
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible O–H bond activation by an intramolecular frustrated Lewis pair†
Petra
Vasko
 *ab,
M. Ángeles
Fuentes
a,
Jamie
Hicks
a and
Simon
Aldridge
*a
*ab,
M. Ángeles
Fuentes
a,
Jamie
Hicks
a and
Simon
Aldridge
*a
aDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: petra.vasko@chem.ox.ac.uk; simon.aldridge@chem.ox.ac.uk
bDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P. O. Box 35, Jyväskylä, FI-40014, Finland
First published on 7th February 2019
Abstract
The interactions of the O–H bonds in alcohols, water and phenol with dimethylxanthene-derived frustrated Lewis pairs (FLPs) have been probed. Within the constraints of this backbone framework, the preference for adduct formation or O–H bond cleavage to give the corresponding zwitterion is largely determined by pKa considerations. In the case of the PPh2/B(C6F5)2 system and p-tBuC6H4OH, an equilibrium is established between the two isomeric forms which allows the thermodynamic parameters associated with zwitterion formation via O–H bond cleavage to be probed.
In recent years, frustrated Lewis pairs (FLPs) have emerged as an attractive new approach in the fields of small molecule activation and catalysis;1 reports of the use of FLPs in the heterolytic cleavage of dihydrogen and other E–H bonds (E = C, N, O, Si, B) continue to rise year-on-year. By extension ‘metal-free’ approaches for carrying out bond modification reactions using FLPs have evolved from proof-of-concept to an active research field in main group catalysis. Most notably, FLP-catalysed hydrogenation reactions have been successfully deployed for a wide range of functional groups, including imines, aldehydes, ketones, alkenes and alkynes.2
We have previously shown that the dimethylxanthene backbone provides a versatile scaffold for intramolecular FLPs (e.g.1a; Scheme 1); the separation between the Lewis acid and base components typically lies between 4.0 and 4.5 Å and is therefore pre-organised for the activation of small molecules such as H2.4 Recently we also showed that FLP 1a reacts readily with the C–H bonds in terminal alkynes and with the B–H bonds in selected boranes, and can act as a pre-catalyst for the hydroboration of alkynes.5
 | ||
| Scheme 1 Reactions of FLP 1a with water, methanol and 2-propanol. | ||
In terms of other E–H bonds, the interactions of FLPs with water and O–H containing systems are important,6–13 not least because they influence strongly their sensitivity to trace impurities present in commercial substrates and solvents; as a consequence, it is often necessary to carry out catalytic processes under the exclusion of moisture. Similar chemistry potentially arises when using alcohol solutions in combination with FLPs as alcohols can have comparable nucleophilicity/basicity to H2O.3 With these issues in mind, we set out to investigate the reactivity of FLP 1a (and related systems) towards common O–H bond containing substrates, namely water, alcohols and phenols.
The reactions of FLP 1a with excess water or methanol/2-propanol in dichloromethane result in a gradual colour change from a bright yellow to colourless (Scheme 1 and ESI†). The products of these reactions (2–4) can be crystallized from dichloromethane/hexane and their solid-state structures determined crystallographically. These confirm that 1a assimilates one equivalent of ROH (R = H (2) Me (3) or iPr (4)) to form the respective O-bound adduct (Fig. 1). In each case, the ROH fragment is bonded to B(1), rendering the boron centre tetrahedral and O(2) trigonal pyramidal. The O–H proton(s) in each case could be located in the difference Fourier map and refined without restraints. The B(1)–O(2) distances fall in the range 1.595(3)–1.611(2) Å, which are comparable to previously reported borane adducts of water/alcohols.6–11
