attack of powerful oxidizing species, i.e. the hydroxyl radical, or by direct attack of the photogenerated holes.75,86 As described before, it is not possible to evaluate mineralization using spectrophotometric techniques. However, if no spectral changes are observed even during long periods of irradiation, % CTOCn will be unaltered, and the photodegradation and mineralization will certainly be negligible. If spectral changes occur that are not associated to adsorption, such as a decrease in Amax, a shift of the absorbance peak (usually to lower wavelengths) or the appearance of new absorbance peaks (also at lower wavelengths), it is worthwhile to perform a total organic carbon (TOC) content analysis. To perform this analysis, it is very important to determine the effective carbon content available for mineralization:| % CTOC1 = % CTOC0 − % θTOCads | (9) |
The percentage of mineralization at a given time is then calculated from the effective carbon content as follows:
| % Mine = % CTOC1 − % CTOCn | (10) |
As observed by these definitions, the fraction of adsorbed molecules must be properly considered to avoid false values for both the photodegradation (% Cdeg)53 and mineralization (% Mine) results.
Following the case study, the mineralization induced by the α/β-Bi2O3 material was evaluated in a complementary experiment by comparing the effective or initial total organic carbon (% CTOC1) of the dye solution with the TOC values after irradiation for extended periods of time (% CTOCn). The dye concentration, photocatalyst load and light illumination conditions were the same as described above; the pH was nearly neutral for the RhB and IC dyes and basic (9.5) for the AB dye in order to limit the adsorption. The TOC analysis was carried out using a Shimadzu TOC-L analyzer with a high sensibility column.
Fig. 11 shows the decrease of the % CTOCn values achieved by the α/β-Bi2O3 photocatalyst for the three dye solutions at different visible light exposure times. The percentages of photodegradation (% Cdeg) measured by the spectrophotometric technique are also included in Fig. 11. The figure shows that during the first 60 min of irradiation, the decrement of % CTOCn is comparable to the adsorption percentage measured by the spectrophotometric technique, suggesting that the decreased carbon content in solution is mainly due to the adsorption (% θTOCads). At longer irradiation times (360 min), % CTOCn values of 52%, 81% and 56% were achieved for RhB, IC and AB, respectively. However, using eqn (9) and (10), the % Mine values actually achieved (Fig. 12) are 32%, 11% and 26% for RhB, IC and AB, respectively. These values are lower than those obtained for the % Cdeg; this suggests that during the spectrophotometric experiments, we observed photodiscoloration of the dye, but the intermediate products (% ITOCi) remained in the solution without being degraded. Therefore, this confirms that the estimated % Cdeg does not necessarily reflect the mineralization of the probe-molecule.
 | ||
| Fig. 11 Percentage of visible-light photodegradation or conversion of the IC and RhB dyes, where a good estimation of the C/C0 data could be obtained, and the TOC decrease percentages of the three dye solutions at longer irradiation times under visible light. For the AB dye at alkaline pH, saturation of % CTOCn was not observed up to 360 min. | ||
