
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium formamidinate derivatives by hydride insertion of carbodiimides†
Joshua
Dyall
,
Michael S.
Hill
 *,
Mary F.
Mahon
,
Luke
Teh
and
Andrew S. S.
Wilson
*,
Mary F.
Mahon
,
Luke
Teh
and
Andrew S. S.
Wilson
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
First published on 26th February 2019
Abstract
The carbodiimides, Ar/RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNAr/R, (Ar = 2,6-di-isopropylphenyl (Dipp), 4-methylphenyl; R = tert-butyl, cyclohexyl, iso-propyl), react with the polar Ca–H bonds of the dimeric β-diketiminato calcium hydride, [(BDI)CaH]2 (BDI = HC{(Me)CN-2,6-i-Pr2C6H3}2), to provide a series of heteroleptic calcium formamidinate derivatives. While reactions with carbodiimides bearing more sterically demanding substituents (Dipp, t-Bu, Cy) are straightforward, and provide four-coordinate compounds, less bulky iso-propyl or 4-methylphenyl substitution results in the sequestration of a further equivalent of the carbodiimide and the isolation of a heteroleptic hydride-bridged dinuclear species. This latter observation is suggested to be a reflection of the robust dimeric constitution of the calcium hydride reagent and the reduced steric demands of the di-para-tolyl carbodiimide reagent.
CNAr/R, (Ar = 2,6-di-isopropylphenyl (Dipp), 4-methylphenyl; R = tert-butyl, cyclohexyl, iso-propyl), react with the polar Ca–H bonds of the dimeric β-diketiminato calcium hydride, [(BDI)CaH]2 (BDI = HC{(Me)CN-2,6-i-Pr2C6H3}2), to provide a series of heteroleptic calcium formamidinate derivatives. While reactions with carbodiimides bearing more sterically demanding substituents (Dipp, t-Bu, Cy) are straightforward, and provide four-coordinate compounds, less bulky iso-propyl or 4-methylphenyl substitution results in the sequestration of a further equivalent of the carbodiimide and the isolation of a heteroleptic hydride-bridged dinuclear species. This latter observation is suggested to be a reflection of the robust dimeric constitution of the calcium hydride reagent and the reduced steric demands of the di-para-tolyl carbodiimide reagent.
Introduction
Amidinate anions are among the most widely applied ligands in contemporary coordination and organometallic chemistry.1,2 Organic amidines are readily available by a variety of routes and are easily metallated by organometallic or amide bases derived from more electropositive elements. These species may, in turn, be applied in onward metathesis reactions with metal salts to provide a panoply of derivatives that encompasses the entirety of the periodic table. A further reliable route to C-alkyl amidinates involves the insertion of a carbodiimide, RNCNR (R = alkyl or aryl), into the M–C bond of a polar organometallic derivative (Scheme 1(a)). In cases where the parent formamidinate species are required, analogous access necessitates the availability of a suitably reactive, molecular metal hydride. Documented examples of this latter process include rare earth metal (Y,3–5 La6 and Lu7), transition metal (Fe,8 Zr,9 Nb, Ta10 and Re11), p-block (B,12–15 Al,16,17 Zn,18–20 Ga16 and Sn21) and s-block metal (Li22 and Mg23,24) hydrides (Scheme 1(b)).
 | ||
| Scheme 1 Carbodiimide insertion into (a) the M–C bond of a metal alkyl or aryl (b) the M–H bond of a metal hydride to provide the respective alkyl- and formamidinate derivatives. | ||
A variety of calcium amidinate species have been synthesised either by salt metathesis of a pre-formed anion with CaI2, by deprotonation of an amidine by [Ca{N(SiMe3)2}2]2 or by carbodiimide insertion into the polar Ca–C bonds of a terminal acetylide.25–34 Of most relevance to the current work, four homoleptic calcium formamidinate derivatives, [Ca{(Ar/RN)2CH}2(THF)] (Ar = 2,6-di-isopropylphenyl (Dipp), o-methylphenyl, 2,6-dimethylphenyl; R = i-Pr) have previously been reported. These latter compounds, however, were synthesised by a redox transmetallation process between the neutral amidine and elemental calcium, either in the presence of Hg(C6F5)2 or in liquid ammonia.35–37 Insertion reactions of carbodiimides with calcium hydride bonds are yet to be reported, a lacuna which may be traced to the relative dearth and only recent provenance of appropriately stable and soluble molecular calcium hydrides.32,38–48
A seminal report in s-block chemistry was provided by Harder's molecular β-diketiminato calcium hydride complex, [(BDI)CaH(THF)]2 (BDI = HC{(Me)CN-2,6-i-Pr2C6H3}2; 1).40 Although compound 1 and related species have been shown to react directly with the polar multiple bonds of small molecules containing CO, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O, CN and CN functionalities to provide the corresponding alkoxide and amide derivatives,41,49–53 its reactivity with carbodiimides appears to have escaped attention. This observation is particularly surprising given the ability of sterically demanding C-alkyl amidinate ligands to impart sufficient kinetic stability in their own right to stabilise hydrido derivatives of the heavier alkaline earth elements.32,54 We have recently reported that a non-THF solvated analogue of compound 1, compound 2 (see Scheme 2), reacts readily with the CC bonds of terminal n-alkenes to provide calcium alkyl species which display sufficient nucleophilicity to allow the direct alkylation of benzene and the heterolysis of dihydrogen under very mild conditions.55,56 In this contribution, we continue to assay the chemistry of compound 2 and report the outcome of its reactions with a representative palette of N,N-dialkyl- and N,N-diarylcarbodiimides.
