
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation of Au(I)-complexes on amorphous substrates governed by aurophilicity†
Petra
Gründlinger
a,
Michael
Györök
a,
Sebastian
Wolfmayr
b,
Tobias
Breuer
 b,
Daniel
Primetzhofer
c,
Barbara
Bruckner
c,
Uwe
Monkowius
*d and
Thorsten
Wagner
*a
b,
Daniel
Primetzhofer
c,
Barbara
Bruckner
c,
Uwe
Monkowius
*d and
Thorsten
Wagner
*a
aJohannes Kepler University Linz, Institute of Experimental Physics, Surface Science Division, Altenberger Straße 69, 4040 Linz, Austria. E-mail: thorsten.wagner@jku.at
bPhillipps University Marburg, Physics Department, Molecular Solids Group, Renthof 7, 35032 Marburg, Germany
cUppsala University, Department of Physics and Astronomy, Box 516, S-75120 Uppsala, Sweden
dJohannes Kepler University Linz, School of Education, Altenberger Straße 69, 4040 Linz, Austria. E-mail: uwe.monkowius@jku.at
First published on 9th September 2019
Abstract
In single crystals of 2-naphthylisonitrile–gold(I)-halide (halide = Cl, Br, I) complexes, Au⋯Au distances are found to be significantly shorter than twice the van der Waals radius, indicating attractive interactions between gold atoms in adjacent molecules. In the particular case of the studied 2-naphthylisonitrile–gold(I) complexes, homodimers are the common structural motifs, in which the linearly coordinated gold exhibits a crossed swords arrangement with the Au atoms of two molecules being at the intersection point. The crossed swords motif is preserved upon physical vapour deposition of both the chlorine and bromine derivatives on amorphous substrates like glass and glassy carbon. The determined activation energies of desorption for the chlorine (0.9 eV) and the bromine (1.2 eV) derivative are comparable to that of unsubstituted naphthalene. Using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and ion scattering (RBS), we confirmed the chemical integrity of the molecules in thin films and revealed the orientation of the crossed swords dimers with respect to the substrate surface.
Introduction
Besides electrochemical procedures, physical and chemical vapour deposition (PVD and CVD) techniques are frequently used in science and technology for gold-plating and the preparation of thin gold films or small structures.1,2 Such techniques play a crucial role in the field of microelectronics, but may also be important for sensing applications or heterogeneous gold catalysis.3 Although a number of gold complexes have been suggested for CVD applications, usually gold(III) complexes are used as gold precursors, particularly of the form Me2AuX (with X = acetate, dithiophosphinates, pivalate, acetylacetonates, etc.).2,4–8 Only a small number of gold(I) complexes have been applied in the past and are mostly of the form L–Au–X (L = phosphine ligands and X = halide, alkyl, carboxylate) but also other gold(I) complexes, e.g. dimeric gold(I) precursors, have been reported.1–16 Usually, the goal of these studies was to obtain pure gold films without contamination with other elements present in the precursors.From the point of view of a gold chemist, the thermal decomposition of a gold complex is an often undesired process during synthesis, handling and storage of such compounds and hard to avoid. In most cases, the storage of gold-containing solutions for some hours or days at room temperature leads to a violet precipitate of colloidal gold. This behaviour can be traced back to the intrinsic thermal instability of many gold complexes due to the high oxidation potential of gold(I) and the general tendency to form elemental gold. Contrary, the evaporation of intact complexes and their deposition on surfaces have been only scarcely investigated. To date – at least to our knowledge – only one systematic study, which could elucidate some structural features necessary to obtain vaporisable gold complexes, has been published.17 Not completely surprisingly, the sublimation temperature increases with increasing aggregation grade and decreasing Au⋯Au distances. The question of the arrangement of such gold complexes on surfaces is still open, although these molecules are often engaged in relatively strong intermolecular attraction. Heavy elements with closed shell configurations (d8, d10, and s2) are capable of forming such so-called metallophilic interactions.18,19 For gold(I) atoms these interactions are particularly strong. This is the reason why the interaction between gold(I) atoms is referred to as aurophilicity.20–23 First evidence of gold atoms attracting each other was found in the 1970s. Gold(I) centres seemed to be drawn together well below the sum of the van der Waals radii (3.32 Å).24 Usually, distances between 2.50 and 3.50 Å are considered as aurophilic bonding.22 At first sight, gold(I) atoms should repel each other due to Coulomb repulsion of the positively charged metal ion. However, it is now widely accepted that these attractive interactions are due to dispersive interactions strongly enhanced by relativistic effects. Strong aurophilic interactions are comparable to average hydrogen bonds regarding the bond energy.22
Meanwhile, aurophilic interactions are an established concept in the structural chemistry of gold(I). They not only determine the arrangement of the gold complexes in the solid state and sometimes also in solution,25–27 but also give rise to multifarious luminescence phenomena.28,29 However, up to now, there has been no information on whether such interactions could also govern the arrangement of the complexes in mono-molecular layers and thin films grown by vapour deposition. This lack of scientific attention is somewhat remarkable as other weak intermolecular interactions (ππ-stacking and hydrogen bonds) have been frequently utilized as the driving force for the preparation of highly structured monolayers.30–32
The aim of the present study was to obtain gold(I) complexes that fulfil the following requirements: (i) the gold(I) complexes should be capable of showing strong aurophilic interactions, i.e. the steric demand of the used ligands has to be relatively small, at least in close proximity to the gold atom. (ii) The complexes should be thermally and chemically stable to avoid undesired decomposition reactions during handling and evaporation. This stability also includes negligible sensitivity to moisture and air. (iii) The volatility of the complexes should match the requirements of the evaporation set-up, which could be in part realized by the right choice of the size of the ligands. However, due to hardly predicable intermolecular interactions in the solid state (ππ-stacking and aurophilic interactions) a direct relation between the molecular size and vapour pressure is difficult to achieve.
In the first part, we report the synthesis and the chemical and the structural characterization of the gold(I) complexes. This also includes a basic photo-physical characterization as we expected photo-luminescence as a result of aurophilic interactions. In the second part, evaporation experiments as well as the characterization of the prepared thin films to ensure the integrity of the complexes on the surface are shown.
Results and discussion
Synthesis
For the preparation of gold(I) complexes, we chose aromatic isonitrile ligands for several reasons: (i) it is known that due to the small steric demand of isonitrile ligands multifarious arrangements via excessive aurophilic interactions in the solid state are realized. In contrast, gold(I) complexes of the frequently used phosphine or N-heterocyclic carbene ligands often exist as monomeric units without strong gold–gold interactions. Indeed, solid state structures of isonitrile gold(I) complexes are rarely found without such interactions. (ii) Isonitrile gold(I) complexes are easy to prepare and stable to air and moisture.33–36 (iii) Many aromatic isonitriles are commercially available or easy to synthesize.37 However, we accept with this choice that ππ-stacking and the aurophilic interactions may affect the arrangement of the complexes.Complexes of the form L–Au–X (L = isonitrile and X = halide) are easily synthesized from (tht)AuCl (tht = tetrahydrothiophene) and the respective isonitrile (Scheme 1) by subsequent metathesis with KBr or KI.38,39 In the first evaporation experiments, it became clear that phenylisonitrile leads to complexes that are too volatile in our evaporation set-up (vide infra). Therefore, 2-naphthylisonitrile (NapNC) was chosen. (NapNC)AuCl has been reported recently,40,41 and the bromide ((NapNC)AuBr) and iodide ((NapNC)AuI) congeners have been synthesized in this study for the first time. The iodide complex is relatively unstable and forms a purple precipitate after some days in solution in the dark. In the IR spectrum, the characteristic stretching band of the –N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of the free ligand is found at 2221 cm−1.40 The corresponding bands for the Au(I) isonitriles are found between 2206 and 2209 cm−1.
C bond of the free ligand is found at 2221 cm−1.40 The corresponding bands for the Au(I) isonitriles are found between 2206 and 2209 cm−1.