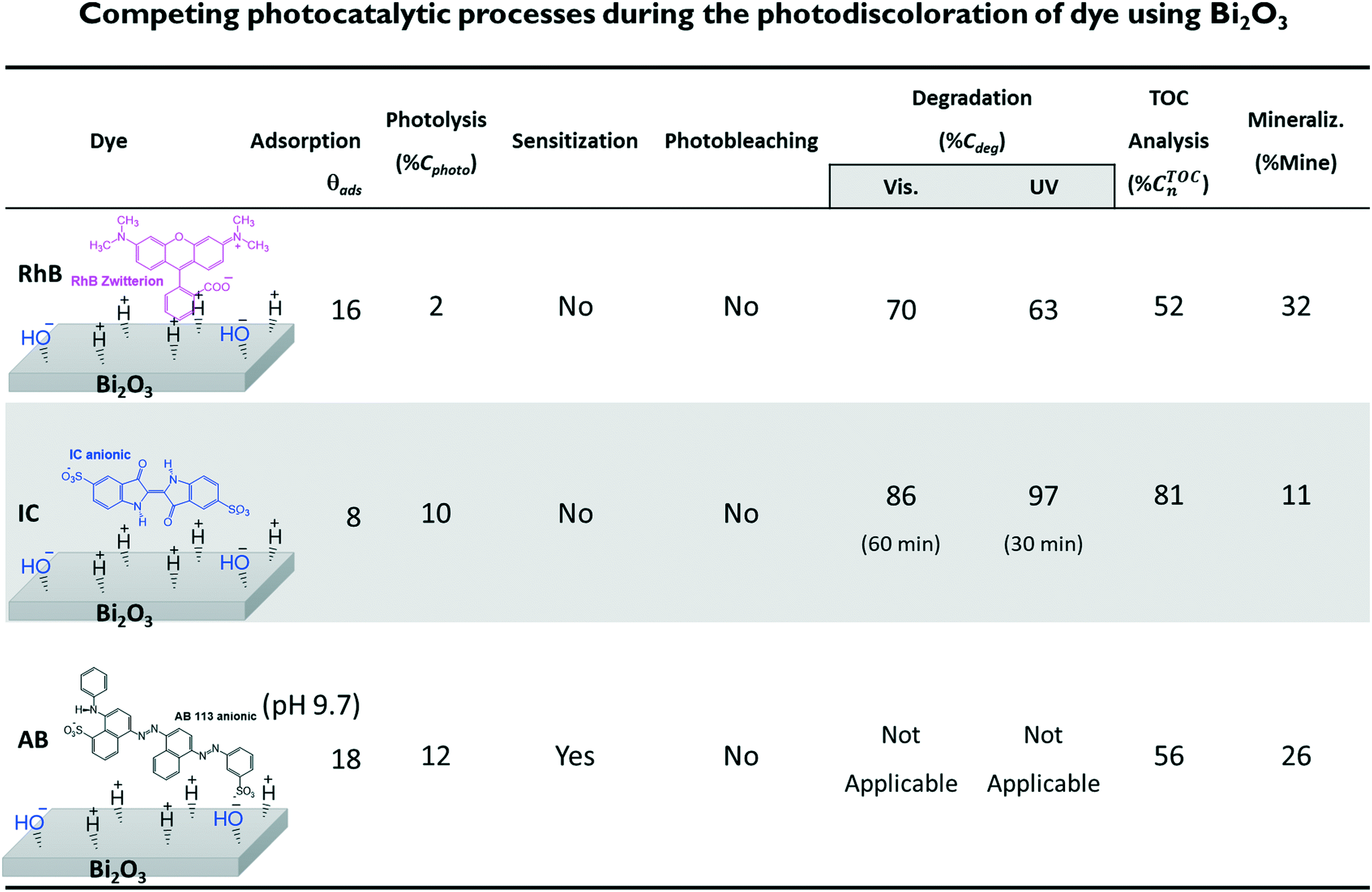 | ||
| Fig. 12 Summary of the results obtained from the competing processes during dye photodiscoloration using the α/β-Bi2O3 composite. The value of the degradation and the final TOC value were estimated correcting the initial concentration according to the percentage of adsorption for each dye. | ||
Fig. 11 also shows that for the RhB and IC dyes, saturation of the % CTOCn occurred at 180 min; this is unexpected because the formation of reactive oxygen species and holes, which are responsible for the mineralization, is typically linear during the irradiation time.87–89 This saturation is an indication that the % CTOCn value obtained at 180 min is probably due to a decrease of the TOC contribution from the intermediate products (% ITOCi) in solution due to their adsorption on the surface of the semiconductor. The spectrophotometric technique provides us with information about the adsorption of the dye molecules but not about the adsorption of the intermediates. Similar results were reported in ref. 90 for the IC dye, in which complete mineralization was not achieved using α-Bi2O3.
The corrected values for % Mine using the effective initial TOC values after adsorption are presented in Fig. 12, including a summary of the data obtained during the different steps. The low % Mine suggests that the intermediates created during the irradiation by sensitization and/or photodegradation remain in the solution.
The message is that TOC analysis, similar to the spectrophotometric technique, also requires correction due to adsorption in order to provide reliable information about the photocatalytic activity of semiconductors.
Discussion
The aim of this paper is to show that by combining theoretical and experimental data, it is possible to distinguish and properly quantify the different processes occurring during the evaluation of the photocatalytic activity of semiconductors using dye solutions, which is of significant importance to reach adequate conclusions. Three dyes were employed to this purpose. For each dye, spectrophotometric measurements of the absorbance spectra were used to monitor the photolysis (% Cphoto) and the photodegradation (% Cdeg) using UV and visible irradiation, while the adsorption–desorption equilibrium was evaluated in the dark. In order to distinguish photodegradation from photobleaching, the theoretical absorbance spectra of the leuco-dye forms and the degradation products were calculated and compared with the experimental data. For the three dyes used, photodegradation was observed; therefore, using a non-spectrophotometric technique, we proceeded to estimate the percentage of decrease of the total organic carbon (% CTOCn).The results are summarized in Fig. 12, where the percentage of photodegradation (% Cdeg), the TOC decrease (% CTOCn) and the mineralization (% Mine) are reported. Moreover, based on the analysis of the spectrum and the response under UV/visible light, we concluded that for the AB dye, the results can be influenced by sensitization and eqn (1) cannot be applied. Therefore, for the AB dye, estimations of the percentage of degradation and reaction rate will be misleading.
