
Efficient alkaline hydrogen evolution on atomically dispersed Ni–Nx Species anchored porous carbon with embedded Ni nanoparticles by accelerating water dissociation kinetics†
Chaojun
Lei
a,
Yu
Wang
b,
Yang
Hou
 *a,
Pan
Liu
cd,
Jian
Yang
a,
Tao
Zhang
e,
Xiaodong
Zhuang
e,
Mingwei
Chen
cd,
Bin
Yang
a,
Lecheng
Lei
a,
Chris
Yuan
f,
Ming
Qiu
*g and
Xinliang
Feng
*e
*a,
Pan
Liu
cd,
Jian
Yang
a,
Tao
Zhang
e,
Xiaodong
Zhuang
e,
Mingwei
Chen
cd,
Bin
Yang
a,
Lecheng
Lei
a,
Chris
Yuan
f,
Ming
Qiu
*g and
Xinliang
Feng
*e
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: yhou@zju.edu.cn
bShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
cWPI Advanced Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan
dCREST, JST, 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan
eCenter for Advancing Electronics Dresden (cfaed) & Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany. E-mail: xinliang.feng@tu-dresden.de
fDepartment of Mechanical and Aerospace Engineering, Case Western Reserve University, 10900 Euclid Ave, Cleveland, OH 44106, USA
gInstitute of Nanoscience and Nanotechnology, College of Physical Science and Technology, Central China Normal University, Wuhan 430079, China. E-mail: qium@mail.ccnu.edu.cn
First published on 21st August 2018
Abstract
Developing inexpensive and efficient electrocatalysts for hydrogen evolution reaction (HER) during alkaline water electrolysis is crucial for renewable and sustainable energy harvesting. Herein, we report a novel hybrid electrocatalyst comprising atomically dispersed Ni–Nx species anchored porous carbon (Ni–N–C) matrix with embedded Ni nanoparticles for HER. This new catalyst is synthesized via pyrolysis of hydrothermally prepared supermolecular composite of dicyandiamide and Ni ions followed by an acid etching treatment. The achieved hybrid exhibits superior catalytic performance toward HER with a small overpotential of 147 mV at 10 mA cm−2 and a low Tafel slope of 114 mV dec−1, comparable to those of state-of-the-art heteroatom-doped nanocarbon catalysts and even outperforming other reported transition-metal-based compounds in basic media. Experimental observations and theoretical calculations reveal that the presence of Ni nanoparticles can optimize surface states of Ni−Nx active centers and reduce energy barriers of dissociated water molecules, which synergistically improve OH− adsorption and promote HER kinetics. When served as electrodes for both cathode and anode, an alkaline water electrolyzer could afford a current density of 10 mA cm−2 at a low cell voltage of 1.58 V, rivalling the sufficiently high overpotentials of integrated Pt/C–Ir/C benchmark electrodes.
Broader contextWater electrolysis is recognized as a promising energy conversion technology for the production of renewable hydrogen energy. Water splitting efficiency is enhanced by the development of efficient earth-enriched electrocatalysts as alternatives to replace precious metal catalysts. Herein, we developed a novel hybrid electrocatalyst comprising atomically dispersed Ni–Nx species anchored porous carbon matrix with embedded Ni nanoparticles, which can efficiently catalyze hydrogen evolution reaction with striking kinetics metrics in basic conditions with excellent long-term stability. The presence of Ni nanoparticles can optimize the surface states of Ni–Nx active centers and reduce the energy barriers of dissociated water molecules, which synergistically improve OH− adsorption and promote reaction kinetics. This work provides a new insight into efficient design of non-noble metal catalysts for large-scale hydrogen production using excess electrical power. |
Electrocatalytic water splitting to produce hydrogen is widely considered as an efficient and attractive technology for the production of renewable energy.1–3 To accelerate the slow reaction dynamics of the hydrogen evolution reaction (HER), particularly in basic electrolytes, tremendous research efforts have been focused on developing high-performance HER electrocatalysts in recent years.4–7 Currently, Pt-based materials remain as the benchmark for HER catalysts,8 but their high cost and low abundance greatly limit large-scale applications. Therefore, great efforts have been made towards the development of inexpensive precious-metal-free alternatives including transition metal alloys,9 phosphides,10 nitrides,11 chalcogenides,12–14 carbides,15,16 and nanocarbon-based materials.5,17,18
Heteroatom-doped carbons, especially the transitional metal–N decorated nanocarbon materials, have attracted immense attention due to their tunable local electronic structures (e.g. charge polarization, difference in electronegativity between carbon atoms and heteroatoms, etc.) and fast electron transfer capacity.19–23 Studies have shown that the heteroatom-doped carbon materials not only possess electrocatalytic activity for HER,24 but also serve as active bifunctional electrocatalysts for alkaline water electrolyzer.25 This functionality is attributed to their strong chemical stability and compatibility in basic medium for both HER and oxygen evolution reaction. However, little progress has been made in the development of efficient transitional metal–N decorated nanocarbon catalysts due to their low overall catalytic activity, and the poor HER kinetics in alkaline electrolytes is still far from satisfactory due to the high energy barrier of initial water molecule dissociation (Volmer step) associated with the subsequent chemisorption of the OH− intermediates formed on the surfaces of nanocarbons.26
Herein, we developed a novel hybrid electrocatalyst for HER comprising atomically dispersed Ni–Nx species anchored porous carbon (Ni–N–C) matrix with embedded Ni nanoparticles (NP) denoted as Ni NP|Ni–N–C. The catalyst was synthesized by thermal treatment of hydrothermally prepared Ni-based precursor under Ar gas and subsequent acid etching treatment. The resulting Ni NP|Ni–N–C hybrid possessed a strong coupling effect and high surface area of 140 m2 g−1. Moreover, it exhibited an excellent HER catalytic activity in basic media, featured by an overpotential of 147 mV to attain 10 mA cm−2 with a low Tafel slope of 114 mV dec−1, comparable to the state-of-the-art heteroatom-doped nanocarbon catalysts, and outperforming even the other reported transition-metal-based compounds. Experimental observations confirmed that a strong chemical coupling interaction between Ni NP and Ni–N–C modulates the electronic structure and facilitates electron transfer at the constructed interfaces, which boosts its HER performance. Theoretical calculations revealed that the incorporation of Ni NP into the Ni–N–C frameworks effectively promoted initial water dissociation process (Volmer step) and simultaneously optimized OH− adsorption free energy of the Ni NP|Ni–N–C, thus resulting in improved HER kinetics in alkaline solutions. A two-electrode alkaline electrolyzer assembled with Ni NP|Ni–N–C as both the cathode and anode was able to approach 10 mA cm−2 at a low cell voltage of 1.58 V.