O, CN and CN functionalities to provide the corresponding alkoxide and amide derivatives,41,49–53 its reactivity with carbodiimides appears to have escaped attention. This observation is particularly surprising given the ability of sterically demanding C-alkyl amidinate ligands to impart sufficient kinetic stability in their own right to stabilise hydrido derivatives of the heavier alkaline earth elements.32,54 We have recently reported that a non-THF solvated analogue of compound 1, compound 2 (see Scheme 2), reacts readily with the CC bonds of terminal n-alkenes to provide calcium alkyl species which display sufficient nucleophilicity to allow the direct alkylation of benzene and the heterolysis of dihydrogen under very mild conditions.55,56 In this contribution, we continue to assay the chemistry of compound 2 and report the outcome of its reactions with a representative palette of N,N-dialkyl- and N,N-diarylcarbodiimides.
 | ||
| Scheme 2 Synthesis of compounds 3–7. | ||
Results and discussion
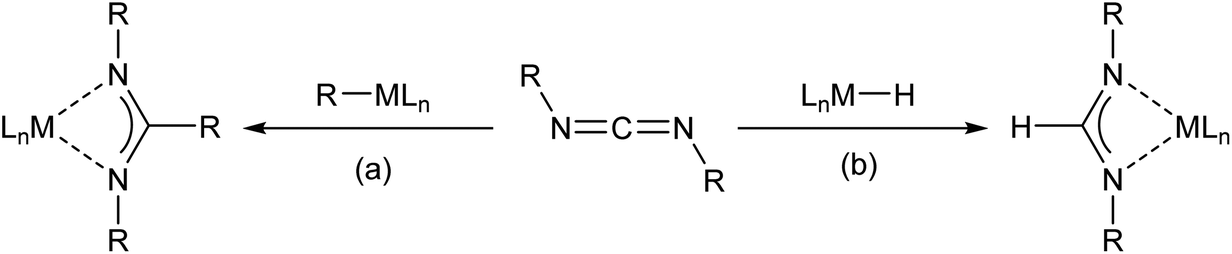
An initial NMR scale reaction between compound 2 and DippNCNDipp afforded clean access to the expected formamidinate, [(BDI)Ca(DippNC(H)NDipp)] (3, Scheme 2). The formation of compound 3 was most clearly evidenced by the appearance of a diagnostic formamidinate methine singlet resonance at δ 8.07 ppm that integrated 1H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1H with a BDI γ-CH methine signal at δ 4.83 ppm in the resultant 1H NMR spectrum. Cooling a saturated pentane solution of 3 to −35 °C afforded single crystals from which its solid state structure was determined by X-ray diffraction analysis (Fig. 1).
:1H with a BDI γ-CH methine signal at δ 4.83 ppm in the resultant 1H NMR spectrum. Cooling a saturated pentane solution of 3 to −35 °C afforded single crystals from which its solid state structure was determined by X-ray diffraction analysis (Fig. 1).
 | ||
| Fig. 1 ORTEP representation of compound 3 with thermal ellipsoids at 25%. H atoms, except for the hydrogen attached to C30, have been removed for clarity. Selected bond distances (Å) and angles (°): Ca1–N1 2.3551(10), Ca1–N2 2.3496(10), Ca1–N3 2.3877(10), Ca1–N4 2.3875(10), C30–N3 1.3250(16), C30–N4 1.3217(16), N1–Ca1–N2 81.21(4), N3–Ca1–N4 57.53(3), N4–C30–N3 120.51(11). | ||
The identity of the formamidinate anion in 3 is clearly indicated by the elongation of the C30–N3 and C30–N4 bonds [1.3250(16), 1.3217(16) Å] and the narrowing of the N4–C30–N3 angle [120.51(11)°] in comparison to the respective measurements in the carbodiimide starting material [1.213(2), 1.221(2) Å and 169.3(2)°].57 Despite the steric constraints imposed by the flanking di-isopropylphenyl groups in both the formamidinate and BDI anions, Ca1 lies in a distorted pseudo-tetrahedral environment provided solely by the four nitrogen donors and is effectively coplanar with both of the ligands [Ca1 is displaced from the mean plane defined by N3, N4, C30 by 0.075 Å and from N1, N2, C1, C2, C3, C4, C5 by 0.06 Å].58 The planes defined by the chelated ligands are not orthogonal, however, and adopt a mutual orientation which is 49.49° away from co-planarity. The Ca–N distances to the formamidinate ligand [Ca1–N3 2.3877(10), Ca1–N4 2.3875(10) Å] fall at the short end of the range reported for terminal calcium amidinates, 2.306(5)–2.525(3) Å.27
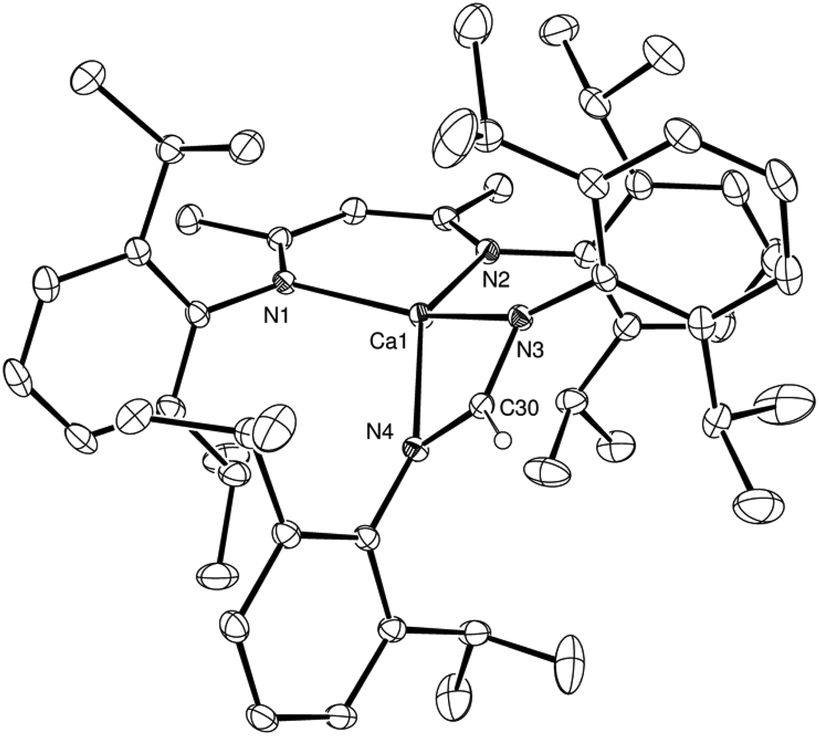
The reactivity of compound 2 was then extended to the N,N-dialkylcarbodiimides, t-BuNCNt-Bu and CyNCNCy, to afford the corresponding formamidinate derivatives, [(BDI)Ca(t-BuNC(H)t-Bu)] and [(BDI)Ca(CyNC(H)NCy)], compounds 4 and 5, respectively (Scheme 2). The formation of both 4 and 5 was again confirmed by the appearance of distinctive formamidinate methine resonances at δ 8.51 and 8.08 ppm in the resultant 1H NMR spectra. The structure of compound 4 was also determined by single crystal X-ray diffraction analysis of crystals grown from a saturated hexane solution at −35 °C (Fig. 2a).