The reported % Cdeg are absolute values corrected according to the effective initial concentration (C1); this was not falsified by the adsorption, as has been observed in many other studies.46,53–55,91 By this, we do not mean that adsorption is not essential, because it is known that photoinduced reactions occur on the surface and, therefore, the adsorption of dye molecules on the photocatalyst is a requirement for photodegradation. However, as C1 decreases, the % Cdeg along with the efficiency of the photocatalyst increase.20 Thus, comparison of the % Cdeg values of different samples requires similar C1 values (equal C0 values is not enough). Otherwise, the material with the larger θads will be falsely selected as the best photocatalyst because it shows a larger decrease in both the absorbance and the TOC due to the lower initial concentration of probe-molecules in solution. For example, Basu et al.5 compared the visible light photodegradation of methylene blue solutions using ZnO and ZnO/CuS nanostructures using the same initial concentration (C0). However, 72% of the dye was adsorbed on the ZnO/CuS in the dark, while no adsorption was observed for the ZnO. Although the authors measured the adsorption in the dark, no corrections were applied to estimate the percentage of photodegradation; therefore, they concluded that the ZnO/CuS nanostructures performed better, although a proper estimation would provide marginal differences. A similar analysis comparing Ag3PO4 and bentonite-supported Ag3PO4 (EB-Ag3PO4) photocatalysts was reported by Ma et al.62 The authors reported that the RhB degradation by the EB-Ag3PO4 composite reached about 95% discoloration under visible light irradiation, which was much higher than that of Ag3PO4 (82%) for the same irradiation time; however, the difference in the dark-adsorption was much larger (50% for EB-Ag3PO4 in comparison to 15% for Ag3PO4). In such a case, it is not appropriate to conclude that the activity of the supported catalyst was enhanced, as the authors did. This adsorption/degradation mechanism has been reported by different authors92–99 as a synergistic decontamination method. However, herein, we are concerned with selecting a semiconductor material or a heterojunction as a good photocatalyst in which the organic molecule is mineralized by the action of sunlight, not adsorbed. Adsorption is clearly important; however, because there is a strong dependence of the photodegradation kinetic rate on the initial pollutant concentration,81,100–102 comparisons between different materials should be performed at similar initial pollutant concentrations.103
In addition to the correct estimation of the adsorption, our results suggest that quantum chemical calculations provide useful information about the degradation or discoloration pathways of dyes of interest. It was shown that IC is a good probe molecule because photobleaching and photodegradation can be clearly distinguished by changes in the spectrum and IC intermediates are easily identified. Meanwhile, for RhB, the distinction between photobleaching and photodegradation requires further experiments; however, it can provide spectral information about sensitization. Finally, AB is not recommended for photocatalysis experiments due to the large number of intermediates whose absorbances interfere with the AB absorbance.
Deep understanding of the reactions involved during photodegradation and mineralization processes requires other measurements, such as the direct detection of ˙OH free radicals,87 the use of hole, electron and radical scavengers104,105 and, from the point of view of materials, the determination of the conduction and valence band positions relative to the standard redox potentials of the adsorbed species.32 Here, we only propose a methodology to perform a proper assessment of the potential photocatalytic activity of a semiconductor material.
From a practical point of view, the aim of wastewater treatment is to diminish its ecological impact by decreasing the light absorption (color removal) and/or inhibiting the toxicity of residual molecules to living organisms. Therefore, after photodiscoloration, evaluation of the total mineralization is important. When the degradation of the parent pollutants and their intermediate compounds into CO2 and H2O is not complete and some residual contaminants remain in the water, it is necessary to assess the potential ecological and health risks of the degradation products.