The synthesis scheme of Ni NP|Ni–N–C is shown in Fig. 1. Firstly, Ni NP|Ni–N–C precursor was prepared by a self-assembly of dicyandiamide and nickel(II) chloride under hydrothermal conditions. The resulting material was then pyrolyzed in Ar atmosphere at an elevated temperature between 700–1000 °C, followed by acid etching treatment with 0.5 M sulfuric acid to remove any accessible Ni species, which finally produces the Ni NP|Ni–N–C. Successful decomposition of the Ni-based precursor to form the Ni NP|Ni–N–C hybrid was confirmed by Fourier transform infrared spectra (Fig. S1, ESI†). The effect of carbonization temperature on HER activity was investigated in detail, and we found that Ni NP|Ni–N–C pyrolyzed at 900 °C yielded the highest HER activity (Fig. S2, ESI†).
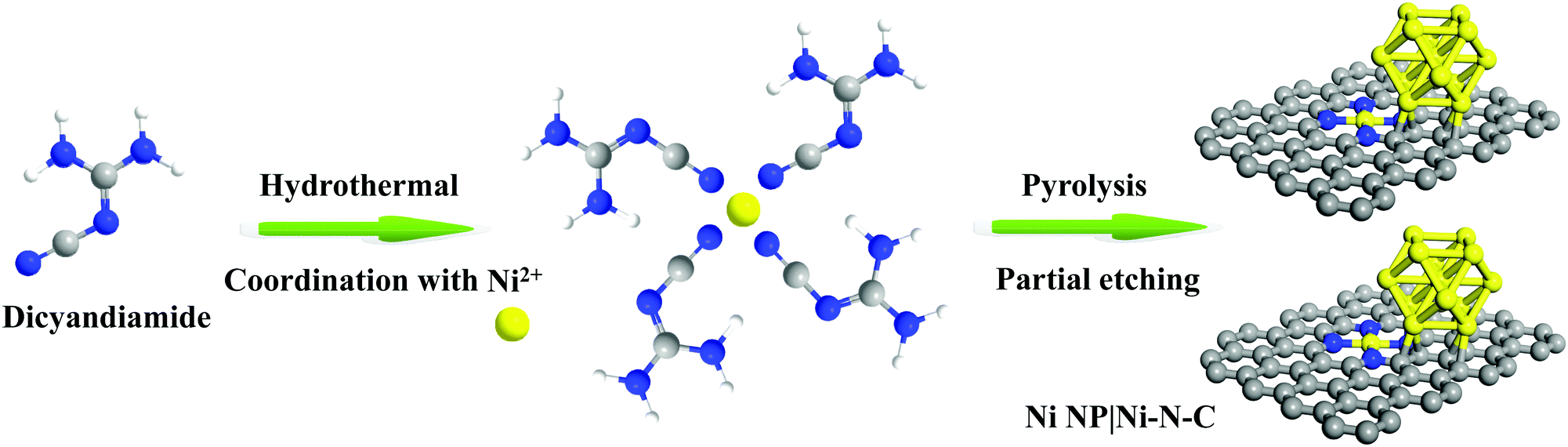 | ||
| Fig. 1 Schematic illustration of the synthesis process of Ni NP|Ni–N–C. | ||
X-ray diffraction (XRD) pattern of Ni NP|Ni–N–C exhibited one typical peak at around 26.4°, which corresponds to the (002) facet of graphitic carbon, and the other peaks located at 44.0° and 51.3° are assigned to the (111) and (200) facets of metallic Ni, respectively (Fig. S3, ESI†). Raman spectrum of Ni NP|Ni–N–C shows three peaks located at 1351, 1576, and 2694 cm−1 (Fig. S4, ESI†) corresponding to the D, G, and 2D bands, respectively. The ID/IG ratio of Ni NP|Ni–N–C is 0.99, which further confirmed the presence of graphitic structure. Transmission electron microscopy (TEM) image of Ni NP|Ni–N–C shows that the hybrid is composed of small Ni NP with particle sizes between 25–50 nm, closely-embedded within a distorted carbon matrix (Fig. 2a). Moreover, high-resolution TEM (HRTEM) images reveal that Ni NPs are crystallized with a lattice distance of 0.20 nm and are encapsulated between a few graphene layers with a lattice distance of 0.34 nm (Fig. 2b and c), which is the result of catalytic graphitization behavior of Ni NP.27 Closer examination of the distorted carbon matrix via high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) discloses the presence of numerous pores in the carbon matrix without any Ni NP (Fig. 2d). This is mainly due to the efficient removal of those accessible Ni species during the acid leaching treatment. The corresponding energy dispersive X-ray spectroscopy (EDX) element mapping demonstrates that the Ni NP|Ni–N–C consists of Ni, N, and C elements (Fig. 2e). Notably, all elements are homogeneously distributed in the Ni NP|Ni–N–C except for the Ni NP, which indicates the formation of Ni–Nx bonding. Additionally, the atomic-resolution HAADF-STEM images present a number of highly dispersed sparking points in the range of 0.2–0.3 nm (Fig. 2f–h), which correspond to the heavier Ni atoms, confirming the uniform dispersion of individual Ni atom across the entire carbon matrix of Ni NP|Ni–N–C. Ni NP|Ni–N–C has nearly 11.8 wt% of Ni species as determined by inductively coupled plasma mass spectrometry, which is well consistent with the results from thermogravimetric analysis (Fig. S5, ESI†).