 | ||
| Fig. 2 (a) ORTEP representation of compound 4 with thermal ellipsoids at 25%. Hexane solvent, disordered carbon atoms and H atoms, except for the hydrogen attached to C34, have been removed for clarity. (b) Space filling representation of compound 4 in the same orientation highlighting the exposed environment of the Ca1 centre. Selected bond distances (Å) and angles (°): Ca1–N1 2.3462(11), Ca1–N2 2.3420(11), Ca1–N3 2.3242(13), Ca1–N4 2.3867(11), N3–C34 1.321(2), N4–C34 1.322(2), N2–Ca1–N1 82.56(4), N3–Ca1–N4 58.57(4), N1–Ca1–N4 139.47(4), N2–Ca1–N4 137.86(4), N3–Ca1–N1 116.83(4), N3–Ca1–N2 112.61(4), N3–C34–N4 121.42(13). | ||
In a similar manner to compound 3, the formation of a formamidinate anion in 4 is indicated by the C34–N3 and C34–N4 bonds [1.321(2), 1.322(2) Å], which are in close correspondence with the analogous measurements of the previously reported tert-butyl formamidinate, [(t-BuNC(H)Nt-Bu)(B{C6F5}2)] [1.321(2), 1.314(2) Å].13 While the calcium centre lies only 0.048 Å out of the mean plane defined by N3, N4 and C30 and is, thus, effectively coplanar with the formamidinate anion, in contrast to compound 3 and presumably as a consequence of the differing steric demands of the N-alkyl substituents, the calcium centre resides some 1.37 Å out of the N1, N2, C1, C2, C3, C4, C5 plane of the BDI ligand. As a result, albeit the four Ca–N interactions [2.3462(11), 2.3420(11), 2.3242(13) and 2.3867(11) Å] lie within the normal expected range, the two chelated ligands adopt an unusual relative disposition. Most notably, the angle between the mean planes containing [Ca1, N1, N2] and [N1, N2, C1, C2, C3, C4, C5] define an angle of 127.4°, which leaves the calcium centre exposed with what appears to be an otherwise vacant coordination site (Fig. 2b). No similar asymmetry of the formamidinate or BDI environments, however, could be discerned in the solution NMR spectra. We are, thus, satisfied to ascribe the unusual coordination environment of the calcium centre in the structure of 4 to a solid state packing effect.
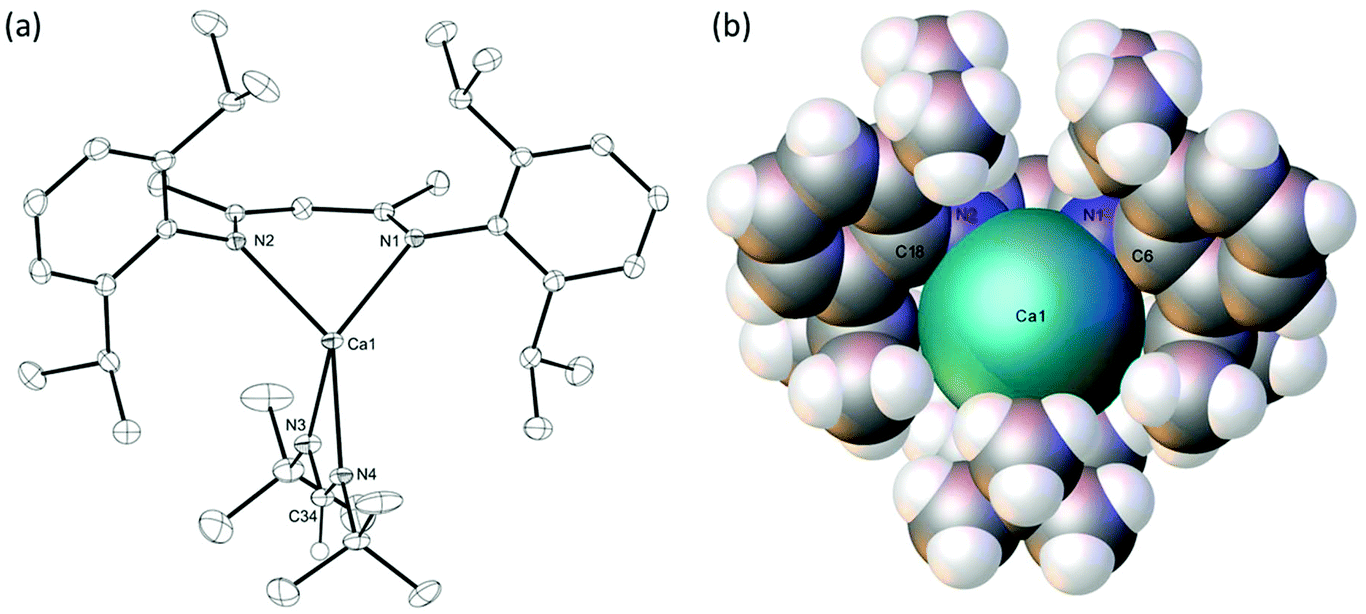
Reduction of the steric bulk of the carbodiimide reaction partner to i-PrNCNi-Pr, also afforded the corresponding formamidinate, compound 6, as evidenced by the appearance of the formamidinate methine signal at δ 8.27 ppm in its 1H NMR spectrum. In this case, however, complete conversion to a single new product necessitated the use of two molar equivalents of the carbodiimide reagent. The origin of this observation was resolved through the crystallisation of compound 6, whereupon the resultant single crystal X-ray diffraction analysis revealed that the formamidinate complex crystallises with the coordination of an equivalent of i-PrNCNi-Pr to afford a 5-coordinate calcium complex, [(BDI)Ca(iPrNC(H)NiPr)(i-PrNCNi-Pr)] (Fig. 3).