Conclusions
The main conclusions from this work are summarized in Fig. 13, where we propose a methodology for the proper evaluation of the photocatalytic properties of semiconductors using standard techniques such as spectrophotometric and TOC analysis. | ||
| Fig. 13 Proposed flow diagram for the photocatalytic evaluation of semiconductors. Numbers indicate the experiment to perform (hexagons) and the corresponding information achieved for each experiment (rectangles). SC refers to the semiconductor material either as a powder or a thin film. | ||
By following this methodology, it is possible to reach correct conclusions about the photocatalytic activity of a material without relying only on the decrease of the absorbance (Amax). As described above, the absorbance reduction can be due to either photodegradation of the molecules or to other non-photocatalytic processes (photobleaching, photolysis, sensitization). From the analysis, we conclude that all four processes discussed are important; however, adsorption appears to be the most critical because it is larger and although it is commonly measured, it is not properly quantified as a non-photocatalytic process. Adsorption should be measured in the dark at different initial concentrations and under the exact experimental conditions (pH, photocatalyst load, stirring rate) of the photodegradation experiments. We suspect that many of the misunderstandings about the photocatalytic activity of semiconductors using dyes as test molecules may arise from neglecting the degree of adsorption because this leads to errors in the effective initial concentration of the dye, which invalidates comparisons between different materials or probe-molecules. Sensitization can be identified by comparing the response of the material using illuminations that are resonant and non-resonant with the electronic absorption of the molecule. Additionally, we propose the use of quantum chemical calculations to distinguish between photobleaching and photodegradation but also to identify the contribution of possible degradation products to the absorbance spectra.
The degradation of the molecule is initiated with the molecular cleavage of the chromophore group due to interactions with the reactive oxygen species (superoxides, hydroxyl radicals or holes) and is identified by a decrease in the maximum absorbance. However, for some molecules, the degradation products or intermediates absorb light in the same spectral region; thus, it is impossible to make a correlation between the absorbance and concentration of the dye in solution. This effect is usually neglected because only the maximum value is recorded. The methodology proposed here, which is based on experimental–theoretical analysis, requires the acquisition and analysis of entire absorbance spectra.
Finally, to distinguish between photodegradation and photodiscoloration and to determine the degree of mineralization, it is necessary to perform quantification of the total organic carbon in solution by an independent technique. The degree of mineralization must be corrected by considering the fraction of adsorbed molecules.
The example material chosen here to illustrate the methodology is the α/β-Bi2O3 heterostructure; according to the spectrophotometric response, it appears to be very effective for the three dyes. However, a careful revision of the evolution of the absorption spectra and the TOC decrease indicated that this material has low photocatalytic activity for water treatment. The conclusion should be that based on the lower values of % Mine obtained by TOC in comparison to the estimated % Cdeg inferred from the absorbance spectra, the tested semiconductor (α/β-Bi2O3) is very effective to induce photodiscoloration of the dye molecules; however, the intermediate products remain in solution without degradation.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
This project was financially supported by the EU project PHOCSCLEEN (FP7-PEOPLE-2012-IRSES reference 318977). S. E. Rodil acknowledges support from the CONACYT 251279 project and UNAM-PAPIIT IN100116. M. Bizarro acknowledges support from UNAM-PAPIIT projects IN106015 and IN108618. Tanveer A. Gadhi personally thanks HEC, Pakistan for the scholarship.Nomenclature
| A | Absorbance according to the Beer–Lambert law (A = ε l C) |
| AB | Acid blue 113 dye |
| A–D | Adsorption–desorption equilibrium |
| A max | Maximum absorbance value of the probe-molecule measured at a fixed wavelength |
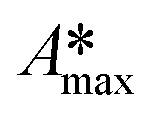
| Maximum absorbance value of the intermediate products measured at a fixed wavelength |
| C n | Concentration of dye in solution acquired from Amax |