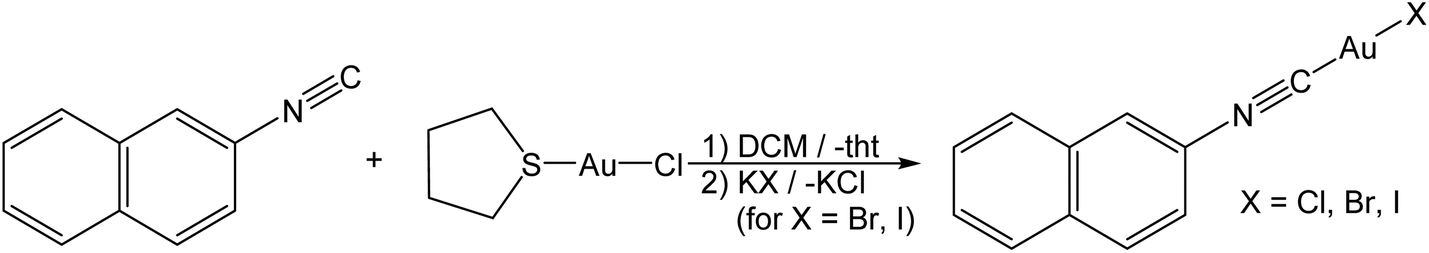 | ||
| Scheme 1 Reaction scheme for the synthesis of 2-naphthylisonitrile–gold(I)-halide ((NapNC)AuX). | ||
Structural studies
For (NapNC)AuBr and (NapNC)AuI, crystals suitable for X-ray diffraction were obtained by slow vapour diffusion of n-pentane into a solution of the respective complex in dichloromethane: (NapNC)AuBr crystallizes in the monoclinic space group C2/c with one molecule in the asymmetric unit. The crystals are isostructural to the chlorido congener (Table 1).40 The gold atom is linearly coordinated to the carbon atom of the isonitrile group and the bromide ligand (C1–Au1–Br1 176.8(4)°). The C1–Au1 and Au1–Br1 distances are 1.95(2) and 2.384(3) Å, respectively. These values are in the expected range for a C–Au–Br moiety. In the crystals, the gold atom features different Au⋯Au distances (in brackets the value for the chlorido congener): the shortest Au⋯Au bonds are formed via the dimerization of the molecules in a crossed swords conformation and amount to 3.239 Å (3.224 Å). These dimers are additionally arranged in columns with a distance of ∼3.6 Å between the naphthyl moieties characteristic of ππ-stacking interactions of aryl groups. The distance between adjacent gold atoms is 4.131 Å (3.943 Å), which is well beyond the sum of the van der Waals radii of gold (3.32 Å),24i.e., the Au⋯Au distance is clearly governed by ππ-stacking interactions of the naphthyl group rather than by aurophilic bonds in this case. The columns are connected to neighbouring columns with a relatively large Au⋯Au distance of 3.595 Å (3.565 Å), which is at the limit of attractive aurophilic forces (Fig. 1).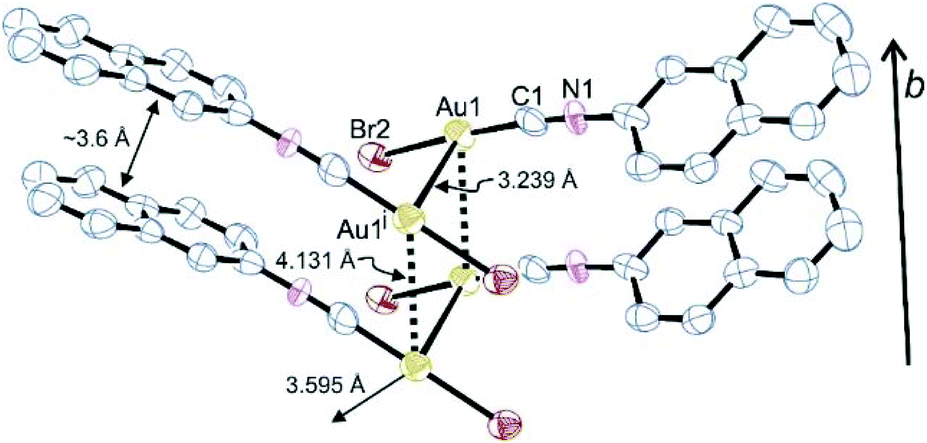 | ||
| Fig. 1 Arrangement of dimers in the crystals of (NapNC)AuBr (H atoms are omitted for clarity, displacement ellipsoids at the 50% probability level; symmetry code: (i) −x + 1, y, −z + 3/2.). Selected bond lengths in Å and angles in °: Au1–Au1i 3.239(3), Au1–Br1 2.384(3), Au1–C1 1.95(2), C1–N1 1.15(2), Br1–Au1–C1 176.8(4), Au1–C1–N1 175(2), Br1–Au1–Au1i–Br1i 124.1(3). | ||
| 2-Naphthylisonitrile–gold(I)- | Chloride40 | Bromide | Iodide |
|---|---|---|---|
| Empirical formula | C11H17AuClN | C11H17AuBrN | C11H17AuIN |
| Mr/g mol−1 | 385.60 | 430.05 | 477.04 |
| Crystal size/mm3 | 0.28 × 0.17 × 0.08 | 0.30 × 0.17 × 0.02 | 0.48 × 0.21 × 0.15 |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | C2/c | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 38.276(5) | 38.18(5) | 4.7397(3) |
| b/Å | 3.9429(3) | 4.131(5) | 9.8038(6) |
| c/Å | 13.178(3) | 13.345(12) | 12.3826(7) |
| α/° | 90 | 90 | 99.575(3) |
| β/° | 96.844(19) | 96.66(5) | 91.594(3) |
| γ/° | 90 | 90 | 98.130(3) |
| V/Å3 | 1974.6(5) | 2092(4) | 560.89(6) |
| ρ calcd/g cm−3 | 2.59 | 2.731 | 2.825 |
| Z | 8 | 8 | 2 |
| μ (MoKα)/mm−1 | 15.123 | 17.84 | 15.83 |
| T/K | 123 | 293 | 296 |
| θ range/° | 2.1–25.7 | 3.1–23.3 | 2.9–30.3 |
| Reflections collected | 7121 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 790 790 |
35373 |
| Unique reflections | 1859 | 1512 | 3424 |
| Observed reflections I > 2σ(I) | 1655 | 1302 | 2380 |
| Absorption correction | Analytical | Multi-scan | Multi-scan |
| Transmission Tmin/Tmax | 0.06/0.15 | 0.08/0.72 | 0.05/0.20 |
| Residual electron density Δρfin(max/min)/e Å−3 | 2.88/−1.96 | 3.73/−3.44 | 1.53/−2.19 |
| Final R-indices | |||
| R 1 (I ≥ 2σ(I)) | 0.036 | 0.072 | 0.046 |
| wR2 | 0.094 | 0.182 | 0.105 |
| CCDC | 1427633 | 1905061 | 1905062 |
Notably, (NapNC)AuI is not isostructural to the lighter congeners (Table 1). It crystallizes in the triclinic space group P with one molecule in the asymmetric unit. Again, the gold atom is linearly coordinated with Au–C (1.95(2) Å) and Au–I (2.5167(7) Å) distances in the expected range.42 The molecules are arranged in columns via a relatively short ππ-stacking distance of ∼3.5 Å, which seems to determine the aggregation in the crystal (Fig. 2). Aurophilic interactions do not play a crucial role: the shortest Au⋯Au distance is 3.539 Å indicative of only a weak contribution of aurophilic bonding. Also the intermolecular Au⋯I distance (3.760 Å) is larger than the sum of the van der Waals radii. A comparable behaviour was also found for t-BuNCAuX (X = Cl, Br, I): whereas the chlorido and bromido analogues are isomorphous with short Au⋯Au bonds, the iodide complex crystallizes in a different space group with very large Au⋯Au distances.43 We can interpret this difference in the molecular packing such that the combination of isonitriles and iodide ligands leads to particularly weak interactions, which we assign mostly to weaker electrostatic interactions in these particular complex geometries due to the less electronegative heavier halogens.42
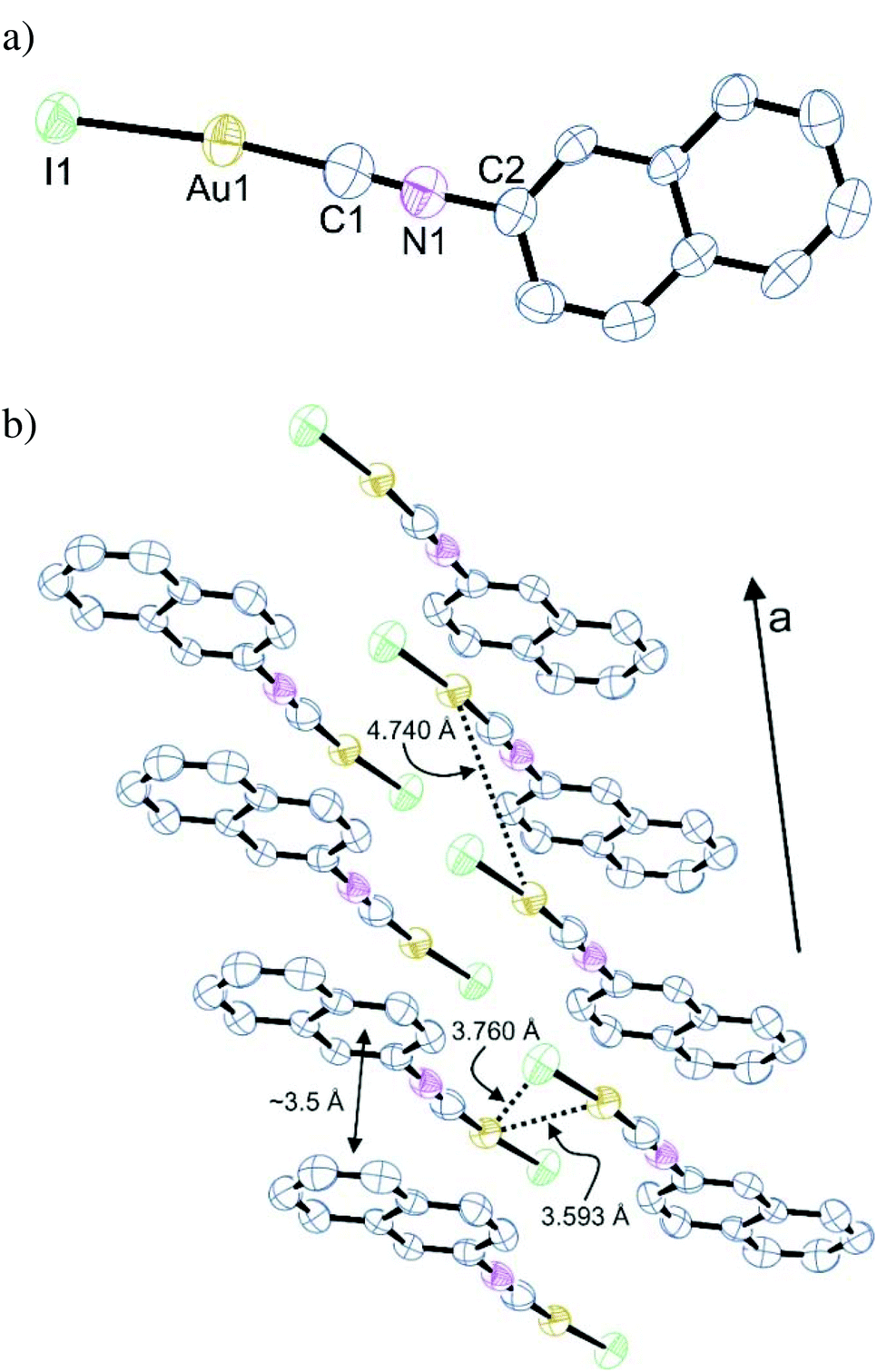 | ||
| Fig. 2 (a) Molecular structure of (NapNC)AuI. Selected bond lengths in Å and angles in °: Au1–I1 2.5167(7), Au1–C1 1.92(1), C1–N1 1.17(1), I1–Au1–C1 173.8(3), Au1–C1–N1 175.5(9). (b) Excerpt from a cell plot of the crystalline phase of compound (NapNC)AuI depicting the arrangement of the complexes. | ||
Photophysical characterisation