 | ||
| Fig. 2 TEM image (a), HRTEM images (b and c), HAADF-STEM image (d) C, N, and Ni elemental EDX mappings (e), and atomic-resolution HAADF-STEM images (f–h) of Ni NP|Ni–N–C. | ||
X-ray photoelectron spectroscopy (XPS) reveals the presence of Ni, N, C, and O elements in Ni NP|Ni–N–C (Fig. S6, ESI†). The high-resolution Ni 2p XPS spectrum of Ni NP|Ni–N–C displays the binding energies of Ni 2p3/2 and Ni 2p1/2 peaks located at 855.5 and 873.2 eV, respectively with two shakeup satellites at 861.4 and 879.8 eV (Fig. 3a) indicating the co-existence of Ni2+ and Ni3+, respectively.28 In comparison to the XPS peak centered at 855.5 eV corresponding to Ni 2p3/2 of Ni NP|Ni–N–C, the noticeable shift of the corresponding peak of Ni–N–C to 856.4 eV suggests the existence of strong coupling interactions between Ni NP and Ni–N–C (Fig. 3a).29 Deconvoluted high-resolution N 1s XPS spectrum of Ni NP|Ni–N–C displays five peaks located at 398.2, 398.8, 399.7, 400.7, and 402.3 eV (Fig. 3b), attributed to pyridinic N, Ni-bonded N, pyrrolic N, graphitic N, and oxidized N, respectively.30–32 This signifies that the N species are coordinated with Ni atoms to form Ni–Nx moieties, with atomic percentage of doped nitrogen at about 4.9 at%. Deconvoluted high-resolution C 1s XPS spectrum of Ni NP|Ni–N–C shows characteristics peaks for C–C (284.5 eV), C–N (285.3 eV), C–O (286.1 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.1 eV), and O–CO (288.8 eV) bonds (Fig. 3c) further validating that the N atoms are successfully doped. To further investigate the local coordination environment of Ni species, X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses of Ni NP|Ni–N–C were performed. Fig. 3d shows the XANES spectra at the Ni K-edge of Ni NP|Ni–N–C with Ni foil and NiO as references. The absorption edge of Ni NP|Ni–N–C is located between Ni and NiO, which is possibly due to coordination of Ni–N/C in Ni NP|Ni–N–C. Fourier-transformed EXAFS curve for Ni NP|Ni–N–C in Fig. 3e exhibits a dominant metallic Ni–Ni contribution with a peak at 2.14 Å. Signals ranging from 1.03 to 1.69 Å correspond to the Ni–N/C scattering pair, which further corroborates the existence of Ni–Nx coordination in Ni NP|Ni–N–C. These Ni–Nx coordination peaks from 1.03 to 1.69 Å are close to the Ni–N bonds in the standard nickel phthalocyanin sample,33 where a single Ni atom is coordinated with four surrounding N atoms. The nitrogen adsorption–desorption isotherm of Ni NP|Ni–N–C shows mesoporous features with a high surface area of 140 m2 g−1 and a total pore volume of 0.34 cm3 g−1 (Fig. 3f), which are favorable for active site exposure and rapid HER-relevant species transport. Furthermore, a small water contact angle of 46.7° is observed for Ni NP|Ni–N–C, indicating its highly hydrophilic nature that can boost electrolyte permeation and affinity (Fig. S7, ESI†).34
O (287.1 eV), and O–CO (288.8 eV) bonds (Fig. 3c) further validating that the N atoms are successfully doped. To further investigate the local coordination environment of Ni species, X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses of Ni NP|Ni–N–C were performed. Fig. 3d shows the XANES spectra at the Ni K-edge of Ni NP|Ni–N–C with Ni foil and NiO as references. The absorption edge of Ni NP|Ni–N–C is located between Ni and NiO, which is possibly due to coordination of Ni–N/C in Ni NP|Ni–N–C. Fourier-transformed EXAFS