 | ||
| Fig. 3 ORTEP representation of compound 6 with thermal ellipsoids at 25%. H atoms, except for the hydrogen attached to C30, have been removed for clarity. Selected bond lengths (Å) and bond angles (°). Ca1–N1 2.352(2), Ca1–N2 2.3691(19), Ca1–N3 2.407(2), Ca1–N4 2.380(2), Ca1–N5 2.538(2), N3–C30 1.324(3), N4–C30 1.317(3), N5–C37 1.235(4), N6–C37 1.219(5), N1–Ca1–N2 81.17(7), N4–Ca1–N3 57.50(7), N1–Ca1–N3 136.05(8), N1–Ca1–N4 107.04(7), N1–Ca1–N5 95.01(7), N2–Ca1–N3 141.17(8), N2–Ca1–N4 108.32(7), N2–Ca1–N5 98.19(7), N3–Ca1–N5 90.09(7), N4–C30–N3 121.3(2), N6–C37–N5 168.8(6). | ||
The C30–N3 and C30–N4 bonds [1.324(3), 1.317(3) Å] and the N4–C30–N3 angle [121.3(2)°] in compound 6 are comparable with the corresponding measurements within Gordon's homoleptic calcium N,N-di-isopropyl formamidinate complex, [(iPrNC(H)NiPr)2Ca]2, [1.28(3)–1.328(6) Å and 118.6(4)–122.3(4)°].37 The calcium centre of 6 is five-coordinate with four nitrogen donor interactions provided by the formally anionic formamidinate and BDI ligands [2.352(2)–2.407(2) Å]. The coordination sphere of the calcium is completed by a longer nitrogen interaction [2.538(2) Å] from the molecule of i-PrNCNi-Pr, that effectively occupies the ‘open’ coordination site of the calcium atom that was a notable feature of the structure of compound 4. This observation is underscored by a calculated τ5 value of 0.09, which is indicative of a coordination geometry approaching square pyramidal with the N1 atom of the BDI ligand providing the apex of the pyramid.59
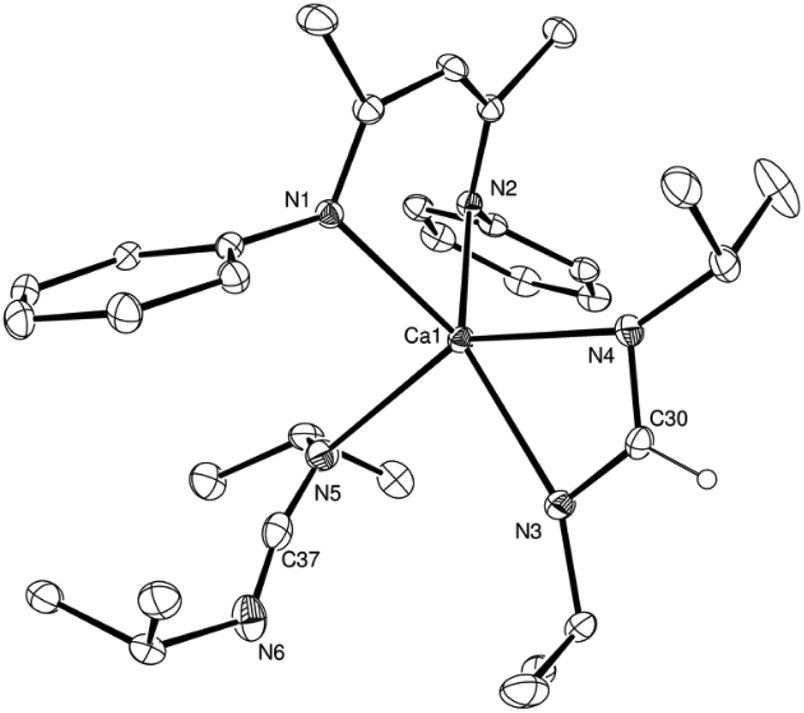
A further reaction at room temperature with the N-aryl carbodiimide, p-tolNCNp-tol, afforded a mixed formamidinate-hydride complex, [{BDI}(H)(p-tolNC(H)Np-tol)] (7, Scheme 2), irrespective of the reaction stoichiometry. The formation of compound 7 was clearly apparent from the appearance of unique formamidinate and BDI methine signals (δ 8.04, 4.95 ppm), which were observed to emerge alongside a further singlet resonance that was assigned to the maintenance of a Ca–H environment at δ 4.41 ppm and which integrated in a respective 1H:2H:1H ratio in the resultant 1H NMR spectrum. The structure of compound 7 was confirmed by a further single crystal X-ray diffraction analysis of crystals formed by cooling a saturated hexane solution to −35 °C (Fig. 4).