| C(t) | Concentration of the probe-molecule in solution at a specific time of irradiation |
| C 0 | Initial concentration of the probe-molecule in solution (equal to unity) |
| C 1 | Concentration of the probe-molecule in solution after adsorption–desorption equilibrium |
| C ads | Relative concentration (C/C0) of the probe-molecule after the adsorption process |
| C photo | Relative concentration (C/C0) of the probe-molecule after the photolysis process |
| C deg | Relative concentration (C/C1) of the probe-molecule after the photodegradation process using the photocatalyst material |
| C TOC | Concentration of the probe-molecule after the photodegradation process using TOC analysis |
| % CTOC0 | Total organic carbon of the original dye solution, equal to 100% |
| % CTOC1 | Theoretical or experimental effective total organic carbon of the dye solution |
| % CTOCn | Total organic carbon of the irradiated dye solution by TOC analysis |
| % Cphoto | Percentage of degradation of the probe-molecule by the photolysis process |
| % Cdisc | Percentage of discoloration of the probe-molecule by the photocatalytic process |
| % Cdeg | Percentage of degradation of the probe-molecule by the photocatalytic process |
| % Mine | Percentage of mineralization of the probe-molecule by the photocatalytic process calculated by the TOC analysis value |
| IC | Indigo carmine dye |
| I i | Concentration of intermediate products formed from dye degradation |
| K app | Apparent kinetic rate constant of the photodegradation of the probe-molecule |
| k r | Rate constant of the photodegradation of the probe-molecule |
| K S | Adsorption constant of the probe-molecule |
| K Si | Adsorption constant of intermediate products formed from dye degradation |
| L–H | Langmuir–Hinshelwood |
| PZC | Point of zero charge of a material |
| r | Rate of the reaction during the photocatalytic process |
| RhB | Rhodamine-B dye |
| ROS | Reactive oxygen species |
| S | Sensitization of the organic molecule by light |
| SMD | Solvation model based on density |
| TD-DFT | Time-dependent-density functional theory |
| TOC | Total organic carbon |
| ε | Absorptivity coefficient of the organic molecule |
| λ | Fixed wavelength of the absorbance of the dye |
| λ max | Wavelength of Amax of each probe-molecule |
| θ ads | Fraction of probe-molecules adsorbed on the photocatalyst surface |
| % θTOCads | Total organic carbon calculated from the fraction of probe-molecules adsorbed on the photocatalyst surface |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors fully acknowledge the contributions of Dr. Pravin Jagdale, Dr. Mauro Giorcelli and Dr. Mauro Raimondo for technical assistance.This work was carried out using a NES supercomputer provided by Dirección General de Cómputo y Tecnologías de Información y Comunicación (DGTIC), Universidad Nacional Autónoma de México (UNAM). We would like to thank the DGTIC of UNAM for their excellent and free supercomputing services.
References
- D. F. Oliveira, P. S. Batista, P. S. Muller, V. Velani, M. D. França, D. R. De Souza and A. E. Machado, Dyes Pigm., 2012, 92, 563–572 CrossRef CAS.
- S. Bagheri, A. TermehYousefi and T.-O. Do, Catal. Sci. Technol., 2017, 7, 4548–4569 RSC.
- M. Rochkind, S. Pasternak and Y. Paz, Molecules, 2014, 20, 88–110 CrossRef PubMed.
- J.-M. Herrmann, Catal. Today, 1999, 53, 115–129 CrossRef CAS.
- M. Basu, N. Garg and A. K. Ganguli, J. Mater. Chem. A, 2014, 2, 7517–7525 RSC.
- C. P. Ireland, R. G. Palgrave, S. C. Bennett, A. W. J. Smith, J. H. Clark, J. R. Darwent, J. B. Claridge, S. Poulston and M. J. Rosseinsky, J. Mater. Chem. A, 2016, 4, 12479–12486 RSC.
- L. Li, J. Deng, R. Yu, J. Chen, Z. Wang and X. Xing, J. Mater. Chem. A, 2013, 1, 11894–11900 RSC.
- L. Mohapatra and K. Parida, J. Mater. Chem. A, 2016, 4, 10744–10766 RSC.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- N. M. Mahmoodi, M. Arami, N. Y. Limaee and N. S. Tabrizi, Chem. Eng. J., 2005, 112, 191–196 CrossRef CAS.
- F. M. D. Chequer, D. P. de Oliveira, E. R. A. Ferraz, G. A. R. de Oliveira, J. C. Cardoso and M. V. B. Zanoni, Textile dyes: dyeing process and environmental impact, INTECH Open Access Publisher, 2013 Search PubMed.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS.
- M. A. Rauf and S. S. Ashraf, Chem. Eng. J., 2009, 151, 10–18 CrossRef CAS.
- M. Vautier, C. Guillard and J.-M. Herrmann, J. Catal., 2001, 201, 46–59 CrossRef CAS.
- B. Ohtani, Phys. Chem. Chem. Phys., 2014, 16, 1788–1797 RSC.
- K. Rajeshwar, M. E. Osugi, W. Chanmanee, C. R. Chenthamarakshan, M. V. B. Zanoni, P. Kajitvichyanukul and R. Krishnan-Ayer, J. Photochem. Photobiol., C, 2008, 9, 171–192 CrossRef CAS.