The (NapNC)AuCl complex was investigated photophysically before.40 The electronic absorption and emission spectra of (NapNC)AuBr and (NapNC)AuI (Fig. 3) are similar to those of (NapNC)AuCl and the free ligand NapNC. In the absorption spectra, intensive bands assignable to π–π* transitions of the naphthalene ligand are present below 275 nm with a structured band between 275 and 310 nm. At room temperature, the emission spectra show two structured bands. The more intense high-energy (HE) band at ∼330 nm ((NapNC)AuBr) and ∼350 nm ((NapNC)AuI) can be assigned to an intraligand singlet state (1IL), and the low-energy (LE) bands above 490 nm for both (NapNC)AuBr and (NapNC)AuI are from the respective triplet state (3IL). Such dual emission composed of fluorescence and phosphorescence is typical for gold-tagged aromatic systems.44–48 Also bromine substituted naphthalene derivatives show comparable dual luminescence behaviour in nitrogen purged solutions with phosphorescence caused by intersystem crossing processes facilitated by the presence of heavy atoms.49 Accordingly, the iodido species features a more intense phosphorescence band in relation to the fluorescence band than the bromido complex due to the enhanced heavy atom effect. At 77 K, the phosphorescence intensifies in relation to the fluorescence bands with a slight bathochromic shift. In the solid state, the room temperature HE and the LE emission bands are broadened and significantly shifted to longer wavelengths. At 77 K, the emission bands become more structured resembling the solution emission. The following effects complicate the assignment of the emission features: (i) aggregation effects lead to excited states aurophilic interactions and (ii) possible excimer formation. The general trend that the phosphorescence band is more intense compared to the fluorescence band can be observed as well for (NapNC)AuI. | ||
| Fig. 3 Electronic absorption, excitation and emission spectra of (NapNC)AuBr (upper panels) and (NapNC)AuI (lower panels) in DCM solution (left column, (NapNC)AuBr: c = 2.5 × 10−5 mol L−1; (NapNC)AuI: c = 1.88 × 10−5 mol L−1) and in the solid state (right column) at RT (red) and 77 K (blue). The black lines correspond to the UV-VIS spectra recorded at RT for the respective solutions. | ||
Evaporation experiments
Evaporation experiments were carried out for all three complexes: the temperature of the quartz crucibles containing powder of the Au(I) complex was increased in a stepwise fashion. After achieving a steady state, the deposition rate was determined via a quartz microbalance. For the conversion between the detuning of the oscillation of the quartz resonator and the film thickness, the known mass density of the corresponding bulk structure – see Table 1 – was used. Fig. 4 shows the corresponding Arrhenius plots, i.e., the natural logarithm of the deposition rate R as a function of inverse temperature 1/T, for (NapNC)AuCl and (NapNC)AuBr. Assuming a constant sticking coefficient of the Au(I) complex at the quartz balance and a large reservoir of molecules in the crucible of the evaporator, the measured deposition rate R at the quartz microbalance is directly proportional to the rate of desorption Rdes of the Au(I) complex. Assuming further a zeroth order desorption process, which is independent of the remaining amount of material, the following equation holds:| R ∝ Rdes ∝ e−Edes/(kBT) |
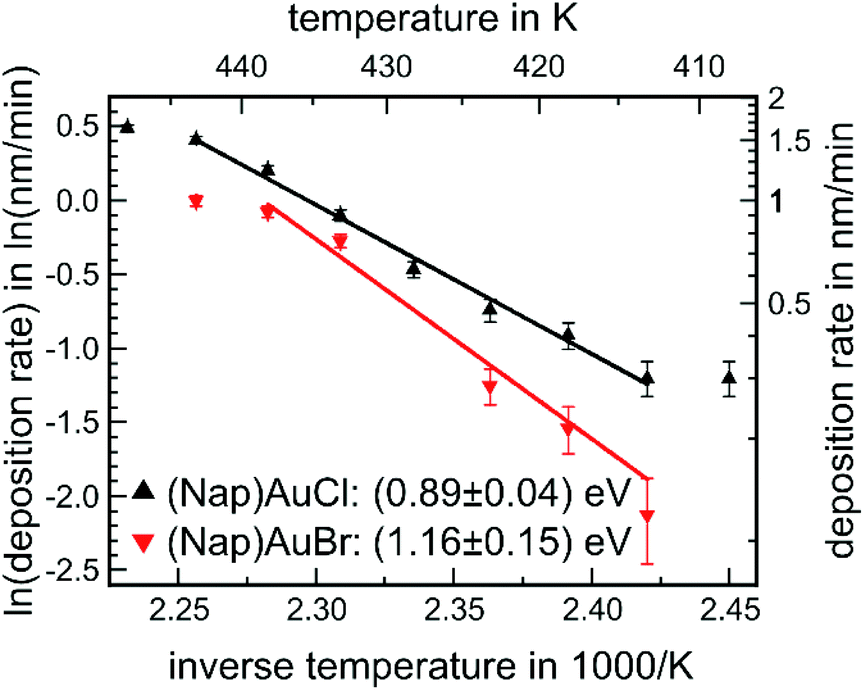 | ||
| Fig. 4 Arrhenius plot of the deposition rate as a function of the inverse temperature of the crucible for (NapNC)AuCl and (NapNC)AuBr. | ||
This equation predicts a linear behaviour of the Arrhenius plot, which was indeed found for the two complexes, (NapNC)AuCl and (NapNC)AuBr. Only the data points for the highest and lowest temperatures deviate from this linear trend. The reasons are obvious: (i) in general, low rates are hard to determine as indicated film thicknesses are in the same order of magnitude as the thermal detuning of the quartz microbalance. (ii) The high rates (at high temperatures) were determined last. Therefore, a lot of material from the crucible had already been sublimed and the crucible was almost empty, and so the assumption of a zeroth order desorption process may not hold any more. In fact, collecting data for the next temperature step was not possible as the rate suddenly dropped to zero indicating consumption of the molecules. Therefore, we excluded these data points from the fits. The slope of the Arrhenius fits yields an activation barrier for desorption of Edes = (0.89 ± 0.04) eV for (NapNC)AuCl and Edes = (1.16 ± 0.15) eV for (NapNC)AuBr. Zacharia et al. measured by thermal desorption spectroscopy (TDS) for naphthalene on graphite an activation barrier for desorption of Edes = (0.90 ± 0.07) eV.50 Chakarova-Käck et al. predicted a slightly lower value by using a van der Waals extension of density functional theory.51 Therefore, we can conclude in the case of (NapNC)AuCl that the activation barrier is mainly determined by the ππ-interaction of the naphthyl groups rather than by the Au(I)⋯Au(I) interaction between the molecules.
