
Understanding the behaviour of UV absorbance of natural waters upon chlorination using model compounds†
Nicolas
Beauchamp
 *a,
Caetano
Dorea
b,
Christine
Beaulieu
c,
Christian
Bouchard
d and
Manuel
Rodriguez
e
*a,
Caetano
Dorea
b,
Christine
Beaulieu
c,
Christian
Bouchard
d and
Manuel
Rodriguez
e
aDépartement de génie civil et de génie des eaux, Université Laval, 1065, Avenue de la Médecine, Québec, QC G1V 0A6, Canada. E-mail: nicolas.beauchamp.1@ulaval.ca
bDepartment of Civil Engineering, University of Victoria, PO Box 1700, STN CSC, 3800 Finnerty Road, Victoria, B.C. V8W 2Y2, Canada. E-mail: caetanodorea@uvic.ca
cDivision de la qualité de l'eau, Service du traitement des eaux, Ville de Québec 214, Ave St-Sacrement, suite 210, Québec, QC G1N 3X6, Canada. E-mail: christine.beaulieu@ville.quebec.qc.ca
dDépartement de génie civil et de génie des eaux, Université Laval, 1065, Avenue de la Médecine, Québec, QC G1V 0A6, Canada. E-mail: christian.bouchard@gci.ulaval.ca
eÉcole supérieure d'aménagement du territoire et de développement régional, Université Laval, 2325, Allée des Bibliothèques, Québec, QC G1V 0A6, Canada. E-mail: manuel.rodriguez@esad.ulaval.ca
First published on 4th December 2018
Abstract
Differential UV absorbance (ΔA) is a promising indicator that could allow operators and utility managers to routinely monitor and manage disinfection by-product (DBP) concentrations without the limitations of regulatory sampling and analyses. So far, empirical relationships between DBP formation and ΔA at 272 nm have been proposed, but these relationships are known to be specific to the waters being analyzed. The objective of this study is qualitative rather than quantitative; it is to identify features of the spectra of simple compounds that are distinguishable in the spectra of natural water and relevant to predict DBP formation empirically. In order to meet this objective, compounds that could model the different components of natural organic matter (NOM) were chlorinated, and their UV spectra were collected at various reaction times, along with samples for DBP analyses. The same procedure was conducted on natural waters that were pretreated using coagulation and filtration. Results show that both diketone and phenolic moieties could be responsible for the initial decrease in absorbance observed around 270 nm, while the continuous gradual decrease that follows is attributable to phenolic structures. This gradual decrease is most notable around 250 nm, which means that ΔA at this wavelength might be more closely related to DBP formation than the usual ΔA at 272 nm. Other compounds such as carboxylic acids and amino acids do not seem to contribute significantly to trihalomethanes, haloacetic acids or ΔA in natural waters, although amino acids probably make up a significant proportion of the haloacetonitrile precursors.
Water impactMonitoring chlorination by-products at high frequency can be costly and time-consuming, but would provide valuable feedback to operators of drinking water facilities. Differential UV absorbance could be an effective tool for this purpose. Our study identifies some of the reactions responsible for specific changes in the absorbance spectra of natural waters, and suggests ways to generalize empirical differential absorbance models. |
1 Introduction
Differential UV absorbance (ΔA) has been studied as a surrogate for disinfection by-product (DBP) analysis in drinking water treatment. The most widely cited wavelength for differential UV spectroscopy is 272 nm, based on the peak commonly found in this region of the differential spectra,1 although other characteristics of the UV-visible absorbance spectra have also been explored and linked to DBP concentrations.2,3 DBP–ΔA relationships are known to be linear or curvilinear (exponential or power functions) in the range of chlorination conditions commonly found in drinking water. This is true for both regulated DBPs such as trihalomethanes (THMs) and haloacetic acids (HAAs),3–7 and unregulated DBPs such as haloketones (HKs), haloacetonitriles (HANs), chloral hydrate (CH) and chloropicrin (CPK).3,8–10 While determination coefficients of the relationships between ΔA at 272 nm and DBP concentrations can be relatively high (R2 > 0.90), the fitting parameters of those relationships vary from one water source to another,4,6,11,12 or for the same water source in different seasons.8 These differences are generally attributed to changes in the characteristics and concentration of the natural organic matter (NOM) that constitute the DBP precursors.So far, little research has been done to provide a deeper understanding of the NOM characteristics that can influence the DBP–ΔA relationship. Changes in UV spectra caused by NOM protonation–deprotonation (pH variations) have been studied,13,14 although associating this behaviour with reactivity towards chlorine or DBP formation has not been attempted yet. What we know about UV spectra and chlorination DBPs is that the absorbance in the spectral region of 250 to 280 nm is usually attributed to aromatic molecules.15 We also know that some of these molecules (namely biphenols, and more generally, dioxygenated moieties with the second oxygenated moiety in meta-position) react promptly with chlorine to produce chloroform (TCM) and other DBPs16–20 at the same timescale observed in drinking water chlorination reactions (a few hours to a few days).