curve for Ni NP|Ni–N–C in Fig. 3e exhibits a dominant metallic Ni–Ni contribution with a peak at 2.14 Å. Signals ranging from 1.03 to 1.69 Å correspond to the Ni–N/C scattering pair, which further corroborates the existence of Ni–Nx coordination in Ni NP|Ni–N–C. These Ni–Nx coordination peaks from 1.03 to 1.69 Å are close to the Ni–N bonds in the standard nickel phthalocyanin sample,33 where a single Ni atom is coordinated with four surrounding N atoms. The nitrogen adsorption–desorption isotherm of Ni NP|Ni–N–C shows mesoporous features with a high surface area of 140 m2 g−1 and a total pore volume of 0.34 cm3 g−1 (Fig. 3f), which are favorable for active site exposure and rapid HER-relevant species transport. Furthermore, a small water contact angle of 46.7° is observed for Ni NP|Ni–N–C, indicating its highly hydrophilic nature that can boost electrolyte permeation and affinity (Fig. S7, ESI†).34
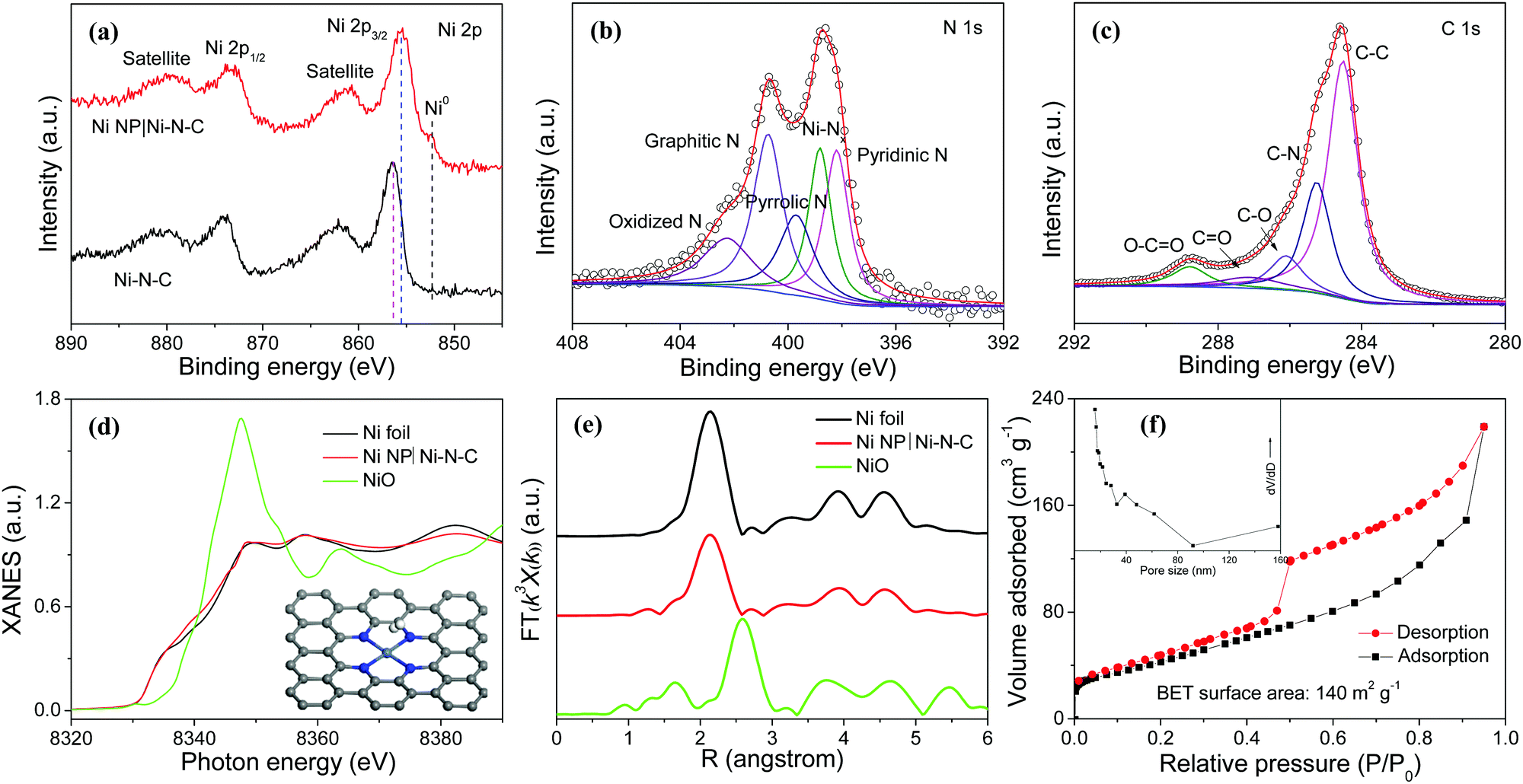 | ||
| Fig. 3 High-resolution Ni 2p (a), N 1s (b), and C 1s (c) XPS spectra, (d) Ni K-edge XANES spectra, (e) Ni K-edge k3-weighted EXAFS spectra, and (f) N2 adsorption–desorption isotherm and corresponding pore size distribution (inset) of Ni NP|Ni–N–C. Data for Ni foil and NiO are also shown. | ||
Electrocatalytic HER activity of the Ni NP|Ni–N–C was assessed in 1.0 M KOH solution using a standard three-electrode cell, in which electrochemically exfoliated graphene (EG) foil served as the conducting support for growth of electrocatalyst (Fig. S8, ESI†). For comparison, the catalytic activities of Ni–N–C/EG prepared with sufficient acid leaching (48 h reaction), Ni NP/EG, EG, physical mixture of EG, Ni NP, and Ni–N–C, and commercial Pt/C were assessed. As expected, Pt/C catalyst exhibits high HER performance (Fig. 4a) while bare EG shows very limited HER activity. The Ni NP|Ni–N–C/EG presents the smallest onset potential of 46 mV towards HER compared with those of the other samples, beyond which a rapid increase in cathodic current is observed under more negative potentials with H2 bubble generation at the electrode surface. The current density on Ni NP|Ni–N–C/EG at an overpotential of −0.3 V can reach 44.6 mA cm−2, which is about 4.85, 10.1, and 19.6 times higher than that of Ni–N–C/EG (9.19 mA cm−2), Ni NP/EG (4.41 mA cm−2), and EG (2.28 mA cm−2), respectively. To achieve a current density of 10 mA cm−2, Ni NP|Ni–N–C/EG requires an overpotential of only 147 mV, while Ni–N–C/EG and Ni NP/EG need larger overpotentials of 307 and 398 mV, respectively, although Ni–N–C catalyst has a higher surface area (208 m2 g−1, Fig. S9, ESI†) than Ni NP|Ni–N–C (140 m2 g−1). Such observation clearly discloses that the Ni NP in Ni NP|Ni–N–C also play a positive role in the HER activity. Additionally, the physical mixture sample also delivers a considerably large overpotential of 236 mV at 10 mA cm−2 compared to Ni NP|Ni–N–C/EG, which demonstrates the presence of strong coupling effects in the Ni NP|Ni–N–C/EG catalyst. Notably, the overpotential of 147 mV at 10 mA cm−2 for Ni NP|Ni–N–C/EG is well comparable to those of the state-of-the-art heteroatom-doped nanocarbon catalysts (Table S1, ESI†). The new hybrid catalyst even outperforms other reported transition-metal-based compounds in basic media, such as Pr0.5BSCF,35 CoP/rGO-400,36 CoP/CC,37 Ni5P4,38 and ONPPGC/OCC39 with overpotentials of >150 mV at 10 mA cm−2.