Our data show a lower rate for the Br-derivative than for the Cl-complex using an identical experimental setup. An offset between the two desorption rates could be related to the pre-exponential factor, but we can assume based on the transition state theory an identical pre-exponential factor for both molecules as they exhibit the same molecular skeleton. Therefore, the offset between the two Arrhenius plots might be related simply to the initial filling level of the crucibles and the grain size of the used powders.
Consistent with the lower desorption rate of (NapNC)AuBr compared to (NapNC)AuCl, the slopes of the Arrhenius plots indicate an increased attractive interaction between the molecules and hence a stronger aurophilic interaction. Contrary to this assumption, the XRD data of the bulk material indicate an almost unchanged Au⋯Au bond length, i.e., 3.224 Å (ref. 40) for the chlorido and 3.239 Å for the bromido complex. In a similar study on isocyanide Au(I) complexes, even a trend for the lowering of the volatility was observed for the series chlorine, bromine, and iodine.17 Therefore, the increase in the barrier of desorption by 0.27 eV has to be related to other effects rather than an increased Au⋯Au interaction.
For (NapNC)AuI, the quartz microbalance did not measure a significant deposition rate even after reaching a crucible temperature of 440 K. Consequently, the quartz glass used as a sample did not show a visible film deposited and the crucible was gilded after the experiment – see the image in the ESI.† This indicates that (NapNC)AuI decomposes rather than subliming. As stated above, (NapNC)AuI was unstable in solution. These observations are in agreement with reports in the literature indicating that other iodido complexes decompose faster than the lighter congeners.17
As we have not been able to sublime (NapNC)AuI, the films grown from (NapNC)AuCl and (NapNC)AuBr could contain at least partly decomposed molecules. Therefore, we checked the integrity of the deposited films by ion beam analysis (IBA), photoelectron spectroscopy, and X-ray fluorescence. Films of (NapNC)AuCl and (NapNC)AuBr were grown using crucible temperatures between 418 K and 423 K resulting in deposition rates between 0.2 nm min−1 and 0.5 nm min−1. A plausible degradation route would be the loss of the halogen atom, hence affecting the 1:1 ratio between gold and halogen in the deposited film. The employed IBA methods are comprised of Rutherford Backscattering Spectrometry (RBS) and complementary Time of Flight Elastic Recoil Detection Analysis (see ESI†). RBS is a method that allows quantifying this ratio without any assumption about sensitivity factors. RBS data for (NapNC)AuCl and (NapNC)AuBr deposited on glassy carbon are shown in Fig. 5.
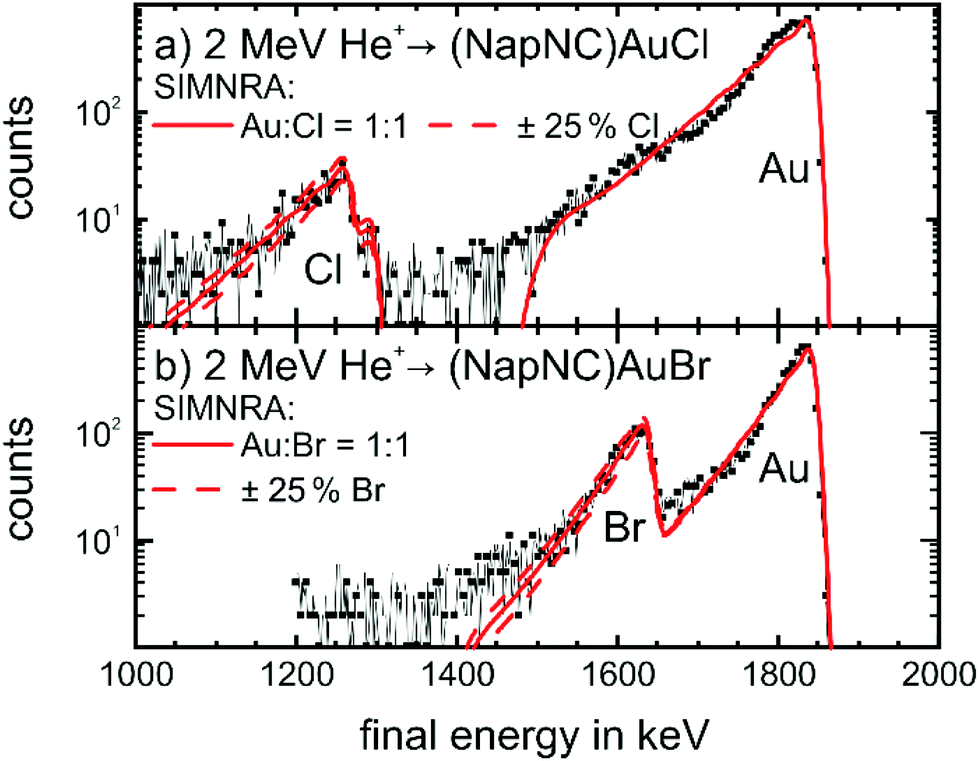 | ||
| Fig. 5 RBS spectra of films of (a) (NapNC)AuCl (25 nm thick) and (b) (NapNC)AuBr (23.8 nm thick) on glassy carbon. The black lines with symbols indicate the experimental data and the solid red lines the curve-fitting with the software SIMNRA68 for a ratio of 1:1 between gold and the respective halogen atom. The dashed red lines indicate the fit with an increased (+25%) and decreased (−25%) amount of halogen atoms. | ||
The spectra show the expected high energy cut-off for gold and the respective halogens, i.e., chlorine and bromine. The fit to the data and the respective area under the peaks confirm the expected 1:1 ratio between the amount of gold and halogen atoms in the thin film with a deviation of clearly less than 25%. Therefore, we can exclude the formation of dimers of the form [(NapNC)Au–X–Au(CNNap)]+ (with X = Cl or Br) – a possible decomposition route of L–Au–X complexes – because this would result in a 2:1 ratio.52–55
We can also determine the integrity of the molecules in the deposited films by X-ray photoelectron spectroscopy. The corresponding data are shown for (NapNC)AuCl and (NapNC)AuBr grown on glassy carbon in Fig. 6. The detailed spectra of Au, N, and the corresponding halogen, i.e., Cl or Br, exhibit just one chemical compound. The absence of other spectral contributions further confirms that there is no mixture of intact and broken molecules in the films but only the intact species. Elemental gold exhibits a doublet due to spin–orbit coupling: the Au 4f7/2 state is found at 84 eV and the Au 4f5/2 state at 87.6 eV.56 For (NapNC)AuCl and (NapNC)AuBr thin films, both of these spectral compounds are shifted by 2.1 eV to higher binding energies. A similar chemical shift for Au(I) was found by Casaletto, which confirms even the expected oxidation state of the Au atoms in the complexes after deposition.57