- J.-M. Herrmann, Appl. Catal., B, 2010, 99, 461–468 CrossRef CAS.
- A. V. Emeline, V. K. Ryabchuk and N. Serpone, J. Phys. Chem. B, 2005, 109, 18515–18521 CrossRef CAS PubMed.
- N. Barbero and D. Vione, Environ. Sci. Technol., 2016, 50, 2130–2131 CrossRef CAS PubMed.
- S. Bae, S. Kim, S. Lee and W. Choi, Catal. Today, 2014, 224, 21–28 CrossRef CAS.
- T. A. Gadhi, A. Hernández-Gordillo, M. Bizarro, P. Jagdale, A. Tagliaferro and S. E. Rodil, Ceram. Int., 2016, 42, 13065–13073 CrossRef CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Revision A.08 Inc., 2009 Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Phys., 2005, 123, 161103 CrossRef PubMed.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. E. Casida, H. Chermette and D. Jacquemin, J. Mol. Struct.: THEOCHEM, 2009, 914, 1–2 CrossRef CAS.
- F. Santoro and D. Jacquemin, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 460–486 CAS.
- Y. Xu and M. A. A. Schoonen, American Mineralogist, 2000, vol. 85, pp. 543–556 Search PubMed.
- S. S. Youji Li, M. Ma, Y. Ouyang and W. Yan, Chem. Eng. J., 2008, 142, 147–155 CrossRef.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2013, 117, 1383–1391 CrossRef CAS.
- Q. Xiang, J. Yu and P. K. Wong, J. Colloid Interface Sci., 2011, 357, 163–167 CrossRef CAS PubMed.
- H. Guan, L. Zhu, H. Zhou and H. Tang, Anal. Chim. Acta, 2008, 608, 73–78 CrossRef CAS PubMed.
- N. G. Asenjo, R. Santamaría, C. Blanco, M. Granda, P. Álvarez and R. Menéndez, Carbon, 2013, 55, 62–69 CrossRef CAS.
- D. F. Ollis, J. Phys. Chem. B, 2005, 109, 2439–2444 CrossRef CAS PubMed.
- Z. Khuzwayo and E. M. N. Chirwa, J. Hazard. Mater., 2015, 300, 459–466 CrossRef CAS PubMed.
- N. S. a. A. V. Emeline, Int. J. Photoenergy, 2002, 4, 91–131 CrossRef.
- K. B. Fontana, G. G. Lenzi, E. C. R. Seára and E. S. Chaves, Ecotoxicol. Environ. Saf., 2018, 151, 127–131 CrossRef CAS PubMed.
- Y. Kohno, S. Kitamura, T. Yamada, K. Sugihara and S. Ohta, Life Sci., 2005, 77, 601–614 CrossRef CAS PubMed.
- J.-X. Ma, H. Yang, S. Li, R. Ren, J. Li, X. Zhang and J. Ma, RSC Adv., 2015, 5, 97520–97527 RSC.
- K. Gude, V. M. Gun'ko and J. P. Blitz, Colloids Surf., A, 2008, 325, 17–20 CrossRef CAS.
- V. Etacheri, R. Roshan and V. Kumar, ACS Appl. Mater. Interfaces, 2012, 4, 2717–2725 CrossRef CAS PubMed.
- H.-Y. Xu, L.-C. Wu, H. Zhao, L.-G. Jin and S.-Y. Qi, PLoS One, 2015, 10, e0142616 CrossRef PubMed.
- S. Merouani, O. Hamdaoui, F. Saoudi and M. Chiha, Chem. Eng. J., 2010, 158, 550–557 CrossRef CAS.
- A. G. S. Prado, L. B. Bolzon, C. P. Pedroso, A. O. Moura and L. L. Costa, Appl. Catal., B, 2008, 82, 219–224 CrossRef CAS.
- S. Mohammadzadeh, M. E. Olya, A. M. Arabi, A. Shariati and M. R. K. Nikou, J. Environ. Sci., 2015, 35, 194–207 CrossRef CAS PubMed.
- E. Kordouli, K. Bourikas, A. Lycourghiotis and C. Kordulis, Catal. Today, 2015, 252, 128–135 CrossRef CAS.
- J. Xiao, H. Zhang, Y. Xia, Z. Li and W. Huang, RSC Adv., 2016, 6, 39861–39869 RSC.
- K. Tanaka, K. Padermpole and T. Hisanaga, Water Res., 2000, 34, 327–333 CrossRef CAS.