 | ||
| Fig. 4 ORTEP representation of the Ca1/Ca2-containing molecule in the asymmetric unit of compound 7 with thermal ellipsoids at 25%. The Ca3/Ca4-containing molecule, H atoms, except for H1 and the hydrogen attached to C37, and i-Pr groups have been omitted for clarity. Selected bond distances (Å) and angles (°): Ca1–N1 2.3289(11), Ca1–N2 2.3360(11), Ca1–N3 2.4055(12), Ca2–N4 2.4207(12), Ca2–N5 2.3485(11), Ca2–N6 2.3264(10), N3–C37 1.321(2), N4–C37 1.321(2), N1–Ca1–N2 80.57(4), N6–Ca2–N5 80.79(4), N4–C37–N3 124.82(13). | ||
The asymmetric unit of compound 7 comprises two similar di-calcium molecules in which dimerization is propagated through a single amidinate anion, which spans both Ca centres, and a residual μ2-Ca–H–Ca bridging interaction. The C–N bonds [C37–N3 1.321(2), C37–N4 1.321(2), N9–C110 1.321(2), N10–C110 1.324(2) Å] of the formamidinate anions within both entities of the asymmetric unit are closely comparable with the analogous measurements within the previously reported magnesium complex of the same ligand, [(p-tolNC(H)Np-tol)2Mg(THF)2] [1.312(3)–1.323(3) Å].60 In contrast, the chelating disposition adopted by the ligands about magnesium in this previously described complex subtend significantly narrower N–C–N angles [116.0(3), 117.9(3)°] than either of the bridging formamidinate anions in the structure of 7 [N4–C37–N3, 124.82(13); N9–C110–N10 125.05(13)°]. The bridging mode adopted also contrasts significantly with that observed in the only other amidinate-bridged calcium complexes, the aforementioned [(iPrNC(H)NiPr)2Ca]2 and a similarly homoleptic species comprising a propargyl-substituted anion.37,61 Whereas the bridging amidinate nitrogen atoms in both of these previously reported species are 4-coordinate, engaging with both calcium centres in a μ2-Ca–N–Ca fashion, N3 and N4 within the structure of compound 7 interact solely with Ca1 and Ca2, respectively. Despite this reduction in the number of nitrogen-to-element bonds from 4 to 3, the resultant Ca1–N3 and Ca2–N4 distances [2.4055(12), 2.4207(12) Å] in 7 are not notably shorter than the ranges established previously [2.400(3)–2.650(3) Å]. Both calcium centres, thus, reside in four-coordinate environments, which are completed by two Ca–N interactions from the BDI ligands and the bridging hydride. Attempts to react the remaining hydride of compound 7 with a further equivalent of p-tolNCNp-tol at elevated temperatures were unsuccessful and merely resulted in ligand redistribution to various species, including the known bis-β-diketiminate derivative, [Ca(BDI)2].62,63 We have previously noted that the reactivity of compound 2 with the CC bonds of terminal alkenes occurs with the retention of the relatively robust hydrido-bridged dimer.55,56 The formation of compounds 3–6 necessarily ensues with the generation of mononuclear calcium species, which is most likely a reflection of the significant steric bulk of the nitrogen-bound hydrocarbon residues. Notably, inspection of the resultant 1H NMR spectrum immediately after addition of a single further equivalent of i-PrNCNi-Pr to an isolated sample of compound 7 demonstrated the disappearance of the residual hydride resonance at δ 4.41 ppm and the generation of an equimolar mixture of compound 6 and a new amidinate species, which we assign as the further amidinate complex [(BDI)Ca(p-tolNC(H)Np-tol)] (Fig. S11†). Although not pursued further, we, thus, tentatively suggest that the dinuclear constitution of compound 7 is a consequence of both the reduced steric demands of the N-p-tolyl substituents and the lower basicity of p-tolNCNp-tol. The consequent inability of a single equivalent of p-tolyl carbodiimide to induce dimer fragmentation during the initial insertion process, thus, restricts access to the remaining Ca–H bond.
In conclusion, reactions between the dimeric calcium hydride, [(BDI)CaH]2 and representative carbodimide small molecules are facile and result in the generation of well-defined formamidinate products. X-ray crystallographic studies reveal that the resultant calcium coordination environments and coordination numbers are strongly dependent upon the relative steric demands of the N-aryl or N-alkyl substituents.