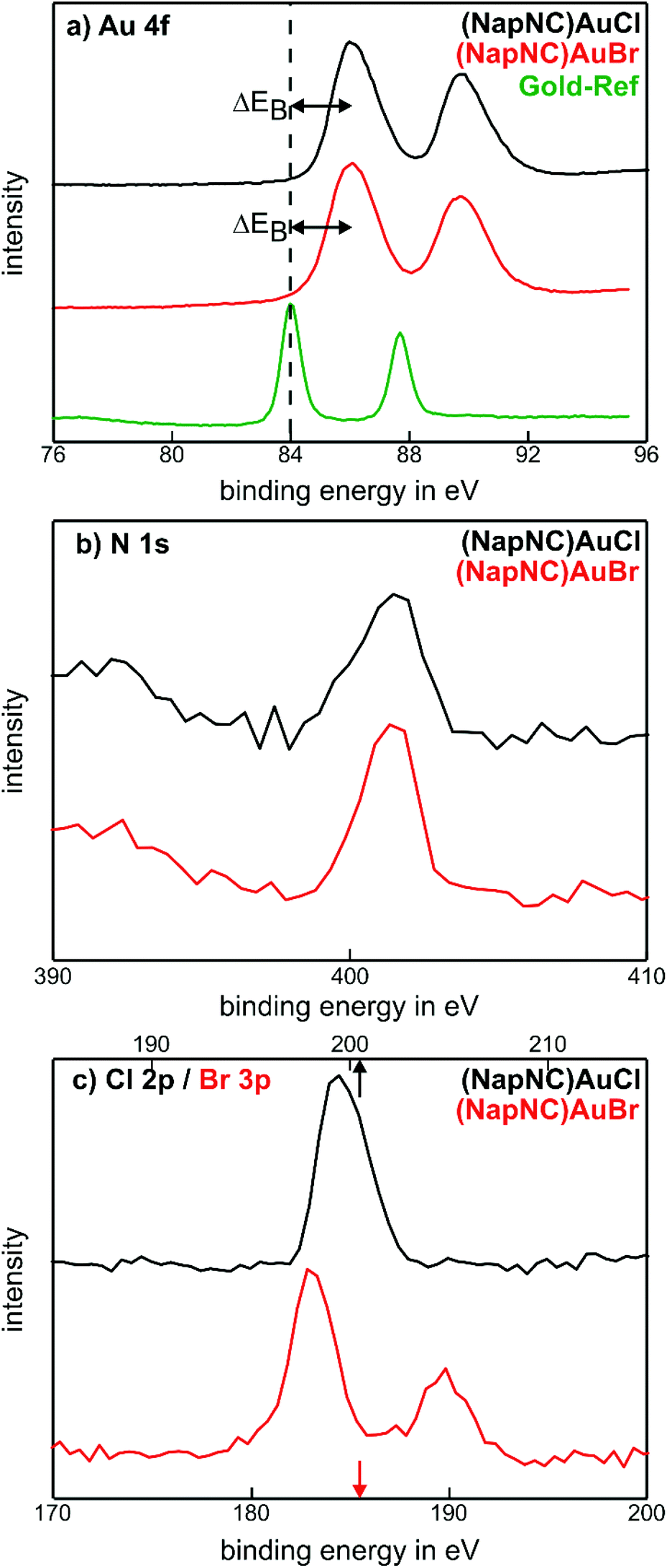 | ||
| Fig. 6 XPS analyses of the (NapNC)AuCl and (NapNC)AuBr films grown on glassy carbon. | ||
As the molecular composition was preserved during the deposition process, we also studied the crystal structure and molecular orientation within the thin films. As precipitation from liquid and physical vapour deposition are physically different processes, the crystal structures of both preparations might be different. In particular for the deposition of thin films, the substrate surface can also influence the structure of the films deposited on top of it. For example, a “thin film phase” occurs for pentacene deposited on alkali halides and SiO2 substrates.58,59 Therefore, we used two different amorphous substrates, namely glass and glassy carbon, to study such a potential influence of the substrate. The diffraction patterns of both substrates are shown in Fig. 7(a) and (b). The absence of sharp diffraction spots and the rather smooth structure in the reference patterns of the bare substrates confirm the amorphous character of both substrates.
 | ||
| Fig. 7 X-ray diffraction patterns of the bare substrates, i.e., (a) microscope slides made of soda lime glass (grey) and (b) glassy carbon (Sigradur D from HTW, grey), 25 nm thick films of (NapNC)AuCl (black) and (NapNC)AuBr (red) on top of these. | ||
After deposition of about 25 nm (NapNC)AuCl on glass substrates, sharp diffraction peaks can be observed as shown in Fig. 7(a). The ω − 2θ scan exhibits a series of peaks, which we assigned to the (n00) planes of the bulk structure – see Table 1. The sharpness and large intensity of these peaks reveal a high degree of order. The presence of the (n00) peaks reveals the orientation of the molecules, in which the (100) plane containing the gold molecules lies parallel to the surface plane. In addition, we have also performed in-plane measurements (GIXD), which further provide insights into lattice planes that are not oriented parallel to the surface – see ESI.† In these measurements, we observed peaks that are perfectly compatible with those predicted from the bulk crystal structure. Hence, we can safely conclude that the crystal structure of the thin (NapNC)AuCl films on glass substrates is equivalent to that found for free-standing crystals.
In contrast to the (NapNC)AuCl films on glass, the diffraction pattern of (NapNC)AuCl deposited on glassy carbon exhibits only very weak diffraction signals indicating a low degree of crystallinity. Furthermore, the direct determination of the molecular orientation is not possible due to the presence of different signals and the aforementioned low signal intensities. Therefore, we applied another experimental technique to determine the molecular orientation even for the films with low crystallinity on glassy carbon. This was enabled by C 1s-NEXAFS (Near Edge X-Ray Absorption Fine Structure) dichroism measurements. By comparing the contribution of the core level excitations into π*- and σ*-orbitals in the spectra obtained for the C K-edge for different angles of incidence of the linearly polarized light, one can determine the average tilt angle of the aromatic systems.60 The signal contributions in the NEXAFS spectra in the energetic range corresponding to π*-excitations are attributed to the naphthalene units in the molecular complexes due to their aromatic state and the spectral similarity as compared to bare naphthalene.61
Fig. 8(a) shows the NEXAFS data for (NapNC)AuCl on glass. Clearly, there is a strong correlation between the angle of incidence and the spectral contribution of the π*- (at the spectral onset) and the σ*-resonances (further away from the onset of absorption). While the contribution of the π*-resonances is huge in the spectra obtained at normal incidence (red curve), the contribution of the σ*-resonances dominates the NEXAFS spectra at grazing incidence (blue curve). A quantitative analysis of this dependence results in a tilt angle of 80° with respect to the surface normal. This angle corresponds exactly to the tilt angle of the naphthyl groups with respect to the (100) plane of the bulk structure. We note here that although the unit cell contains two (NapNC)AuCl molecules – see Table 1 – both exhibit an identical tilt angle with respect to the (100) plane of the monoclinic unit cell. In this case, the XRD data and the NEXAFS data suggest the same molecular orientation of (NapNC)AuCl on glass substrates.