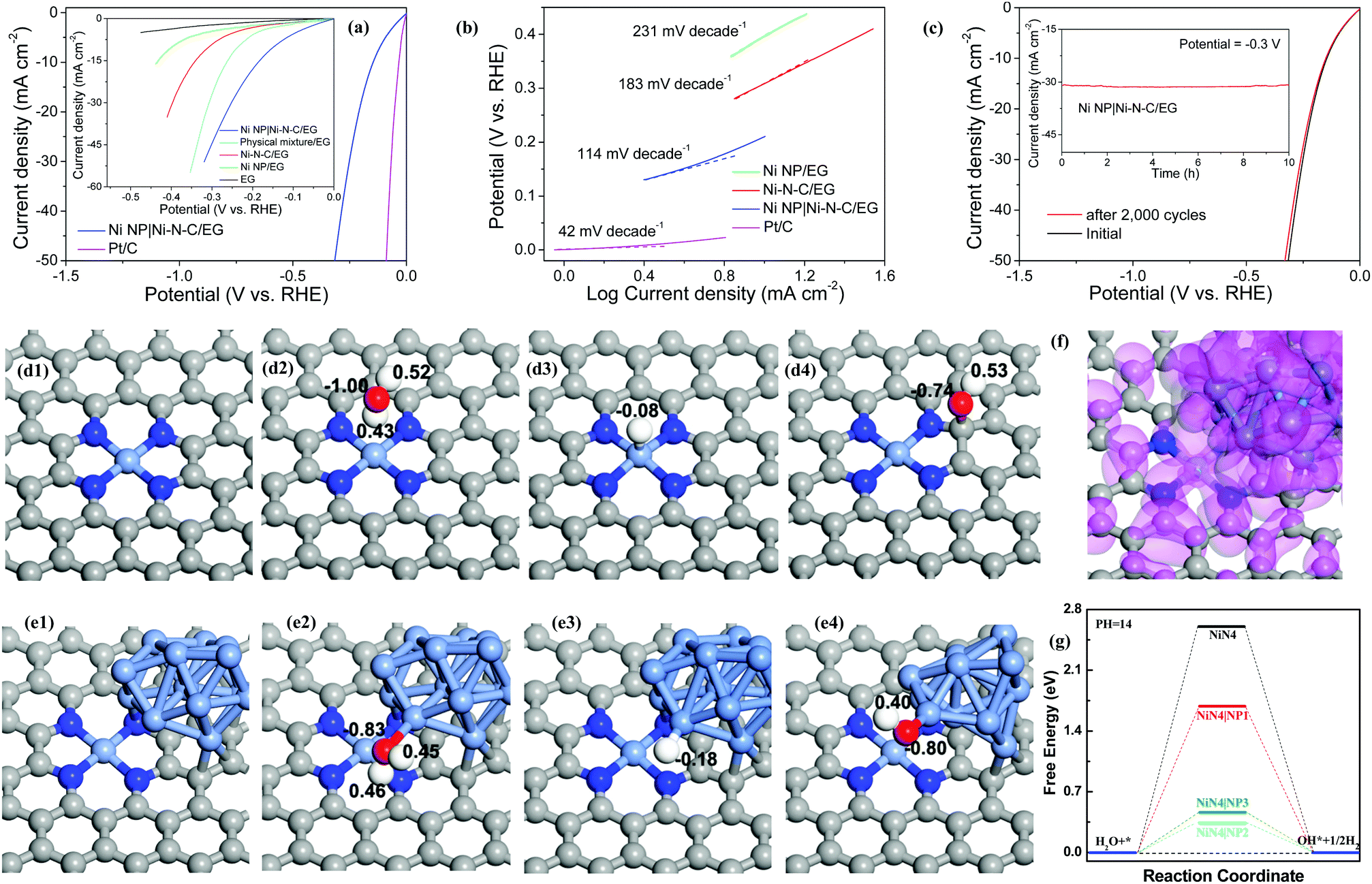 | ||
| Fig. 4 (a) Polarization curves of EG, Ni NP/EG, Ni–N–C/EG, physical mixture, Ni NP|Ni–N–C/EG, and Pt/C for HER. (b) Tafel plots of Ni NP/EG, Ni–N–C/EG, Ni NP|Ni–N–C/EG, and Pt/C for HER. (c) Polarization curves of Ni NP|Ni–N–C/EG before and after 2000 cycles. Inset: Chronoamperometry curve of Ni NP|Ni–N–C/EG at a potential of −0.3 V for 10 h without iR correction. (d and e) DFT calculated models (Volmer–Tafel–Heyrovsky mechanism) for NiN4 and NiN4|NP2 and corresponding population distributions of the groups adsorbed. Gray atom: carbon, light blue atom: nitrogen, blue atom: nickel, red atom: oxygen, and white atom: hydrogen. (f) Distribution of the highest occupied molecular orbital of NiN4|NP2. (g) Free-energy diagrams of the water dissociation process for NiN4 with and without Ni NP. | ||
High HER activity of Ni NP|Ni–N–C/EG is further supported by comparing their Tafel plots. A Tafel slope of 114 mV dec−1 is obtained for Ni NP|Ni–N–C/EG (Fig. 4b), which demonstrates that its electrocatalytic HER kinetics was determined by a combined Volmer–Heyrovsky process in alkaline condition.40 This slope is much smaller than that of Ni–N–C/EG (183 mV dec−1) and Ni NP/EG (231 mV dec−1), which indicates that the Ni NP|Ni–N–C/EG catalyst follows a more efficient Volmer–Heyrovsky mechanism.41 In other words, the slow Volmer step is significantly accelerated by introduction of Ni NP into Ni–N–C matrix. The exchange current density of Ni NP|Ni–N–C/EG, determined by extrapolation of the Tafel plots, was calculated to be 0.21 mA cm−2. The stability of Ni NP|Ni–N–C/EG was evaluated by continuous sweep measurements that show a high operational stability for 2000 cycles (Fig. 4c). We further examined the long-term durability of Ni NP|Ni–N–C/EG and observed no obvious activity decay after continuous electrolysis for 10 h (inset of Fig. 4c). The excellent structural stability of Ni NP|Ni–N–C/EG after electrocatalysis was corroborated by electronic microscopy, XRD and XPS analysis (Fig. S10, ESI†). The electrochemical results, combined with HAADF-STEM analyses, XPS results and X-ray absorption spectroscopy observations confirm that the excellent HER activity of Ni NP|Ni–N–C originated from Ni–N–C, while Ni NP synergistically promoted initial water dissociation process (Volmer step). To further clarify the nature of catalytically active sites of Ni NP|Ni–N–C, the effect of thiocyanate ion (SCN−) on HER activity was studied. After treatment with SCN−, the onset potential of Ni NP|Ni–N–C/EG cathodically shifted by approximately 210 mV with a serious drop in the current density (Fig. S11, ESI†). The significant decrease in catalytic activity can be attributed to the blocking of Ni–Nx sites by SCN− ions.28,42 However, no obvious decay of HER activity is found for Ni NP/EG catalyst under same test condition, which clarifies the crucial role of atomically dispersed Ni–Nx species as active sites for HER.