Experimental details
General considerations and starting materials
All manipulations were carried out using standard Schlenk line and glovebox techniques under an inert atmosphere of argon. NMR experiments were conducted in J Young tap NMR tubes prepared and sealed in a Glovebox. NMR spectra were collected on a Bruker AV300 spectrometer operating at 300.2 MHz (1H), 75.5 MHz (13C) or an Agilent ProPulse spectrometer operating at 500 MHz (1H), 126 MHz (13C). The spectra were referenced relative to residual protio solvent resonances. Solvents (toluene, hexane, pentane) were dried by passage through a commercially available (MBraun) solvent purification system, under nitrogen and stored in ampoules over 4 Å molecular sieves. C6D6 was purchased from Sigma-Aldrich Corp., dried over a potassium mirror before vacuum distilling under argon and storing over molecular sieves. Calcium iodide (99.95%) and carbodiimide reagents were purchased from Sigma-Aldrich Corp. and used without further purification. [(BDI)CaH]2 (2) was synthesised by a literature procedure.55[(BDI)Ca(DippNC(H)NDipp)] (3)
Toluene (10 ml) was added to 2 (0.92 g, 1.00 mmol) and DippNCNDipp (0.73 g, 2.01 mmol) then stirred overnight at room temperature. The resulting solution was evaporated to dryness, redissolved in pentane (10 ml), cannula filtered and concentrated. Colourless crystals were deposited overnight at −35 °C and collected via cannula filtration to yield compound 3. (0.94 g, 57%). 1H NMR (500 MHz, benzene-d6) δ 8.07 (s, 1H, NC(H)N), 7.06–6.98 (m, 12H, Ar–H), 4.83 (s, 1H, NC(CH3)CH), 3.20 (hept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 3.04 (hept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 1.58 (s, 6H, NC(CH3)CH), 1.17 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.12 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.05 (d, 3JHH = 6.8 Hz, 24H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, benzene-d6) δ 170.0 (NC(H)N), 167.5 (NC(CH3)CH), 146.5, 145.8 (Cipso), 141.8, 141.7 (Cortho), 125.3, 124.2 (Cpara), 123.4, 123.3 (Cmeta), 94.1 (NC(CH3)CH), 28.9, 28.8 (CH(CH3)2), 25.3 (CH(CH3)2), 24.9 (NC(CH3)CH), 24.4, 24.0 (CH(CH3)2) ppm. Elemental analysis for C54H76CaN4: C, 78.97; H, 9.33; N, 6.82%. Found: C, 78.81; H, 9.48, N, 6.73%.
[(BDI)Ca(t-BuNC(H)t-Bu)] (4)
Toluene (10 ml) was added to 2 (0.92 g, 1.00 mmol) and tBuNCNtBu (386 μl, 2.00 mmol) then stirred at room temperature for ca. 1 hour. The resulting solution was evaporated to dryness, redissolved in hexane (10 ml), cannula filtered and concentrated. Colourless crystals were deposited overnight at −35 °C and collected via cannula filtration to yield compound 4. (0.55 g, 45%). 1H NMR (500 MHz, benzene-d6) δ 8.51 (s, 1H, NC(H)N), 7.21–7.11 (m, 6H, Ar–H), 4.87 (s, 1H, NC(CH3)CH), 3.26 (hept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 1.74 (s, 6H, NC(CH3)CH), 1.29 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.27 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 0.99 (s, 18H, NC(CH3)3) ppm. 13C{1H} NMR (126 MHz, benzene-d6) δ 166.1 (NC(CH3)CH), 164.3 (NC(H)N), 146.8 (Cipso), 141.4 (Cortho), 124.6 (Cpara), 123.9 (Cmeta), 91.4 (NC(CH3)CH), 51.3 (NC(CH3)3), 32.5 (NC(CH3)3), 28.8 (CH(CH3)2), 25.8 (CH(CH3)2), 24.8 (NC(CH3)CH), 24.3 (CH(CH3)2) ppm. Elemental analysis for C38H60CaN4: C, 74.46; H, 9.87; N, 9.14%. Found: C, 74.21; H, 9.98, N, 9.22%.
[(BDI)Ca(CyNC(H)NCy)] (5)
Toluene (10 ml) was added to 2 (0.92 g, 1.00 mmol) and CyNCNCy (0.41 g, 1.99 mmol) then stirred at room temperature for ca. 1 hour. The resulting solution was evaporated to dryness, redissolved in hexane (10 ml), cannula filtered and concentrated. Colourless crystals were deposited overnight at −35 °C and collected via cannula filtration to yield compound 5. (0.62 g, 47%). 1H NMR (500 MHz, benzene-d6) δ 8.08 (s, 1H, NC(H)N), 7.18–7.11 (m, 6H, Ar–H), 4.94 (s, 1H, NC(CH3)CH), 3.29 (hept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 2.57–2.51 (m, 2H, NCH(CH2)5), 1.77 (s, 6H, NC(CH3)CH), 1.72–1.64 (m, 4H, NCH(CH2)5), 1.61–1.54 (m, 4H, NCH(CH2)5), 1.51–1.43 (m, 4H, NCH(CH2)5), 1.29 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.26 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.14–1.03 (m, 4H, NCH(CH2)5), 0.92–0.81(m, 4H, NCH(CH2)5) ppm. 13C{1H} NMR (126 MHz, benzene-d6) δ 168.7 (NC(H)N), 165.9 (NC(CH3)CH), 146.4 (Cipso), 141.5 (Cortho), 124.6 (Cpara), 124.1 (Cmeta), 93.0 (NC(CH3)CH), 61.7 (NCH(CH2)5), 37.2 (NCH(CH2)5), 28.7 (CH(CH3)2), 26.4, 26.3 (NCH(CH2)5), 25.6 (CH(CH3)2), 24.5 (NC(CH3)CH), 24.3 (CH(CH3)2) ppm. Elemental analysis for C42H64CaN4: C, 75.85; H, 9.70; N, 8.42%. Found: C, 75.71; H, 9.87, N, 8.37%.