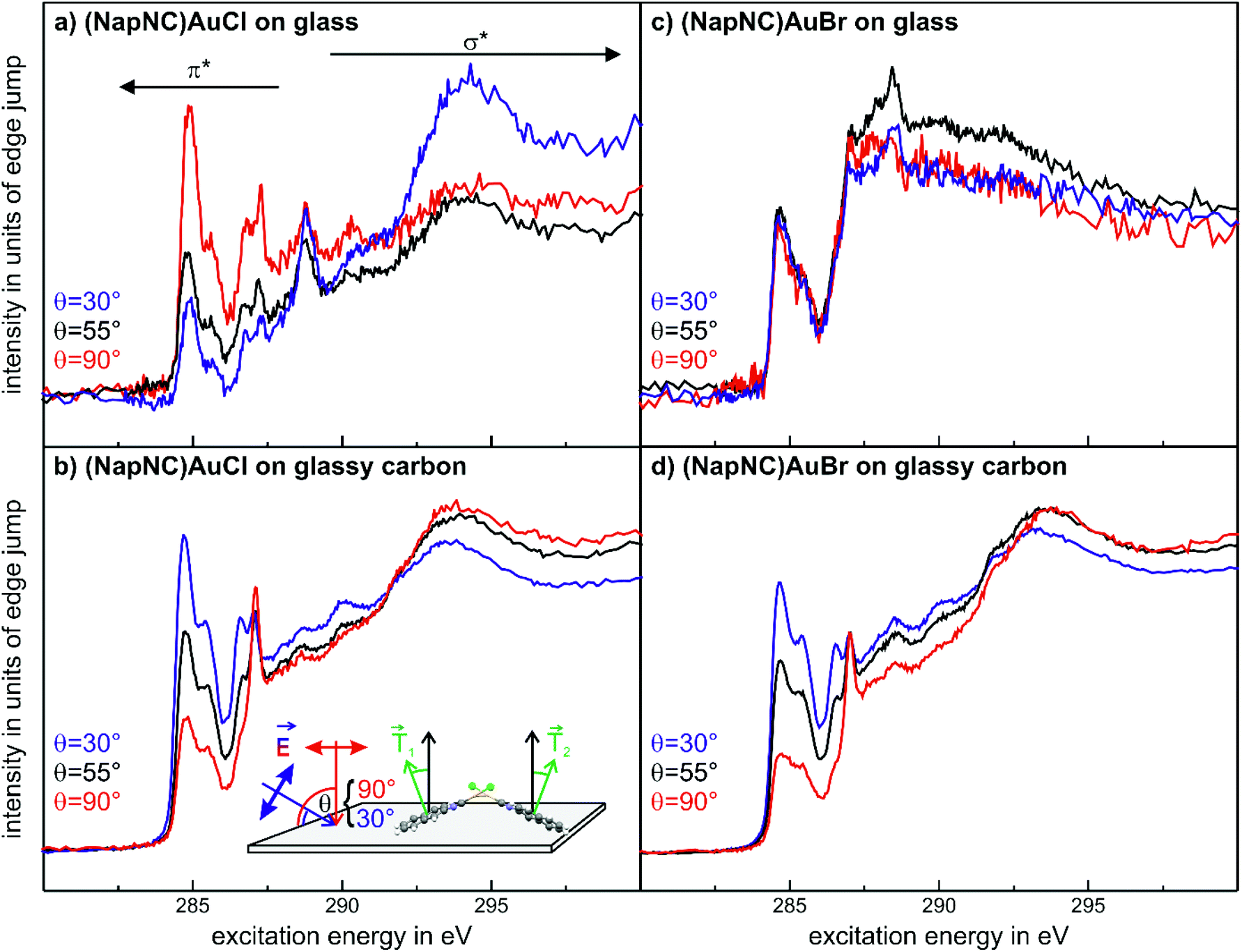 | ||
| Fig. 8 C 1s-NEXAFS data for (NapNC)AuCl (a and b) and of (NapNC)AuBr (c and d) deposited on glass and on glassy carbon, respectively. The thickness of all films was about 25 nm. | ||
In the case of (NapNC)AuCl on glassy carbon, the XRD data suggest a poor crystallinity of the film, but the NEXAFS data exhibit a strong dichroism (cf.Fig. 8(b)). Since the intensity contrast is inverted as compared to the films grown on glass here, this shows a rather lying molecular orientation instead of uprightly oriented molecules. The quantitative analysis of the dichroism yields an effective tilt angle of 38°. This angle does not resemble any tilt angle of an individual molecule against a low index plane of the bulk unit cell. Assuming the known bulk structure, a configuration in which the (010) plane is parallel to the surface results in a tilt angle of 28° of the naphthyl groups with respect to the substrate surface. At least for the first monolayer, this would maximize the van der Waals interaction of the π orbitals of the naphthyl groups with the substrate. On the other hand, this configuration still allows an aurophilic interaction between adjacent molecules forming homodimers resembling the crossed swords motif.40 This arrangement would allow the two molecules, which form a dimer, to exhibit the same tilt angle of the naphthyl group with respect to the substrate, although their transition dipole moments are not parallel to each other (see also the detailed discussion of a similar situation for acenes in crystalline herringbone arrangements in ref. 62). Therefore, the minimum tilt angle determined by NEXAFS amounts to 26–28° for this compound, which is rather close to the determined value of 38°, especially when considering the quasi-amorphous configuration as well as the (faint) presence of upright crystalline phases observed in the XRD measurements.
The spectra obtained for (NapNC)AuBr deposited on glassy carbon exhibit a similar characteristic fingerprint and dichroism as found for (NapNC)AuCl (Fig. 8(c)) on the same substrate. This indicates that the electronic structure of the naphthyl groups is not affected by the Cl and Br atoms and that the molecular orientation is similar. In agreement with these findings, the bulk structures for both derivatives are almost the same.
Interestingly, we do not observe a distinct dichroism for (NapNC)AuBr samples prepared on glass (cf.Fig. 8(d)), thus indicating a rather amorphous growth unlike the highly uniform molecular orientation observed for (NapNC)AuCl on glass. This is in line with our XRD data for this sample, where also no clear diffraction signals are observed in strong contrast to the well-defined XRD pattern for (NapNC)AuCl on glass.
Summary and conclusion
Aurophilic interactions in the solid state and in deposited films have been investigated. The respective Au(I) complexes bearing naphthylisonitrile ligands have been synthesized and characterised. While for (NapNC)AuCl and (NapNC)AuBr a monoclinic unit cell with almost identical lattice parameters was found, the bulk structure of (NapNC)AuI is triclinic. The complexes feature dual emission composed of both fluorescence and phosphorescence assignable to intraligand π–π* excited states.The respective complexes were evaporated under high vacuum on different substrate surfaces. The activation energy for evaporation of (NapNC)AuCl and (NapNC)AuBr is slightly higher than the value for unsubstituted naphthalene indicating that the ππ stacking of the naphthyl groups also dominates the structure formation of the obtained bulk structures. In contrast to the chlorido and bromido derivatives, we were not able to grow films by physical vapour deposition for the iodido derivative, which apparently decomposes upon heating instead of subliming.
The analysis of films, which were prepared by physical vapour deposition of (NapNC)AuCl and (NapNC)AuBr on glass and glassy carbon substrates, shows that the molecules are chemically intact on the surface. This allows the further use of (NapNC)AuCl and (NapNC)AuBr in coatings and as active materials in electronic devices based on organic thin films. The structural analysis furthermore points out that crystallites obtained by precipitation and thin films adopt an identical structure, in which the aurophilic interaction contributes to dimer formation between adjacent molecules.
The deposition of (NapNC)AuCl and (NapNC)AuBr on amorphous glass and glassy carbon did not yield an obvious trend with respect to the influence of the substrate: on the one hand, (NapNC)AuCl films on glass are highly crystalline and the molecules have an “upright” orientation with respect to the substrate. On the other hand, the (NapNC)AuBr films grown on the same substrate are quasiamorphous showing neither a crystalline signature nor a preferred orientation of the molecules. On glassy carbon, both molecules exhibit a preferred “flat lying” orientation with respect to the substrate even though the films do not exhibit a high crystallinity. To this end, the substrate–adsorbate interaction should be studied for more defined substrates like de facto defect free single crystal surfaces.