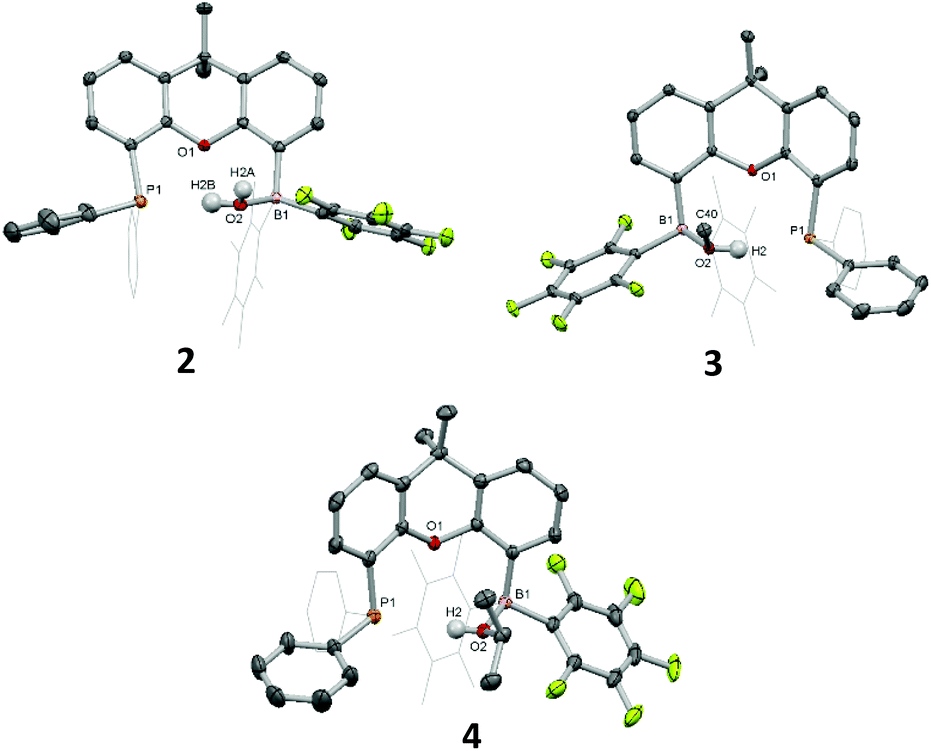 | ||
| Fig. 1 Molecular structures of 2, 3 and 4. Parts of the FLPs are shown in a wireframe format, and solvent molecules and most hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at 40% probability. Selected bond lengths (Å) and angles (°): 2: B1–O2 1.611(2), O2–H2A 0.76(3), O2–H2B 0.92(3), P1–H2B 2.32(3), B1–O2–P1 115.7(1); 3: B1–O2 1.596(2), O2–H2 0.91(2), P1–H2 2.25(2), B1–O2–P1 115.3(1); 4: B1–O2 1.595(4), O2–H2 0.82(3), P1–H2 2.49(3), B1–O2–P1 112.4(1). | ||
Further support for the assignment of 2–4 as simple Lewis adducts can be obtained from solution state NMR studies. The 1H NMR spectra of 2 and 4 show broad singlets for the O-bound proton(s) at 8.10 and 9.17 ppm, respectively. In the case of 3, the corresponding 1H NMR signal (at 10.80 ppm) is actually a doublet, although the coupling constant (JPH = 16.8 Hz) is significantly smaller than that expected for a direct P–H bond in systems of this type (>500 Hz; vide infra). In similar fashion, the respective proton-coupled 31P NMR spectra feature singlet resonances in each case (at ca. −20 ppm) rather than the wide doublet expected for a bona fide P–H single bond. The boron centre in each adduct gives rise to a broad signal in the 11B{1H} spectrum in the range characteristic of four-coordinate boron species (δB = 4.5 (2), 5.4 (3), and 3.7 ppm (4)), consistent with the solid state structures.5
Earlier literature reports detailing the reactivity of FLPs towards water favour heterolytic O–H bond cleavage;6–11 for example the tBu3P/B(C6F5)3 system is reported to activate one molecule of H2O to give the phosphonium borate [tBu3PH][(HO)B(C6F5)3].7 The difference in this system compared to 1a can be understood (primarily) in terms of the relative basicities of the two phosphine components. More difficult to rationalize based on simple pKa values is the contrasting behaviour of 1a and 1,2-C6H4(NPh2){B(C6F5)2};6 the latter has been shown to deprotonate the bound H2O molecule (to give a zwitterionic anilinium borate) despite the fact that systems of the type ArNPh2 are typically very weak Brønsted bases (Scheme 2).14
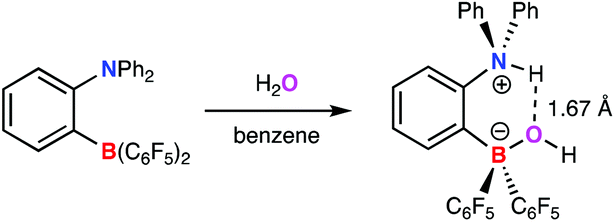 | ||
| Scheme 2 O–H bond cleavage in water by Roesler and Piers’ aniline/borane FLP.6 | ||
Presumably, the difference relates to the presence of a strong intramolecular N–H⋯O hydrogen bond (which is reported to feature an O⋯H contact shorter than those in water) of a type which is precluded in the putative analogue derived from 1a, on the basis of the greater separation of the Lewis acid and base components. The P⋯O separation associated with the bound water molecule in 2 is 3.237(2) Å.
Reasoning that the use of a more acidic O–H bond might bring about O–H bond cleavage, we investigated the reactions of 1a (and the related –PMes2 derivative 1b) with phenols; p-tBuC6H4OH was chosen given the presence of the tBu group as a convenient 1H NMR handle. Accordingly, the reaction between 1a and excess p-tBuC6H4OH in dichloromethane leads to immediate discharge of the yellow colour of the FLP; filtration, concentration and layering with n-hexane yielded colourless single crystals suitable for X-ray diffraction studies.