[(BDI)Ca(iPrNC(H)NiPr)(iPrNCNiPr)] (6)
Toluene (10 ml) was added to 2 (0.92 g, 1.00 mmol) and iPrNCNiPr (621 μl, 4.01 mmol) then stirred at room temperature for ca. 1 hour. The resulting solution was evaporated to dryness, redissolved in hexane (10 ml), cannula filtered and concentrated. Colourless crystals were deposited overnight at −35 °C and collected via cannula filtration to yield compound 6. (0.56 g, 39%). 1H NMR (500 MHz, benzene-d6) δ 8.27 (s, 1H, NC(H)N), 7.18–7.07 (m, 6H, Ar–H), 4.92 (s, 1H, NC(CH3)CH), 3.44 (hept, 3JHH = 6.8 Hz, 4H, CH(CH3)2), 3.14 (hept, 3JHH = 6.4 Hz, 2H, NC(H)N{CH(CH3)2}2), 3.09 (hept, 3JHH = 6.4 Hz, 2H, NCN{CH(CH3)2}2) 1.76 (s, 6H, NC(CH3)CH), 1.32 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.29 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 1.09 (d, 3JHH = 6.4 Hz, 12H, NC(H)N{CH(CH3)2}2), 0.86 (d, 3JHH = 6.4 Hz, 12H, NCN{CH(CH3)2}2) ppm. 13C{1H} NMR (126 MHz, benzene-d6) δ 169.0 (NC(H)N), 165.4 (NC(CH3)CH), 148.0 (Cipso), 141.8 (Cortho), 124.1 (Cpara), 123.8 (Cmeta), 93.6 (NC(CH3)CH), 53.9 (NC(H)N{CH(CH3)2}2), 49.5 (NCN{CH(CH3)2}2), 28.3 (CH(CH3)2), 26.8 (NC(H)N{CH(CH3)2}2), 25.8 (CH(CH3)2), 24.9 (NC(CH3)CH), 24.6 (CH(CH3)2), 24.2 (NCN{CH(CH3)2}2.) ppm. Elemental analysis for C43H70CaN6: C, 72.62; H, 9.92; N, 11.82%. Found: C, 72.43; H, 9.95, N, 11.69%.[{(BDI)Ca}2(H)(p-tolNC(H)Np-tol)] (7)
Toluene (10 ml) was added to 2 (0.92 g, 1.00 mmol) and p-tolNCNp-tol (0.22 g, 0.99 mmol) then stirred at room temperature for ca. 1 hour. The resulting solution was evaporated to dryness, redissolved in hexane (10 ml), cannula filtered and concentrated. Colourless crystals were deposited overnight at −35 °C and collected via cannula filtration to yield compound 7. (0.60 g, 53%). 1H NMR (500 MHz, benzene-d6) δ 8.04 (s, 1H, NC(H)N), 7.15–7.05 (m, 16H, Ar–H), 6.91–6.89 (m, 4H, Ar–H), 4.95 (s, 2H, NC(CH3)CH), 4.41 (s, 1H, CaH), 3.25–2.77 (br, 8H, CH(CH3)2), 2.11 (s, 6H, NC6H4CH3), 1.66 (s, 12H, NC(CH3)CH), 1.23–0.71 (br, 48H, CH(CH3)2) ppm. 13C{1H} NMR (126 MHz, benzene-d6) δ 166.4 (NC(CH3)CH), 166.1 (NC(H)N), 149.3 (p-tolCipso), 144.3 (Cipso), 142.3 (Cortho), 131.9 (p-tolCpara), 130.9 (Cpara), 124.9 (p-tolC), 124.1 (Cmeta), 120.5 (p-tolC), 94.3 (NC(CH3)CH), 28.4 (CH(CH3)2), 25.2 (CH(CH3)2), 24.2 (NC(CH3)CH), 24.2 (CH(CH3)2), 20.6 (NC6H4CH3) ppm. Elemental analysis for C73H98CaN6: C, 76.93; H, 8.67; N, 7.37%. Found: C, 76.80; H, 8.87, N, 7.48%.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EPSRC (UK) for the provision of a DTP studentship (ASSW) and the University of Bath.References
- (a) F. T. Edelmann, in Adv. Organometal. Chem, ed. A. F. Hill and M. J. Fink, 2013, vol. 61, pp. 55–374 Search PubMed; (b) F. T. Edelmann, Adv. Organometal. Chem., 2008, vol. 57, 183–352 Search PubMed.
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219 CrossRef CAS.
- E. Lu, Y. Chen and X. Leng, Organometallics, 2011, 30, 5433–5441 CrossRef CAS.
- Z. Jian, N. K. Hangaly, W. Rong, Z. Mou, D. Liu, S. Li, A. A. Trifonov, J. Sundermeyer and D. Cui, Organometallics, 2012, 31, 4579–4587 CrossRef CAS.
- B. M. Schmiege, M. E. Fieser, J. W. Ziller and W. J. Evans, Organometallics, 2012, 31, 5591–5598 CrossRef CAS.
- C. Wang, L. Xiang, X. Leng and Y. Chen, Dalton Trans., 2017, 46, 1218–1227 RSC.
- J. Cheng and Z. Hou, Chem. Commun., 2012, 48, 814–816 RSC.
- Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624–6638 CrossRef CAS PubMed.
- S. Gambarotta, S. Strologo, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Am. Chem. Soc., 1985, 107, 6278–6282 CrossRef CAS.
- J. F. Leboeuf, J. C. Leblanc and C. Moïse, J. Organomet. Chem., 1987, 335, 331–337 CrossRef CAS.
- G. Albertin, S. Antoniutti and G. Roveda, Inorg. Chim. Acta, 2005, 358, 3093–3105 CrossRef CAS.
- R. Boese, R. Köster and M. Yalpani, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 1453 CAS.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559–13568 CrossRef CAS PubMed.
- Y. Yuan, X. Wang, Y. Li, L. Fan, X. Xu, Y. Chen, G. Li and W. Xia, Organometallics, 2011, 30, 4330–4341 CrossRef CAS.
- C. Wang, X. Leng and Y. Chen, Organometallics, 2015, 34, 3216–3221 CrossRef CAS.
- M. L. Cole, C. Jones, C. Junk Peter, M. Kloth and A. Stasch, Chem. – Eur. J., 2005, 11, 4482–4491 CrossRef CAS PubMed.
- J. Hellmann, I. Rhotert, H. Westenberg, R. Fröhlich, B. Wibbeling, W. Uhl and E. U. Würthwein, Eur. J. Org. Chem., 2013, 3356–3368 CrossRef CAS.