Experimental section
General
All reactions were carried out under ambient conditions. Solvents and 2-naphthylisonitrile were commercially available and used without further purification. (tht)AuCl (tht = tetrahydrothiophene) was prepared according to a reported procedure.63 NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer. UV/vis spectra were measured on a Cary 300 spectrophotometer and emission spectra on a Jobin Yvon Fluorolog 3.Synthesis of Au(I)-complexes
Evaporation experiments
For the growth of the thin films, an ultrahigh vacuum system was used – see Fig. 9. A turbomolecular pumping system maintained a base pressure below 1 × 10−6 mbar. A Bayard-Alpert type ionization gauge monitored the pressure. For evaporation, the Au(I) complexes were filled in quartz glass crucibles, which were heated by Ta filaments embedded in ceramic carriers. The temperature of the crucibles was monitored via type K thermocouples in direct contact with the quartz glass crucibles. The temperature of the crucibles was controlled via a PID controller. Up to four samples can be loaded onto the manipulator. At any time, only one sample was exposed to the molecular beam of the evaporator, while the other three samples were blocked by an aperture. The thickness of the films during deposition was determined via a quartz microbalance. A quartz oscillator is placed only with a minimal lateral displacement beside the sample so that no correction for the rate had to be applied. Based on the Sauerbrey equation64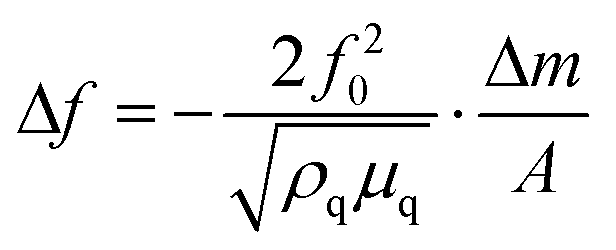 and the relation
and the relation 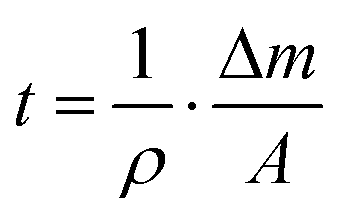 the equivalent thickness of a homogenous film with density ρ can be determined. Here, f0 is the resonance frequency of the quartz oscillator with active area A, density ρq, and shear modules μq. The deposited film changes the resonance frequency of the quartz oscillator by Δf due to the additional mass Δm. The error bars in Fig. 4 correspond to the quantization error of the digital display of the controller. As substrates optical microscope slides (made of soda lime glass) and glassy carbon (Sigradur G purchased from HTW, Germany) were used. The substrates were cleaned before insertion into the vacuum in a supersonic bath using iso-propanol and drying in a nitrogen stream. No further in situ cleaning procedure was applied.
the equivalent thickness of a homogenous film with density ρ can be determined. Here, f0 is the resonance frequency of the quartz oscillator with active area A, density ρq, and shear modules μq. The deposited film changes the resonance frequency of the quartz oscillator by Δf due to the additional mass Δm. The error bars in Fig. 4 correspond to the quantization error of the digital display of the controller. As substrates optical microscope slides (made of soda lime glass) and glassy carbon (Sigradur G purchased from HTW, Germany) were used. The substrates were cleaned before insertion into the vacuum in a supersonic bath using iso-propanol and drying in a nitrogen stream. No further in situ cleaning procedure was applied.
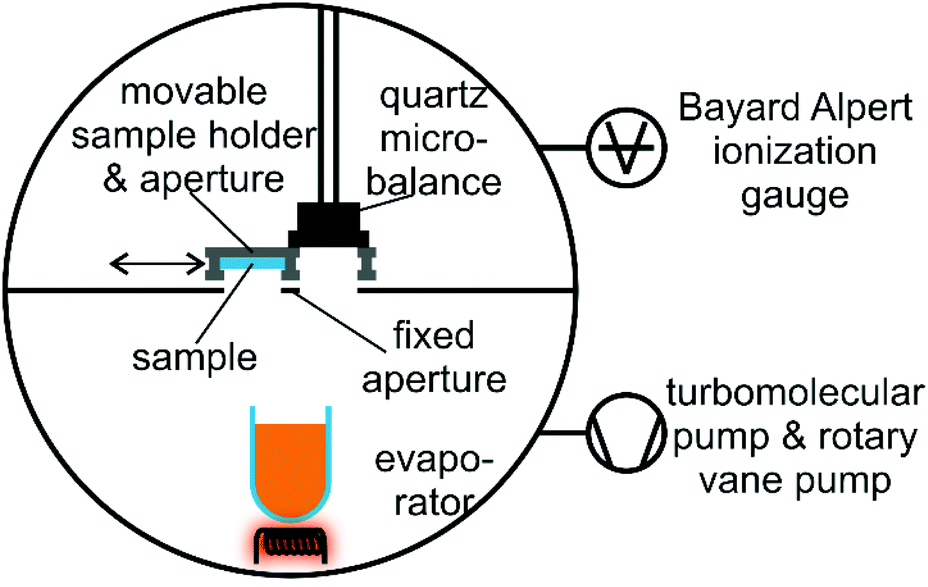 | ||
| Fig. 9 Schematic representation of the vacuum system used for the evaporation experiments. | ||
The vacuum was achieved and maintained via a combination of a turbomolecular pump and a rotary vane pump. The manipulator head also includes an aperture for the quartz microbalance, which is mounted at an almost identical distance to the quartz crucible containing the Au(I) complexes. A fixed aperture can block the molecular beam during the outgassing procedure. The sketch of the vacuum system is not to scale.
Single crystal X-ray diffraction analysis of bulk materials
Crystals suitable for single crystal X-ray diffraction were obtained for (NapNC)AuBr and (NapNC)AuI by slow vapour diffusion of n-pentane into a solution of the respective complex in dichloromethane: colourless needles of (NapNC)AuBr/I were obtained. Diffraction data were collected on a Bruker Smart X2S diffractometer operating with Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods (SHELXS-97)65 and refined by full-matrix least squares on F2 (SHELXL-97).66 The H atoms were calculated geometrically, and a riding model was applied in the refinement process.X-ray analysis of thin films
X-ray diffraction was utilized to characterize the crystal structure of the prepared thin films with a Bruker D8 Discover diffractometer using Cu Kα radiation (λ = 1.54056 Å). C 1s NEXAFS and X-ray photoelectron spectroscopy (XPS) measurements were performed at the HE-SGM dipole beamline of the synchrotron storage ring BESSY II of the Helmholtz Center Berlin (Germany) providing linearly polarized light (polarization factor = 0.91) and an energy resolution at the carbon K-edge of about 300 meV. X-ray photoemission spectra were recorded at an incident angle of 45° and at normal emission using a hemispherical electron energy analyzer (Scienta R3000) with typical pass energies of 100 eV and photon energies between 300 and 800 eV depending on the substrate and the studied region chosen such that parasitic contributions by, e.g., Auger lines were omitted. To provide a precise energy calibration, XPS binding energies were referenced to the characteristic peaks of reference samples. NEXAFS spectra were recorded in partial electron-yield (PEY) mode for films prepared on glassy carbon and in total electron-yield (TEY) mode for films prepared on glass (to achieve satisfactory signal-to-noise ratios despite the partially charged sample surfaces). To determine the average molecular orientation relative to the sample surface, NEXAFS spectra were recorded at different angles of incidence (30°, 55°, 70°, and 90°). Details of the experimental setup and data evaluation are provided in the literature.62Ion beam analysis
Rutherford backscattering spectrometry (RBS) measurements using 2 MeV He+ primary ions were performed on a 5 MV NEC 15SDH-2 Pelletron accelerator at the Uppsala University. Details of the employed setup are described in the supplementary information of ref. 67. The experimental spectra were compared to simulations using SIMNRA68 in order to obtain quantitative information without the need for standards with a focus on the concentration ratios of Au/Br or Cl/Br in the samples of interest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Austrian Science Fund (FWF): P28216-N36. NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”). VR-RFI (contract #821-2012-5144) and the Swedish Foundation for Strategic Research (SSF, contract RIF14-0053) supported the beam time at the tandem laboratory at Uppsala University. We want to thank the Helmholtz-Zentrum Berlin für Materialien und Energie (electron storage ring BESSY II) for providing beam time at the synchrotron beamline HE-SGM. The authors want to thank Markus Himmelsbach for measuring mass spectra and Carina Grill for recording XRF spectra. The authors also want to thank Peter Zeppenfeld, Marko Hapke, Christian Paulik, Antonia Reisecker and Heinz Peirlberger for being huge supporters of this project.References
- A. Grodzicki, I. Łakomska, P. Piszczek, I. Szymańska and E. Szłyk, Coord. Chem. Rev., 2005, 249, 2232–2258 CrossRef CAS.
- T. H. Baum and P. B. Comita, Chemical Vapor Deposition of Gold and Silver, in The Chemistry of Metal CVD, ed. T. Kodas and M. Hampden-Smith, VCH, Weinheim, 1994 Search PubMed.
- G. J. Hutchings, ACS Cent. Sci., 2018, 4, 1095–1101 CrossRef CAS.
- R. J. Puddephatt, Gold metal and gold alloys in electronics and thin film technology, in Gold: Progress in Chemistry, Biochemistry and Technology, ed. H. Schmidbaur, John Wiley & Sons, Chichester, UK, 1999, pp. 237–256 Search PubMed.