First-principles-based density functional theory calculations (DFT) can help to explore and explain the fundamental HER mechanisms for the excellent catalytic performance demonstrated by the Ni NP|Ni–N–C. Theoretical models are investigated to clarify the synergistic effect of Ni NP and NiN4/C for the entire catalytic process. As shown in Fig. 4d–f and Fig. S12, S13 (ESI†), three different deposition sites of Ni NP on the NiN4/C catalyst are studied and the models are named as NiN4|NP1, NiN4|NP2, and NiN4|NP3. Ni atoms, as the typical active centers, and their d-orbital electrons have primary contributions to the HER mechanism.43 Thus, the Ni NP can provide more active sites for H+, which is one of the main reasons to boost their HER kinetics.44,45 To explain the complete HER mechanism, the water dissociation process is first shown in Fig. 4g. For the Volmer step, free energies of the water dissociation process on the active centers of NiN4|NP1, NiN4|NP2, and NiN4|NP3 are 1.69, 0.34, and 0.48 eV, respectively, which are much lower than that on NiN4 (ΔG = 2.60 eV). This result indicates that the states of Ni NP can hybridize with NiN4 (Fig. 4f), optimize the surface states of active centers, and reduce the formation energies of water molecules and OH− adsorption on the active sites, thus lowering the energy barriers of water molecules dissociated in the alkaline solution.
Catalytic reaction profiles of Volmer–Heyrovskey mechanisms are shown in Fig. 5a and b. In Fig. 5a, there are obvious energy barriers for the water adsorption, water dissociation, and hydrogen desorption when no voltage is applied on the surface of NiN4. Further DFT calculations elucidate that the –OH groups always block water adsorption and the active sites can trap H2 molecules in alkaline solutions. When a potential of 1.50 V is applied, these barriers are overcome and H2 can be generated continuously. Considering that NiN4|NP2 demonstrated the best performance during the water dissociation process, we take it as an example to study its HER mechanism in detail. In Fig. 5b, comparing with NiN4, the barriers for water adsorption and dissociation are reduced while the barrier for hydrogen production is enhanced due to the electron transfer between Ni NP and NiN4. In NiN4|NP2, free energy of the –OH group adsorbed (−0.19 eV) is lower than that of H group (−0.10 eV) on the active sites, which indicates the energy barrier exists for the initial hydrogen adsorption. Because of the hybridization states of NiN4 and Ni NP, the surface potential of NiN4|NP2 can trap H2 molecules stronger than that of NiN4. As a result, the energy barrier is increased from 0.56 eV to 1.41 eV for H2 desorption (Fig. 5a). Overall, H2 can be continuously produced only when the applied potential is about 0.50 V. For H adsorption, the bond length of Ni–H is 1.439 Å on NiN4 and the average bond lengths are 1.435 Å (Ni–H on the Ni NP) and 1.452 Å (Ni–H on the NiN4 of NiN4|NP). Meanwhile, average population distributions of the H atoms adsorbed are −0.08e on NiN4, and are −0.06e and −0.18e on the NiN4 and Ni NP of NiN4|NP on average (Fig. 4d and e). Ni NP near NiN4 increases the bond length and reduces the orbital hybridization between H and Ni atoms of NiN4 on the NiN4|NP. Moreover, from Fig. 5c, Ni NP increases the density of states of the highest occupied molecular orbital below the Fermi energy, which enhances the d electron contributions to HER. Briefly, benefiting from the Ni NP introduced, the NiN4|NP can provide more active sites, lower the energy barrier of water dissociation, reduce the hybridization states between H and Ni atom of NiN4, and boost the HER performance significantly (Fig. 5d).