The NOM in natural and treated waters and its fractions (humic, fulvic, etc.) are constituted of multiple moieties forming large and sometimes complex molecules.21 Because of this, it is hard to interpret the changes occurring in their UV spectra upon chlorination and to associate these changes with the contribution of a given moiety. Therefore, in the research presented herein, model compounds of simpler molecular structures have been chlorinated. The resulting absorbance spectra have then been studied at different reaction times and compared to the changes occurring in the spectra of treated (coagulated and filtered) water upon chlorination. The objective of this experiment is different from previous literature on DBP–ΔA relationships. While previous research has focused mostly on establishing empirical models for predicting DBP concentrations in natural waters using ΔA as the only predictor, this research has a more qualitative approach. Specifically, it does not aim at establishing quantitative empirical models of DBP formation of model compounds, as they would have little relevance for the prediction of DBP concentrations in natural waters were the nature and size of the NOM molecules are much more varied. Instead, it seeks to identify features of the spectra of model compounds that are distinguishable in the spectra of natural water, e.g. the ΔA peak at 272 nm, and postulates on the relevance of these features for the prediction of DBP concentrations in natural waters. In a sense, it is another step in the search for a theoretical explanation of the empirical observations of ΔA spectra in natural waters.
2 Methodology
2.1 Choice of model compounds
The choice of the model compounds was based on the classification by Leenheer and Huffman22 and Leenheer and Croué23 who categorized NOM compounds based on polarity (hydrophobic/hydrophilic) and acidity (acid/neutral/base). Also, we selected model compounds that have a variety of known or expected UV absorbances in the region of 270 nm. Although the UV-visible absorbance spectra are already known for most of these model compounds, the information about ΔA in the context of DBP formation in drinking water is still lacking. The compounds studied were the phenols resorcinol and p-cresol (hydrophobic/acid), acetylacetone (hydrophobic/neutral), tyrosine (hydrophobic/base), malonic and citric acids (hydrophilic/acid), methylcellulose (hydrophilic/neutral) and histidine (hydrophilic/base). It should be pointed out that the amino acids tyrosine and histidine, the carboxylic acids malonic and citric acids, and methylcellulose (in the form of cellulose) are actual constituents of NOM found in natural waters, and are therefore not “models” of NOM. They have been chosen to represent their respective NOM fractions, even though they obviously do not mimic all of the compounds comprised in each fraction. Also, results from phenols and acetylacetone should be interpreted with the care associated to extrapolation from a simple model compound to a complex, undefined mixture of much larger humic substances. The experiment for these compounds was designed to elucidate absorbance spectra behaviour of their moieties, rather than to perfectly mimic absorbance spectra of humic substances. Published research supports the idea that activated aromatic moieties are important THM and HAA precursors in natural waters,15,20,21,24,25 which justifies the choice of these compounds in the present study.Lastly, information on the reactivity of the compounds with chlorine in aqueous solution was gathered from a targeted literature review in order to plan the chlorination experiments. Table S1 and supplementary text 1, in the ESI,† summarize the literature review that supports the use of the model compounds chosen. The chemical structures of the compounds are shown in Fig. S.1–S.8 in the ESI.†
2.2 Experimental design
The UV-visible absorbance spectra between 200–600 nm were then measured for each concentration using cells of 1, 5 or 10 cm, depending on the resulting absorbance, with the objective of obtaining absorbance measurements lower than 2.00 absorbance units (a.u.) at every wavelength. The molar absorptivity spectrum could then be calculated for each ionic form of all model compounds examined by dividing the absorbance spectrum obtained by the molar concentration of the solution and the optical path length. Results from each of the concentrations were compared to identify any inconsistencies or contamination and then the valid results were averaged. The molar absorptivities obtained are presented in Fig. S1–S8 in the ESI.† The absorbances are presented for wavelengths between 200 and 400 nm only, because none of the compounds exhibited absorbances in the visible (>380 nm) region of the spectrum.