The molecular structure shows that 1a reacts with one equivalent of the phenol to give the (O–H activated) phosphonium borate zwitterion 5a (Scheme 3 and Fig. 2). The P-bound proton H(1) could be located in the difference Fourier map and refined without restraints. With the usual caveats concerning the location of hydrogen atoms by X-ray methods, there appears to be little residual interaction between H(1) and O(2) (d(H(1)–O(2) = 2.19(1) Å cf. d(P(1)–H(1)) = 1.28(2) Å). The location of the hydrogen atom at P(1) is also consistent with the observed widening of the C–P–C angles in 5a compared to 1a itself (108.5(1)–112.6(1)° cf. 101.4(1)–102.7(1)°).
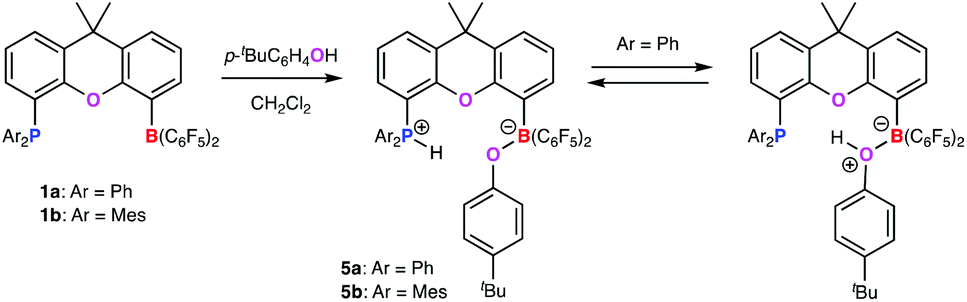 | ||
| Scheme 3 Reactions of FLPs 1a and 1b with p-tBuC6H4OH. | ||
 | ||
| Fig. 2 Molecular structures of 5a (left) and 5b (right). Parts of the FLPs are shown in a wireframe format, solvent molecules and most hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at 40% probability. Selected bond lengths (Å) and angles (°): 5a: B1–O2 1.495(1), P1–H1 1.28(2), O2–H1 2.19(1), P1–O2 3.458(1), C2–P1–C16 108.1(1), C2–P1–C22 112.6(1), C16–P1–C22 109.3(1); 5b: B1–O2 1.498(3), P1–H1 1.39(3), O2–H1 2.31(3), P1–O2 3.615(2), C2–P1–C16 106.0(1), C2–P1–C25 116.8(1), C16–P1–C25 112.4(1). | ||
Intriguingly, however, a solution made by re-dissolving crystals of 5a in benzene-d6 features a singlet resonance in the 1H spectrum at δH = 9.69 ppm, and in the proton-coupled 31P NMR spectrum (at δP = −13.0 ppm). These signals contrast markedly with the wide doublet resonances typically associated with P–H groups in systems of this sort (vide infra). In addition, the 19F NMR signals associated with the boron-bound C6F5 groups in 5a are more consistent with a neutral borane adduct RO(H)·BAr(C6F5)2 than an anionic system of the type [RO·BAr(C6F5)2]−. In particular, the separation between the resonances associated with the meta and para CF groups (Δδm,p = 6.5 ppm) is similar to those measured for water/alcohol adducts 2–4 (7.5–8.0 ppm). These observations suggest that 5a adopts a different structure in solution to the zwitterionic form seen in the solid state – namely a simple donor/acceptor adduct akin to 2–4.
With this apparent inconsistency in mind, we carried out variable temperature NMR measurements on 5a in dichloromethane-d2. Cooling the sample to 257 K results in the appearance of a well-resolved doublet in the 31P NMR spectrum at δP = −11.9 ppm (1JPH = 554 Hz) and a change in the low-field part of the 1H NMR spectrum, with a complementary doublet appearing at δH = 10.48 ppm. The 19F NMR spectrum in the low temperature limit is characterized by a reduced value of Δδm,p (4.9 ppm), similar to that measured for the related PMes2 system (4.2 ppm) which adopts the zwitterionic O–H activated structure under all conditions examined (vide infra). These changes are reversible, and are consistent with an equilibrium involving isomeric species related through O–H bond cleavage; the solution-phase structure of 5a at low temperatures resembles the zwitterion found in the solid state, while at higher temperatures a structure similar to those determined for adducts 2–4 predominates. A van't Hoff analysis carried out in CD2Cl2 at temperatures in the range 259–281 K allows for the determination of the enthalpic (ΔH° = −69 kJ mol−1) and entropic terms (ΔS° = −201 J mol−1 K−1) associated with O–H bond breakage in this equilibrium. The relatively large magnitude of ΔS° is consistent with the narrow temperature window over which the transformation occurs, and reflects the more ordered nature of the zwitterionic form and its influence on the solvent sphere (presumably driven by electrostatic considerations).