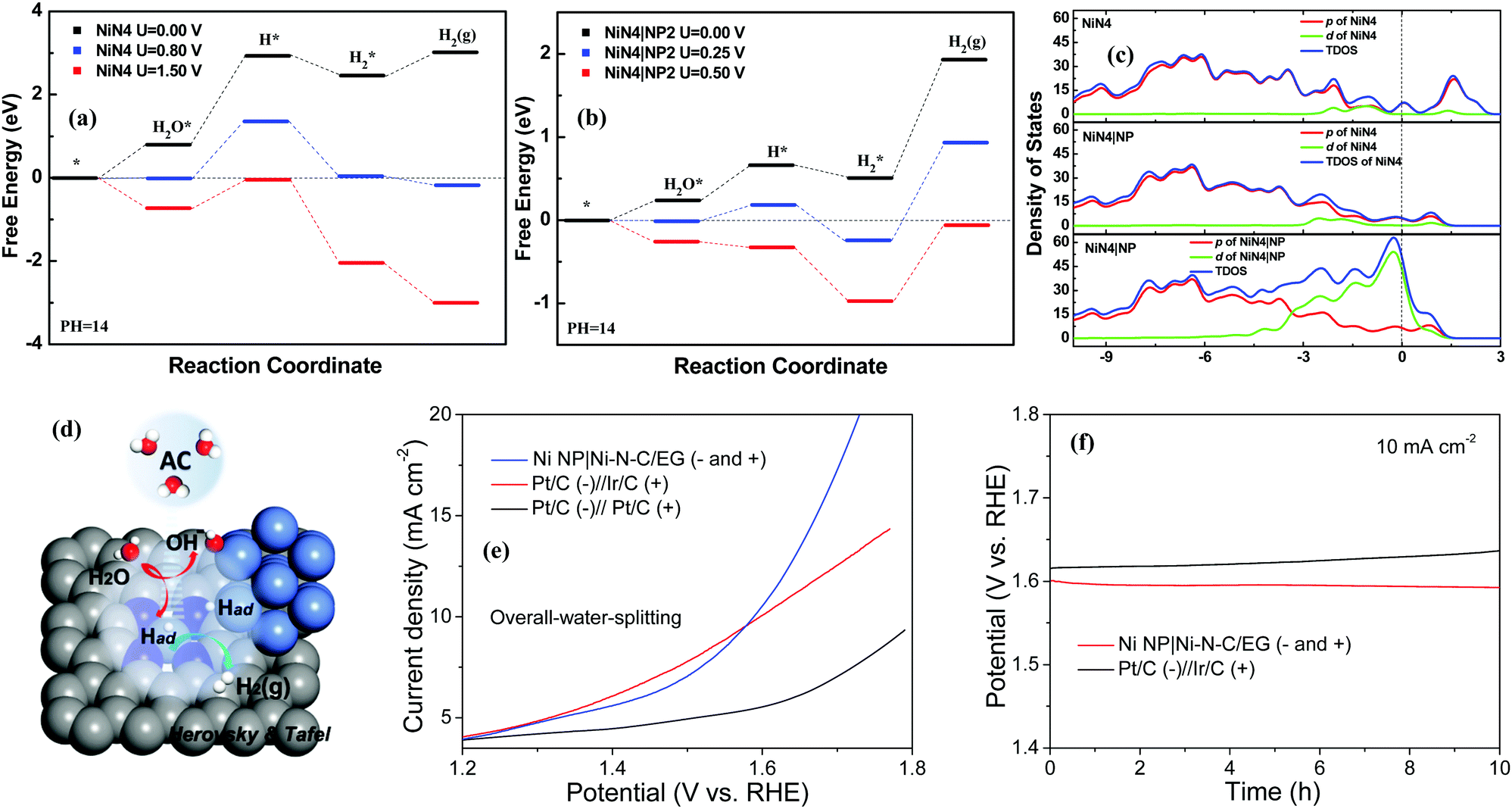 | ||
| Fig. 5 (a) Free-energy diagrams of the Volmer–Heyrovsky mechanism for NiN4. (b) Free-energy diagrams of the Volmer–Heyrovsky mechanism for NiN4|NP2. (c) Density of states for NiN4, NiN4 of NiN4|NP2, and NiN4|NP2. (d) Schematic illustration of the HER mechanism of NiN4|NP. (e) Polarization curves of Ni NP|Ni–N–C/EG (− and +), Pt/C (−)‖Ir/C (+), and Pt/C (−)‖Pt/C (+) for overall-water-splitting without iR correction. (f) Chronopotentiometry curves of Ni NP|Ni–N–C/EG (− and +) and Pt/C (−)‖Ir/C (+) at a current density of 10 mA cm−2 without iR correction. | ||
As transition metal-based materials are identified as bifunctional electrocatalysts,46 an alkaline water electrolyzer was established by integrating the Ni NP|Ni–N–C as both the cathode and anode in 1.0 M KOH solution using a two-electrode setup. A low cell voltage of 1.58 V is required for Ni NP|Ni–N–C to reach 10 mA cm−2, which surpass that of the integrated Pt/C−Ir/C benchmark electrodes (1.60 V at 10 mA cm−2) under sufficiently high overpotentials (Fig. 5e). Furthermore, Ni NP|Ni–N–C shows a high long-term stability with negligible decay of current density over 10 h of continuous operation in catalyzing overall-water-splitting (Fig. 5f).
Conclusions
In summary, we developed a novel Ni NP|Ni–N–C hybrid electrocatalyst for HER via self-assembly of dicyandiamide and Ni ions under hydrothermal treatment, followed by pyrolysis and an acid etching process. Benefiting from its high surface area and strong coupling interaction of Ni NP and Ni–N–C, the optimized Ni NP|Ni–N–C possessed a high HER catalytic activity. An overpotential of 147 mV was required to drive a current density of 10 mA cm−2, comparable to state-of-the-art heteroatom-doped nanocarbon catalysts, and even outperforming other reported transition-metal-based compounds in basic media. Meanwhile, its overall water-splitting performance outperformed the integrated Pt/C−Ir/C benchmark electrodes. Experimental observations and theoretical studies confirmed that the synergistic effect of Ni NP and Ni–N–C in Ni NP|Ni–N–C efficiently lowers the kinetic energy barrier of the water dissociation process and facilitates the OH− adsorption and thus benefits the HER. The novel metal NP embedded in the metal–N–C hybrid electrocatalysts described in this work will pave a promising pathway to develop highly active cost-effective catalysts for other potential electrochemical applications including nitrogen fixation, CO2 reduction, and oxygen reduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. Hou thanks the support of NSFC 51702284, Fundamental Research Funds for the Central Universities (112109*172210171) and the Startup Foundation for Hundred-Talent Program of Zhejiang University (112100-193820101/001/022). X. L. Feng acknowledges the support of the ERC Grant 2DMATER, UP-GREEN, the CFAED, the EC under the Graphene Flagship (CNECT-ICT-604391). M. Qiu thanks the support of Hubei Natural Science Foundation of China (2018CFB531). Dr Qiang Zheng is sincerely acknowledged for help in HAADF-STEM measurements.Notes and references
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- K. H. Liu, H. X. Zhong, S. J. Li, Y. X. Duan, M. M. Shi, X. B. Zhang, J. M. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64–111 CrossRef CAS.
- Y. Hou, M. Qiu, G. Nam, M. G. Kim, T. Zhang, K. Liu, X. Zhuang, J. Cho, C. Yuan and X. Feng, Nano Lett., 2017, 17, 4202–4209 CrossRef CAS PubMed.