| Initial compound concentration | Chlorine dose | Average pH | 30 min Cl2 consumption | 24 h Cl2 consumption | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| μmol L−1 | mg L−1 | mg C L−1 | mg L−1 | mg mg−1 C | mol mol−1 | mg L−1 | mol mol−1 | mg L−1 | mol mol−1 | ||
| a All of the chlorine was consumed during this experiment; hence chlorine consumption ratios and DBP yields are not necessarily comparable. b Considering the uncertainty of the chlorine measurement (9.6 ± 0.2 mg L−1) and chlorine dose (10 ± 0.3 mg L−1), chlorine consumption can be considered nil. | |||||||||||
| Resorcinol | 9.8 | 1.08 | 0.71 | 10 | 14.1 | 14.3 | 6.93 | 3.15 | 4.51 | 3.95 | 5.66 |
| p-Cresol | 7.9 | 0.86 | 0.48 | 10 | 21 | 17.8 | 6.96 | 1.50 | 2.67 | 3.00 | 5.33 |
| Acetylacetone | 40.2 | 4.02 | 2.41 | 10 | 4.1 | 3.5 | n.a. | 7.78 | 2.75 | 7.08 | 2.49 |
| Citric acid | 28.2 | 5.43 | 2.03 | 10 | 4.9 | 5.0 | 6.81 | 0.00 | 0.00 | 0.28 | 0.14 |
| Malonic acid | 111.5 | 11.61 | 4.01 | 10 | 2.5 | 1.27 | n.a. | 0.00 | 0.00 | 3.35 | 0.42 |
| Tyrosine | 14.9 | 2.71 | 1.61 | 10 | 6.2 | 9.5 | 6.99 | 4.85 | 4.62 | 9.98a | 9.43a |
| Histidine 1 | 8.0 | 1.25 | 0.58 | 10 | 17.3 | 17.5 | 6.84 | 1.50 | 2.63 | 2.05 | 3.60 |
| Histidine 2 | 0.8 | 0.125 | 0.058 | 1.0 | 17.3 | 17.5 | n.a. | 0.15 | 2.63 | 0.30 | 5.26 |
| Methylcellulose | 44.4 | 7.21 | 3.2 | 10 | 3.1 | 3.2 | 6.94 | n.a. | n.a. | 0.40b | 0.13b |
| Pretreated water | n.a. | n.a. | 1.96 | 3.0 | 1.5 | n.a. | 6.94 | 1.99 | n.a. | 2.25 | n.a. |
| Blank | 0.0 | 0.0 | 0.0 | 3 | n.a. | n.a. | 6.94 | n.a. | n.a. | 0.00 | n.a. |
The bottles were incubated in the dark at 20 °C for 0.5, 1, 2, 6, and 24 hours (and 48 hours for the natural water). When an incubation time was reached, glass vials pre-dosed with buffer and a quenching agent for DBP quantification were filled for further analyses, and the free chlorine residual concentration was measured. The residual chlorine was then quenched with a 10 g L−1 sodium sulphite solution in the volume of water left in the 1 L bottles. Sodium sulphite was preferred to ammonium chloride because the product of its reaction with chlorine does not interfere with UV measurements. In contrast, ammonium chloride forms monochloramines that exhibit high UV absorbance,26–28 thus causing interference with UV measurements. The volume of sodium sulphite solution was adjusted to neutralize the free chlorine residual measured previously. A small amount in excess of the stoichiometric dose was used, varying between 10 and 20%, in order to minimize UV interference from the extra sulphite (see ESI,† Fig. S9). UV-visible absorbance spectra were measured after at least 20 minutes to allow the quenching agent to reduce the free chlorine completely.