- S. Schulz, T. Eisenmann, S. Schmidt, D. Blaser, U. Westphal and R. Boese, Chem. Commun., 2010, 46, 7226–7228 RSC.
- G. Bendt, S. Schulz, J. Spielmann, S. Schmidt, D. Bläser and C. Wölper, Eur. J. Inorg. Chem., 2012, 3725–3731 CrossRef CAS.
- A. Rit, P. Spaniol Thomas and J. Okuda, Chem. – Asian J., 2014, 9, 612–619 CrossRef CAS.
- A. W. Jana, H. C. Roesky, C. Schulzke and A. Döring, Angew. Chem., Int. Ed., 2009, 48, 1106–1109 CrossRef CAS PubMed.
- A. Stasch, Dalton Trans., 2014, 43, 7078–7086 RSC.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973–2977 CrossRef CAS PubMed.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2016, 22, 7158–7162 CrossRef CAS PubMed.
- M. Westerhausen and W. Schwarz, Z. Naturforsch., B: J. Chem. Sci., 1992, 47, 453–459 CAS.
- H. F. Hu and C. M. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS.
- C. Glock, C. Loh, H. Görls, S. Krieck and M. Westerhausen, Eur. J. Inorg. Chem., 2013, 3261–3269 CrossRef CAS.
- C. Loh, S. Seupel, H. Görls, S. Krieck and M. Westerhausen, Eur. J. Inorg. Chem., 2014, 2014, 1312–1321 CrossRef CAS.
- C. Loh, S. Seupel, H. Görls, S. Krieck and M. Westerhausen, Organometallics, 2014, 33, 1480–1491 CrossRef CAS.
- C. Loh, S. Seupel, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Dalton Trans., 2014, 43, 14440–14449 RSC.
- M. He, M. T. Gamer and P. W. Roesky, Organometallics, 2016, 35, 2638–2644 CrossRef CAS.
- A. Causero, G. Ballmann, J. Pahl, H. Zijlstra, C. Farber and S. Harder, Organometallics, 2016, 35, 3350–3360 CrossRef CAS.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock, S. L. Lomas, M. F. Mahon, P. A. Procopiou and K. Suntharalingam, Organometallics, 2008, 27, 6300–6306 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill, S. L. Lomas, D. J. MacDougall and M. F. Mahon, Organometallics, 2013, 32, 4961–4972 CrossRef CAS.
- M. L. Cole and P. C. Junk, New J. Chem., 2005, 29, 135–140 RSC.
- M. L. Cole, G. B. Deacon, C. M. Forsyth, K. Konstas and P. C. Junk, Dalton Trans., 2006, 3360–3367 RSC.
- B. Kim Sang, C. Yang, T. Powers, M. Davis Luke, X. Lou and G. Gordon Roy, Angew. Chem., Int. Ed., 2016, 55, 10228–10233 CrossRef PubMed.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS PubMed.
- S. Harder, Chem. Commun., 2012, 48, 11165–11177 RSC.
- S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS PubMed.
- J. Spielmann and S. Harder, Chem. – Eur. J., 2007, 13, 8928–8938 CrossRef CAS PubMed.
- V. Leich, T. P. Spaniol and J. Okuda, Inorg. Chem., 2015, 54, 4927–4933 CrossRef CAS PubMed.
- V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 4794–4797 CrossRef CAS PubMed.
- A. Causero, G. Ballmann, J. Pahl, C. Farber, J. Intemann and S. Harder, Dalton Trans., 2017, 46, 1822–1831 RSC.
- A. Causero, H. Elsen, J. Pahl and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 6906–6910 CrossRef CAS PubMed.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed.
- B. Maitland, M. Wiesinger, J. Langer, G. Ballmann, J. Pahl, H. Elsen, C. Farber and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 11880–11884 CrossRef CAS PubMed.
- H. Bauer, M. Alonso, C. Farber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nat. Catal., 2018, 1, 40–47 CrossRef.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- J. Spielmann and S. Harder, Eur. J. Inorg. Chem., 2008, 1480–1486 CrossRef CAS.
- J. Intemann, H. Bauer, J. Pahl, L. Maron and S. Harder, Chem. – Eur. J., 2015, 21, 11452–11461 CrossRef CAS PubMed.
- D. Mukherjee, T. Hollerhage, V. Leich, T. P. Spaniol, U. Englert, L. Maron and J. Okuda, J. Am. Chem. Soc., 2018, 140, 3403–3411 CrossRef CAS PubMed.
- C. Ruspic and S. Harder, Inorg. Chem., 2007, 46, 10426–10433 CrossRef CAS PubMed.
- C. N. de Bruin-Dickason, T. Sutcliffe, C. A. Lamsfus, G. B. Deacon, L. Maron and C. Jones, Chem. Commun., 2018, 54, 786–789 RSC.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed.
- M. Findlater, N. J. Hill and A. H. Cowley, Dalton Trans., 2008, 4419–4423 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Nat. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Vanrijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. L. Cole, D. J. Evans, P. C. Junk and L. M. Louis, New J. Chem., 2002, 26, 1015–1024 RSC.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill, S. L. Lomas, M. S. Heng, P. B. Hitchcock and G. Kociok-Köhn, Dalton Trans., 2014, 43, 14249–14256 RSC.
- S. Harder, Organometallics, 2002, 21, 3782–3787 CrossRef CAS.
- A. G. Avent, M. R. Crimmin, M. S. Hill and P. B. Hitchcock, Dalton Trans., 2005, 278–284 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of compounds 3–7 and details of the crystallographic analysis of compounds 3, 4, 6 and 7. CCDC 1886241–1886244 for 3, 4, 6 and 7, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt05107k |
| This journal is © The Royal Society of Chemistry 2019 |