- Y. Hou, M. Qiu, T. Zhang, X. Zhuang, C. S. Kim, C. Yuan and X. Feng, Adv. Mater., 2017, 29, 1701589 CrossRef PubMed.
- H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- G. Zhang, G. Wang, Y. Liu, H. Liu, J. Qu and J. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- P. Cai, Y. Li, G. Wang and Z. Wen, Angew. Chem., Int. Ed., 2018, 130, 3974–3979 CrossRef.
- P. Wang, K. Jiang, G. Wang, J. Yao and X. Huang, Angew. Chem., Int. Ed., 2016, 55, 12859–12863 CrossRef CAS PubMed.
- F. H. Saadi, A. I. Carim, W. S. Drisdell, S. Gul, J. H. Baricuatro, J. Yano, M. P. Soriaga and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 12927–12930 CrossRef CAS PubMed.
- H. Yan, C. Tian, L. Wang, A. Wu, M. Meng, L. Zhao and H. Fu, Angew. Chem., Int. Ed., 2015, 54, 6325–6329 CrossRef CAS PubMed.
- J. X. Feng, J. Q. Wu, Y. Tong and G. R. Li, J. Am. Chem. Soc., 2017, 140, 610–617 CrossRef PubMed.
- Y. Hou, M. R. Lohe, J. Zhang, S. H. Liu, X. D. Zhuang and X. L. Feng, Energy Environ. Sci., 2016, 9, 478–483 RSC.
- Y. Hou, M. Qiu, T. Zhang, X. Zhuang, C. S. Kim, C. Yuan and X. Feng, Adv. Mater., 2017, 29, 1701589 CrossRef PubMed.
- Q. Gong, Y. Wang, Q. Hu, J. Zhou, R. Feng, P. N. Duchesne, P. Zhang, F. Chen, N. Han, Y. Li, C. Jin, Y. Li and S. T. Lee, Nat. Commun., 2016, 7, 13216 CrossRef CAS PubMed.
- Q. Yan, H. Fan, H. Yu, Y. Zhang, Y. Zheng, Z. Dai, Y. Luo, B. Li and Y. Zong, Angew. Chem., Int. Ed., 2017, 56, 12566–12570 CrossRef PubMed.
- J. Lai, S. Li, F. Wu, M. Saqib, R. Luque and G. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 RSC.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- H. W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Mullen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- K. Yuan, S. Sfaelou, M. Qiu, D. Lützenkirchen-Hecht, X. Zhuang, Y. Chen, C. Yuan, X. Feng and U. Scherf, ACS Energy Lett., 2018, 3, 252–260 CrossRef CAS.
- Z. L. Wang, X. F. Hao, Z. Jiang, X. P. Sun, D. Xu, J. Wang, H. X. Zhong, F. L. Meng and X. B. Zhang, J. Am. Chem. Soc., 2015, 137, 15070–15073 CrossRef CAS PubMed.
- C. Lei, H. Chen, J. Cao, J. Yang, M. Qiu, Y. Xia, C. Yuan, B. Yang, Z. Li, X. Zhang, L. Lei, J. Abbott, Y. Zhong, X. Xia, G. Wu, Q. He and Y. Hou, Adv. Energy Mater., 2018, 1801912 CrossRef.
- Y. Hou, M. Qiu, T. Zhang, J. Ma, S. Liu, X. Zhuang, C. Yuan and X. Feng, Adv. Mater., 2017, 29, 1604480 CrossRef PubMed.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W. C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16086–16090 CrossRef CAS PubMed.
- M. S. Balogun, W. Qiu, H. Yang, W. Fan, Y. Huang, G. R. Li, H. Ji and Y. Tong, Energy Environ. Sci., 2016, 9, 3411–3416 RSC.
- J. Zhang, T. Wang, P. Liu, S. Liu, R. Dong, X. Zhuang, M. Chen and X. Feng, Energy Environ. Sci., 2016, 9, 2789–2793 RSC.
- Y. Hou, S. Cui, Z. Wen, X. Guo, X. Feng and J. Chen, Small, 2015, 11, 5940–5948 CrossRef CAS PubMed.
- J. Yin, Q. Fan, Y. Li, F. Cheng, P. Zhou, P. Xi and S. Sun, J. Am. Chem. Soc., 2016, 138, 14546–14549 CrossRef CAS PubMed.
- W. J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L. J. Zhang, J. Q. Wang, J. S. Hu, Z. Wei and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- L. Ye, G. L. Chai and Z. H. Wen, Adv. Funct. Mater., 2017, 27, 1606190 CrossRef.
- L. Z. Liu, G. Zeng, J. X. Chen, L. L. Bi, L. M. Dai and Z. H. Wen, Nano Energy, 2018, 49, 393–402 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- H. Zhong, J. Wang, F. Meng and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9937–9941 CrossRef CAS PubMed.
- X. Xu, Y. Chen, W. Zhou, Z. Zhu, C. Su, M. Liu and Z. Shao, Adv. Mater., 2016, 28, 6442–6448 CrossRef CAS PubMed.
- L. Jiao, Y. X. Zhou and H. L. Jiang, Chem. Sci., 2016, 7, 1690–1695 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- M. Ledendecker, S. Krick Calderon, C. Papp, H. P. Steinruck, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- J. P. Lai, S. P. Li, F. X. Wu, M. Saqib, R. Luque and G. B. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 RSC.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- P. Cai, J. Huang, J. Chen and Z. Wen, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 CrossRef CAS PubMed.
- J. A. Varnell, E. C. M. Tse, C. E. Schulz, T. T. Fister, R. T. Haasch, J. Timoshenko, A. I. Frenkel and A. A. Gewirth, Nat. Commun., 2016, 7, 12582 CrossRef CAS PubMed.
- Y. Hou, X. D. Zhuang and X. L. Feng, Small Methods, 2017, 1, 1700090 CrossRef.
- R. Parsons, Trans. Faraday Soc., 1958, 54, 1053–1063 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee01841c |
| This journal is © The Royal Society of Chemistry 2019 |