- L. Shan, G. Wang, D. Li, X. San, L. Liu, L. Dong and Z. Wu, Dalton Trans., 2015, 44, 7835–7843 RSC.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505 CrossRef CAS PubMed.
- S. Wang, Y. Zhang, T. Zhang, F. Dong and H. Huang, Readily attainable spongy foam photocatalyst for promising practical photocatalysis, 2017 Search PubMed.
- J. Piella, F. Merkoci, A. Genc, J. Arbiol, N. G. Bastus and V. Puntes, J. Mater. Chem. A, 2017, 5, 11917–11929 RSC.
- T. Bechtold, A. Turcanu, S. Geissler and E. Ganglberger, Bioresour. Technol., 2002, 81, 171–177 CrossRef CAS PubMed.
- N. Srividya, G. Paramasivan, K. Seetharaman and P. Ramamurthy, J. Chem. Soc., Faraday Trans., 1994, 90, 2525–2530 RSC.
- A. Hernández-Gordillo, V. Rodríguez-González, S. Oros-Ruiz and R. Gómez, Catal. Today, 2016, 266, 27–35 CrossRef.
- C. Pan, J. Xu, Y. Chen and Y. Zhu, Appl. Catal., B, 2012, 115–116, 314–319 CrossRef CAS.
- R. Pol, M. Guerrero, E. García-Lecina, A. Altube, E. Rossinyol, S. Garroni, M. D. Baró, J. Pons, J. Sort and E. Pellicer, Appl. Catal., B, 2016, 181, 270–278 CrossRef CAS.
- J. Ma, Q. Liu, L. Zhu, J. Zou, K. Wang, M. Yang and S. Komarneni, Appl. Catal., B, 2016, 182, 26–32 CrossRef CAS.
- Z. Wu, X. Yuan, G. Zeng, L. Jiang, H. Zhong, Y. Xie, H. Wang, X. Chen and H. Wang, Appl. Catal., B, 2018, 225, 8–21 CrossRef CAS.
- N. U. Silva, T. G. Nunes, M. S. Saraiva, M. S. Shalamzari, P. D. Vaz, O. C. Monteiro and C. D. Nunes, Appl. Catal., B, 2012, 113–114, 180–191 CrossRef CAS.
- S. Rasalingam, R. Peng and R. T. Koodali, Appl. Catal., B, 2015, 174–175, 49–59 CrossRef CAS.
- G. Liu and J. Zhao, New J. Chem., 2000, 24, 411–417 RSC.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- R. Camarillo and J. Rincón, Chem. Eng. Technol., 2011, 34, 1675–1684 CrossRef CAS.
- D. Chatterjee and A. Mahata, J. Photochem. Photobiol., A, 2002, 153, 199–204 CrossRef CAS.
- T. Watanabe, T. Takizawa and K. Honda, J. Phys. Chem., 1977, 81, 1845–1851 CrossRef CAS.
- S.-K. Lee, A. Mills and C. O'Rourke, Chem. Soc. Rev., 2017, 46, 4877–4894 RSC.
- R. Quesada-Cabrera, A. Mills and C. O'Rourke, Appl. Catal., B, 2014, 150–151, 338–344 CrossRef CAS.
- W. Kuo and P. Ho, Chemosphere, 2001, 45, 77–83 CrossRef CAS PubMed.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6284–6292 CrossRef CAS PubMed.
- B. Liu, X. Zhao, C. Terashima, A. Fujishima and K. Nakata, Phys. Chem. Chem. Phys., 2014, 16, 8751–8760 RSC.
- S. Sarkar, R. Das, H. Choi and C. Bhattacharjee, RSC Adv., 2014, 4, 57250–57266 RSC.
- F. Chen, J. Zhao and H. Hidaka, Int. J. Photoenergy, 2003, 5, 209–217 CrossRef CAS.
- X. Hu, T. Mohamood, W. Ma, C. Chen and J. Zhao, J. Phys. Chem. B, 2006, 110, 26012–26018 CrossRef CAS PubMed.
- Z. Youssef, L. Colombeau, N. Yesmurzayeva, F. Baros, R. Vanderesse, T. Hamieh, J. Toufaily, C. Frochot, T. Roques-Carmes and S. Acherar, Dyes Pigm., 2018, 159, 49–71 CrossRef CAS.
- H. Liu, N. Gao, M. Liao and X. Fang, Sci. Rep., 2015, 5, 7716 CrossRef CAS PubMed.