2.3 Analytical procedures
Free chlorine residuals were measured using the DPD colorimetric method (Hach 8021) with a Hach DR-900 colorimeter. When needed, a dilution of 5 to 25 mL of the incubated solution was used in a total volume of 50 mL of nanofiltered demineralized water (dilution factor of 2 to 10) to adjust the residual chlorine to the measurable range (0.00–2.20 ± 0.02 mg Cl2 L−1) associated with this method. For all samples, the UV-visible absorbance spectra (0.000–2.000 ± 0.002 a.u.) between wavelengths of 200 and 600 nm were obtained using 1, 5 and 10 cm quartz cells and a double-beam Agilent Cary 100 UV-vis spectrophotometer. Blanks and baseline samples consisted of MilliQ water with the same phosphate buffer concentration as the model compound solutions.Samples for THM and HAA analyses were collected in 40 mL glass vials pre-dosed with 4.98 g of ammonium chloride used as a quenching agent. THMs were extracted using solid-phase microextraction (SPME) fibres, and analysed with gas chromatography with ion-trap mass spectrometry (GC-MS). HAAs were extracted using liquid–liquid extraction in MTBE, and the extracts were analysed using gas chromatography with electron capture detection (GC-ECD). The THMs analysed were the chlorinated and brominated THMs, namely chloroform (TCM), bromodichloromethane (BDCM), dibromochloromethane (DBCM) and bromoform (TBM), all of which are commonly known as THM4. The HAAs analysed were the 5 regulated HAAs (HAA5) in the province of Québec: mono-, di- and tri-chloroacetic acids (MCAA, DCAA, TCAA), mono- and di-bromoacetic acids (MBAA, DBAA). An unregulated HAA, bromochloroacetic acid (BCAA), was also analysed. These are collectively known as HAA6. The method quantification limits were 1.1 μg L−1, 0.6 μg L−1, 1.0 μg L−1, 0.8 μg L−1, and 1.0 μg L−1 for TCM, BDCM, DBCM, TBM, and all HAAs, respectively. Samples for HANs, CPK and HKs were collected in 60 mL glass vials. The glass vials were pre-dosed with 6.45 g of ammonium chloride and 1.00 g of phosphate buffer to adjust the sample pH to approximately 5 to preserve the HANs. These DBPs were extracted using liquid–liquid extraction and were analysed with GC-ECD. The method quantification limit was 0.01 μg L−1 for all non-regulated DBPs. Method recovery for all DBPs varied between 80 and 120%.