- A. A. Bessonov, N. B. Morozova, N. V. Gelfond, P. P. Semyannikov, S. V. Trubin, Yu. V. Shevtsov, Yu. V. Shubin and I. K. Igumenov, Surf. Coat. Technol., 2007, 201, 9099–9103 CrossRef CAS.
- R. G. Parkhomenko, A. E. Turgambaeva, N. B. Morozova, S. V. Trubin, V. V. Krisyuk and I. K. Igumenov, Chem. Vap. Deposition, 2013, 19, 38–44 CrossRef CAS.
- A. E. Turgambaeva, G. Zharkova, P. Semyannikov, V. V. Krisyuk, T. Koretskaya, S Trubin, B. Kuchumov and I. Igumenov, Gold Bull., 2011, 44, 177–184 CrossRef CAS.
- A. Turgambaeva, R. Parkhomenko, V. Aniskin, V. Krisyuk and I. Igumenov, Phys. Procedia, 2013, 46, 167–173 CrossRef CAS.
- M. M. Banaszak Holl, P. F. Seidler, S. P. Kowalczyk and F. R. McFeely, Inorg. Chem., 1994, 33, 510–517 CrossRef CAS.
- P. D. Tran and P. Doppelt, J. Electrochem. Soc., 2007, 154, D520–D525 CrossRef CAS.
- T. J. J. Whitehorne, J. P. Coyle, A. Mahmood, W. H. Monillas, G. P. A. Yap and S. T. Barry, Eur. J. Inorg. Chem., 2011, 3240–3247 CrossRef CAS.
- D. J. Mandia, M. B. E. Griffiths, W. Zhou, P. G. Gordon, J. Albert and S. T. Barry, Phys. Procedia, 2013, 46, 12–20 CrossRef CAS.
- P. R. Norton, P. A. Young, Q. Cheng, N. Dryden and R. J. Puddephatt, Surf. Sci., 1994, 307–309, 172–176 CrossRef CAS.
- N. H. Dryden, J. G. Shapter, L. L. Coatsworth, P. R. Norton and R. J. Puddephatt, Chem. Mater., 1992, 4, 979–981 CrossRef CAS.
- J. Messelhäuser, E. B. Flint and H. Suhr, Appl. Surf. Sci., 1992, 54, 64–68 CrossRef.
- J. Puddephatt and I. Treurnicht, J. Organomet. Chem., 1987, 319, 129–137 CrossRef.
- W. G. Carden, J. Pedziwiatr, K. A. Abboud and L. McElwee-White, ACS Appl. Mater. Interfaces, 2017, 9, 40998–41005 CrossRef CAS.
- P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef.
- S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741–2760 RSC.
- F. Scherbaum, A. Grohmann, B. Huber, C. Krüger and H. Schmidbaur, Angew. Chem., Int. Ed., 1988, 27, 1544–1546 CrossRef.
- H. Schmidbaur, Gold Bull., 1990, 23, 11–21 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. A. Rawashdeh-Omary, M. A. Omary, H. H. Patterson and J. P. Fackler, Jr., J. Am. Chem. Soc., 2001, 123, 11237–11247 CrossRef CAS.
- M. A. Rawashdeh-Omary, M. A. Omary and H. H. Patterson, J. Am. Chem. Soc., 2000, 122, 10371–10380 CrossRef CAS.
- O. Elbjeirami, S. Yockel, C. F. Campana, A. K. Wilson and M. A. Omary, Organometallics, 2007, 26, 2550–2560 CrossRef CAS.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- J. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375–407 CrossRef CAS.
- A. Kühnle, Curr. Opin. Colloid Interface Sci., 2009, 14, 157–168 CrossRef.
- X. Bouju, C. Mattioli, G. Franc, A. Pujol and A. Gourdon, Chem. Rev., 2017, 117, 1407–1444 CrossRef CAS.
- M. A. Malwitz, S. H. Lim, R. L. White-Morris, D. M. Pham, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 10885–10893 CrossRef CAS.
- O. Elbjeirami and M. A. Omary, J. Am. Chem. Soc., 2007, 129, 11384–11393 CrossRef CAS.
- R. E. Bachman, M. S. Fioritto, S. K. Fetics and T. M. Cocker, J. Am. Chem. Soc., 2001, 123, 5376–5377 CrossRef CAS.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS.
- I. Ugi, U. Fetzer, U. Eholzer, H. Knupfer and K. Offermann, Angew. Chem., Int. Ed., 1965, 4, 472–484 CrossRef.
- D. Schneider, A. Schier and H. Schmidbaur, Dalton Trans., 2004, 1995–2005 RSC.
- A. Bayler, A. Bauer and H. Schmidbaur, Chem. Ber./Recl., 1997, 130, 115–118 CrossRef CAS.
- E. Hobollahi, M. List, G. Redhammer, M. Zabel and U. Monkowius, Inorg. Chem. Commun., 2016, 65, 24–27 CrossRef.
- A. S. K. Hashmi, Y. Yu and F. Rominger, Organometallics, 2012, 31, 895–904 CrossRef CAS.
- R.-Y. Liau, T. Mathieson, A. Schier, R. J. F. Berger, N. Runeberg and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 881–889 CAS.
- W. Schneider, K. Angermaier, A. Sladek and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 790–800 CAS.
- M. Kriechbaum, G. Winterleitner, A. Gerisch, M. List and U. Monkowius, Eur. J. Inorg. Chem., 2013, 5567–5575 CrossRef CAS.
- M. Kriechbaum, M. List, R.J.F. Berger, M. Patzschke and U. Monkowius, Chem. – Eur. J., 2012, 18, 5506–5509 CrossRef CAS.
- B. David, U. Monkowius, J. Rust, C.W. Lehmann, L. Hyzak and F. Mohr, Dalton Trans., 2014, 43, 11059–11066 RSC.
- M. Kriechbaum, D. Otte, M. List and U. Monkowius, Dalton Trans., 2014, 43, 8781–8791 RSC.
- M. Kriechbaum, D. Otte, M. List and U. Monkowius, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 1188–1198 CAS.
- N. J. Turro, K.-C. Liu, M.-F. Chow and P. Lee, Photochem. Photobiol., 1978, 27, 523–529 CrossRef CAS.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
- S. D. Chakarova-Käck, E. Schröder, B. I. Lundqvist and D. C. Langreth, Phys. Rev. Lett., 2006, 96, 146107 CrossRef.
- T.G. Gray, Inorg. Chem., 2007, 28, 181–212 CAS.
- U. Monkowius, M. Zabel, M. Fleck and H. Yersin, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 1513–1524 CAS.
- U. V. Monkowius, S. D. Nogai and H. Schmidbaur, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 751–758 CAS.
- Note that the corresponding complexes [(LAu)2X]Y (L = R3P, Y = weakly coordinating anion) have previously been prepared and structurally characterized: H. Schmidbaur, A. Hamel, N. W. Mitzel, A. Schier and S. Nogai, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4916–4921 CrossRef CAS.
- X-ray data booklet accessed online viahttp://xdb.lbl.gov on 13.02.2019.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
- T. Kiyomura, T. Nemoto, K. Yoshida, T. Minari, H. Kurata and S. Isoda, Thin Solid Films, 2006, 515, 810–813 CrossRef CAS.
- I. P. M. Bouchoms, W. A. Schoonveld, J. Vrijmoeth and T.M. Klapwijk, Synth. Met., 1999, 104, 175–178 CrossRef CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer, Berlin, 1992 Search PubMed.
- G. Tzvetkov, N. Schmidt, T. Strunskus, C. Wöll and R. Fink, Surf. Sci., 2007, 601, 2089–2094 CrossRef CAS.
- T. Breuer, M. Klues and G. Witte, J. Electron Spectrosc. Relat. Phenom., 2015, 204, 102–115 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, Inorg. Synth., 2007, 26, 85–91 CrossRef.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- M. to Baben, M. Hans, D. Primetzhofer, S. Evertz, H. Ruess and J. M. Schneider, Mater. Res. Lett., 2017, 5, 158–169 CAS.
- M. Mayer, SIMNRA, a simulation program for the analysis of NRA, RBS and ERDA, in AIP Conf. Proc., AIP, 1999, pp. 541–544 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1905061 and 1905062. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9DT03049B |
| This journal is © The Royal Society of Chemistry 2019 |