To complement these experimental studies, we also sought to probe the thermodynamics of the two structural isomers of 5a computationally by DFT. Both the Lewis adduct and zwitterionic forms were optimized using the PBE1PBE hybrid exchange–correlation functional in combination with Def-TZVP basis set;15 we also included a polarizable continuum model16 (PCM, dichloromethane) to describe the difference in optimized energies of the two isomers more accurately. The computed free energies show that the adduct represents the more stable structure at 298 K, but only by 8.3 kJ mol−1. We were also able to locate a transition state for the migration of the proton at 9.2 kJ mol−1 in dichloromethane. These calculated values are consistent with our experimental findings and confirm the lability of the O–H bond in 5a; proton transfer occurs very readily and the solid-state structure is presumably stabilised by favourable packing forces relating to its greater degree of charge separation.
For comparison we also wanted to examine the reactivity of p-tBuC6H4OH towards the related FLP 1b, which features PMes2 (Mes = C6H2-2,4,6-Me3), rather than PPh2 as the Lewis basic component.4 We hypothesized that the enhanced basicity of the phosphine donor would bias the thermodynamics of O–H bond cleavage in favour of the zwitterionic form. The reaction between 1b and excess p-tBuC6H4OH proceeds instantly and the molecular structure of the product, 5b, can be shown by X-ray crystallography to be closely related to that of 5a (Fig. 2). Electron density close to P(1) and the geometry of the C3P heavy atom skeleton strongly suggest the presence of a P–H bond. Importantly – in this case – the solid state-structure appears to be retained in solution at all temperatures. Thus, both the proton coupled 31P and 1H spectra at room temperature feature a doublet with 1JPH = 542 Hz (δP = −27.3 ppm; δH = 9.74 ppm). Consistently, DFT structural optimisations show that the zwitterionic form of 5b is more stable than the corresponding adduct by 34.6 kJ mol−1 in dichloromethane.
In conclusion, we have studied the interactions of the O–H bonds in alcohols, water and phenol with the dimethyl-xanthene based FLPs 1a and 1b. Within the constraints of this particular intramolecular framework, the preference for adduct formation or O–H cleavage to give the corresponding zwitterion is largely determined by pKa considerations. In the case of the 1a/p-tBuC6H4OH system, an equilibrium is established between the two isomeric forms which allows the thermodynamic parameters associated with O–H activation to be probed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Advanced Research Computing (ARC) facilities for providing computational resources and Dr Nick Rees for help with the NMR experiments. PV would like to thank the Magnus Ehrnrooth and Finnish Cultural Foundations for funding. MAF would like to thank the EPSRC for funding (EP/K014714/1)Notes and references
-
(a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed
; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed
-
(a) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018 CrossRef CAS PubMed
-
(a) V. Fasano and M. J. Ingleson, Synthesis, 2018, 50, 1783 CrossRef CAS
-
(a) Z. Mo, E. L. Kolychev, A. Rit, J. Campos, H. Niu and S. Aldridge, J. Am. Chem. Soc., 2015, 137, 12227 CrossRef CAS PubMed
- P. Vasko, I. A. Zulkifly, M. Á; Fuentes, Z. Mo, J. Hicks, P. C. J. Kamer and S. Aldridge, Chem. – Eur. J., 2018, 24, 10531 CrossRef CAS PubMed
- R. Roesler, W. E. Piers and M. Parvez, J. Organomet. Chem., 2003, 680, 218 CrossRef CAS
-
(a) M. Klahn, A. Spannenberg and U. Rosenthal, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o1549 CrossRef CAS PubMed
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581 CrossRef CAS
- A. Di Saverio, F. Focante, I. Camurati, L. Resconi, T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2005, 44, 5030 CrossRef CAS PubMed
- T. Wang, G. Kehr, L. Liu, S. Grimme, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302 CrossRef CAS PubMed
- D. J. Scott, N. A. Phillips, J. S. Sapsford, A. C. Deacy, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2016, 55, 14738 CrossRef CAS PubMed
- T. Xu and E. Y.-X. Chen, J. Am. Chem. Soc., 2014, 136(5), 1774 CrossRef CAS PubMed
- M. J. Drewitt, M. Niedermann and M. C. Baird, Inorg. Chim. Acta, 2002, 340, 207 CrossRef CAS
- See, for example, http://evans.rc.fas.harvard.edu/pdf/evans_pka_table.pdf; retrieved 15/01/19.
-
(a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, X-ray crystallographic and characterisation data. CCDC 1872780–1872784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00228f |
| This journal is © The Royal Society of Chemistry 2019 |