- G. Cinelli, F. Cuomo, L. Ambrosone, M. Colella, A. Ceglie, F. Venditti and F. Lopez, Journal of Water Process Engineering, 2017, 20, 71–77 CrossRef.
- M. M. Sousa, C. Miguel, I. Rodrigues, A. J. Parola, F. Pina, J. S. S. de Melo and M. J. Melo, Photochem. Photobiol. Sci., 2008, 7, 1353–1359 RSC.
- M. Coelho, F. de Andrade, G. de Lima, R. Augusti, M. Ferreira, D. Maria and J. Ardisson, Appl. Organomet. Chem., 2011, 25, 220–225 CrossRef CAS.
- M. Coelho, G. De Lima, R. Augusti, D. Maria and J. Ardisson, Appl. Catal., B, 2010, 96, 67–71 CrossRef CAS.
- I. Dalmázio, A. P. F. M. de Urzedo, T. M. A. Alves, R. R. Catharino, M. N. Eberlin, C. C. Nascentes and R. Augusti, J. Mass Spectrom., 2007, 42, 1273–1278 CrossRef PubMed.
- H. Liu, A. Imanishi and Y. Nakato, J. Phys. Chem. C, 2007, 111, 8603–8610 CrossRef CAS.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2013, 117, 1383–1391 CrossRef CAS.
- K.-i. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- Q. Xiang, J. Yu and P. K. Wong, J. Colloid Interface Sci., 2011, 357, 163–167 CrossRef CAS PubMed.
- D. Sánchez-Martínez, I. Juárez-Ramírez, L. M. Torres-Martínez and I. de León-Abarte, Ceram. Int., 2016, 42, 2013–2020 CrossRef.
- M.-S. Gui, W.-D. Zhang, Q.-X. Su and C.-H. Chen, J. Solid State Chem., 2011, 184, 1977–1982 CrossRef CAS.
- A. Sandoval, C. Hernández-Ventura and T. E. Klimova, Fuel, 2017, 198, 22–30 CrossRef CAS.
- M. Zhao, Q. Yuan, H. Zhang, C. Li, Y. Wang and W. Wang, J. Alloys Compd., 2019, 782, 1049–1057 CrossRef CAS.
- J. Lyu, Z. Hu, Z. Li and M. Ge, J. Phys. Chem. Solids, 2019, 129, 61–70 CrossRef CAS.
- O. Sacco, M. Matarangolo, V. Vaiano, G. Libralato, M. Guida, G. Lofrano and M. Carotenuto, Sci. Total Environ., 2018, 644, 430–438 CrossRef CAS PubMed.
- X. Wang, J. Zhou, S. Zhao, X. Chen and Y. Yu, Appl. Surf. Sci., 2018, 453, 394–404 CrossRef CAS.
- X. Bing, X. Jian, J. Chu, J. Li and C. Guo, Mater. Res. Bull., 2019, 110, 1–12 CrossRef CAS.
- S. K. Mandal, K. Dutta, S. Pal, S. Mandal, A. Naskar, P. K. Pal, T. S. Bhattacharya, A. Singha, R. Saikh, S. De and D. Jana, Mater. Chem. Phys., 2019, 223, 456–465 CrossRef CAS.
- S. Jiao, Y. Zhao, C. Li, B. Wang and Y. Qu, Green Energy & Environment, 2019, 4, 66–74 Search PubMed.
- F. Venditti, F. Cuomo, A. Ceglie, P. Avino, M. V. Russo and F. Lopez, Langmuir, 2015, 31, 3627–3634 CrossRef CAS PubMed.
- K. Wetchakun, N. Wetchakun and S. Sakulsermsuk, J. Ind. Eng. Chem., 2019, 71, 19–49 CrossRef CAS.
- C.-H. Wu, H.-W. Chang and J.-M. Chern, J. Hazard. Mater., 2006, 137, 336–343 CrossRef CAS PubMed.
- A. Chakraborty, D. A. Islam and H. Acharya, J. Solid State Chem., 2019, 269, 566–574 CrossRef CAS.
- C.-Y. Chu and M. H. Huang, J. Mater. Chem. A, 2017, 5, 15116–15123 RSC.
- D. Wang, L. Guo, Y. Zhen, L. Yue, G. Xue and F. Fu, J. Mater. Chem. A, 2014, 2, 11716–11727 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization and optical properties of the tested semiconductor material. See DOI: 10.1039/c9cy00038k |
| ‡ Present Address: CONACYT Research Fellow. |
| This journal is © The Royal Society of Chemistry 2019 |