3 Results
3.1 Reactivity with chlorine check
The resulting chlorine consumptions after 30 minutes and 24 h are shown in Table 1. Comparing the chlorine consumptions (and DBP formation in the following section) with previous results from the literature allows for a rapid assessment of the validity of the experiments. Overall, the results indicate that the chlorination reactions occurred as expected based on the reviewed literature. For resorcinol and p-cresol, the rapid halogenation of the aromatic ring, and possibly chlorine reduction, consumed 4.51 and 2.67 mol Cl2 per mol of compound, respectively, which is consistent with results from de Laat et al.16 and Gallard and von Gunten.18 Acetylacetone rapidly consumed 2.75 mol Cl2 per mol of compound, forming the HKs 1,1,1-trichloropropanone (TCP) and 1,1-dichloropropanone (DCP) as stated by de Laat et al.16 Citric and malonic acids exhibited no rapid chlorine consumption. The citric acid had very low chlorine consumption at 24 h, which is consistent with previously published results.16,29,30 When accounting for uncertainties (see Table 1), the chlorine consumption of citric acid is negligible. However, the DBPs formed indicate that at least some of the chlorine was consumed. Both amino acids had an instantaneous chlorine demand higher than 2.0 mol mol−1, consistent with the rapid primary demand of the amine structure (2.0 mol mol−1) and with the extra reactivity conferred by their side-chain structures (secondary amine for histidine, and phenolic structure for tyrosine). Finally, when accounting for uncertainties, the chlorine consumption of methylcellulose is negligible, as supported by the very low DBP concentrations in this case.3.2 DBP formation
DBP concentrations at 24 h, unless otherwise noted, are shown in Table 2. The complete DBP data for all reaction times studied, in micrograms per liter and as molar yields (mol DBP per mol compounds), is available in ESI† Tables S2 and S3. Monobrominated species of the corresponding chlorinated DBPs were sometimes detected at very low concentrations. These may result from unreported very low bromide concentrations in the MilliQ water, in the stock chlorine solution, in the ammonium chloride used as a quenching agent, or from an impurity in the compounds or on the glassware. Nitrogenous DBPs resulting from compounds that do not contain nitrogen atoms are reported, but are considered to be the result of glassware contamination (nitrile gloves, human skin, sweat, etc.). The ammonium chloride used as the quenching agent for DBP analyses could also be the source of nitrogen in these cases.| THMs | HAAs | HANs | HNMs | HKs | |||||
|---|---|---|---|---|---|---|---|---|---|
| TCM | MCAA | DCAA | TCAA | DCAN | TCAN | CPK | DCP | TCP | |
| μg L−1 | μg L−1 | μg L−1 | μg L−1 | μg L−1 | μg L−1 | μg L−1 | μg L−1 | μg L−1 | |
| a Probable contamination of the solution or of the analysed sample. b DCP and TCP concentrations from acetylacetone chlorination are at their peak at the shortest reaction time, 0.5 h. c After a reaction time of 6 h. | |||||||||
| Resorcinol | 1010 | <1 | 3 | 60 | 1.8a | <0.01 | <0.01 | <0.01 | 0.10 |
| p-Cresol | 2 | <1 | 2 | 11 | 0.19a | <0.01 | <0.01 | 0.40 | 4.8 |
| Acetylacetone | 1340 | 57 | 127 | 1 | <0.01 | <0.01 | <0.01 | >200.0b | >200.0b |
| Citric acid | 46 | <1 | 2 | 46 | 0.08a | <0.01 | 0.03a | 0.17 | 0.58 |
| Malonic acid | 4 | 10c | 303 | 26c | 0.39a | 0.00 | 0.04a | 0.16 | 0.21 |
| Tyrosine | 48 | <1 | 26 | 54 | 35 | 0.50 | <0.01 | <0.01 | <0.01 |
| Histidine 1 | 2 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| Histidine 2 | n.a. | 0 | 1 | 1 | 1.9 | <0.01 | <0.01 | 0.02 | 0.04 |
| Methylcellulose | 2 | 3 | 2 | 1 | <0.01 | <0.01 | 0.03a | 0.13 | 0.10 |
| Pretreated water | 30 | <1 | 11 | 17 | 1.74 | 0.10 | 0.22 | 0.55 | 2.08 |
| Blank | 1 | <1 | 2 | <1 | 0.03 | 0.01 | 0.02 | 0.09 | 0.06 |
As expected, resorcinol formed mostly TCM (0.86 mol mol−1), while for similar chlorine consumption, p-cresol produced almost no TCM (0.0025 mol mol−1). The same is true for TCAA, although to a lesser extent (0.037 mol per mol of resorcinol, 0.0085 mol per mol of p-cresol). A somewhat notable amount of TCP formed from the chlorination of p-cresol, that was more than twice the amount of TCM from resorcinol on a mass basis. This means that cleavage of the aromatic ring can occur with p-cresol although it releases more TCP than TCM, while resorcinol and other meta-dihydroxibenzene release very little TCP.
Acetylacetone formed high concentrations of DCP and TCP upon chlorination. The TCP concentration was too high to quantify at all reaction times (>200 μg L−1). Considering that almost all acetylacetone rapidly produces TCP when an excess of chlorine (>3 mol mol−1) is available, a concentration of about 40 μmol per L of TCP (5336 μg L−1) formed and further released TCM for the rest of the reaction time. Because only 11.2 μmol per L of TCM (1340 μg L−1) were released after 24 h, there were about 29 μmol per L of TCP (3869 μg L−1) still in solution, which is a concentration that is still too high for us to quantify. DCP concentration was also >200 μg L−1 at a reaction time of 30 minutes, but was quantified at 139 μg L−1 after 1 h of reaction time and steadily decreased to 6.1 μg L−1 after 24 h. We suppose the DCP halogenated further to form TCP, or was the precursor for the increasing DCAA concentration. DCP decreased by 133 μg L−1 (1.05 μmol L−1) between the reaction times of 1 and 24 h, while DCAA increased by 121 μg L−1 (0.94 μmol L−1), which supports this hypothesis. A significant amount of MCAA was also formed from acetylacetone, which to our knowledge, is a novel result.
Citric acid formed the trichlorinated by-products TCM, TCAA and TCP. Together, these DBPs represent 0.671 μmol per L of trichlorinated species, 2.01 μmol per L of the chlorine atoms, or 0.28 mg per L of free chlorine. This was the exact chlorine consumption observed (Table 1), which means that these are the dominant DBPs formed from citric acid. DCAA and DCP were found in much lower concentrations. This is consistent with the results from de Laat et al.16 who found that about half of the chlorine consumed was incorporated into TCM. Our results suggest that the other half is mostly incorporated into TCAA. However, considering that the uncertainty of the chlorine consumption is more than 100% (0.28 ± 0.40 mg L−1), this conclusion should be confirmed by future experiments. Malonic acid formed mostly HAAs, with DCAA as the dominant species, and formed HKs in low concentrations. However, contrary to the reaction with citric acid, only about one-tenth of the total chlorine consumption was incorporated into the measured DBPs. Other unidentified DBPs, or the reduction of free chlorine into chloride during the reaction are possible explanations for this result.
The amino acid tyrosine produced significant amounts of TCM, DCAA, TCAA and DCAN, which is consistent with previously published results.16,31,32 The yield of DCAN for histidine (0.0213 mol mol−1) was almost equal to the yield of DCAN for tyrosine (0.0215 mol mol−1). However, tyrosine yielded much more TCM than histidine (0.027 mol mol−1vs. 0.0019 mol mol−1), likely due to its aromatic moiety. The HAA yields of tyrosine and histidine were closer to each other, between 0.0075 and 0.022 mol mol−1. As expected, no CPK was found from the chlorination of amino acids.
As indicated by the low chlorine consumption, methylcellulose did not react very much with chlorine. Contrary to citric acid, however, the low concentrations of TCM, HAAs and HKs measured at 24 h represented yields of less than 0.0006 mol mol−1 in all cases. These concentrations suggest that the actual chlorine consumption could very well be null when considering uncertainty (0.40 ± 0.40 mg L−1).
3.3 Changes in UV-visible absorbance spectra during chlorination
Fig. 1–8 show the absorbance spectra, ΔA spectra, and incremental ΔA spectra (ΔA spectra with the spectrum from the previous reaction time used as the reference spectrum instead of the spectrum at t = 0) for the 8 model compounds. Because sodium sulphite was added in excess of the free chlorine and because it exhibits some absorbance at wavelengths <250 nm, the absorbance spectra shown are corrected based on an approximate excess concentration of sodium sulphite and its absorbance spectrum (see Fig. S1†). However, not all interference could be removed, which must be kept in mind when interpreting the ΔA spectra. The absorbance spectra, ΔA spectra, peak-normalized ΔA spectra and incremental ΔA spectra of the pretreated natural water are shown in Fig. 9 so that they can be compared to the spectra of the model compounds. Due to an operator error using the spectrophotometer, the spectrum at the reaction time of 6 h is not available for the natural water. Table S4 in the ESI,† compares yields of DBPs on a ΔA basis (μg of DBP per L per cm of differential absorbance at specified wavelength) for model compounds and the pretreated water.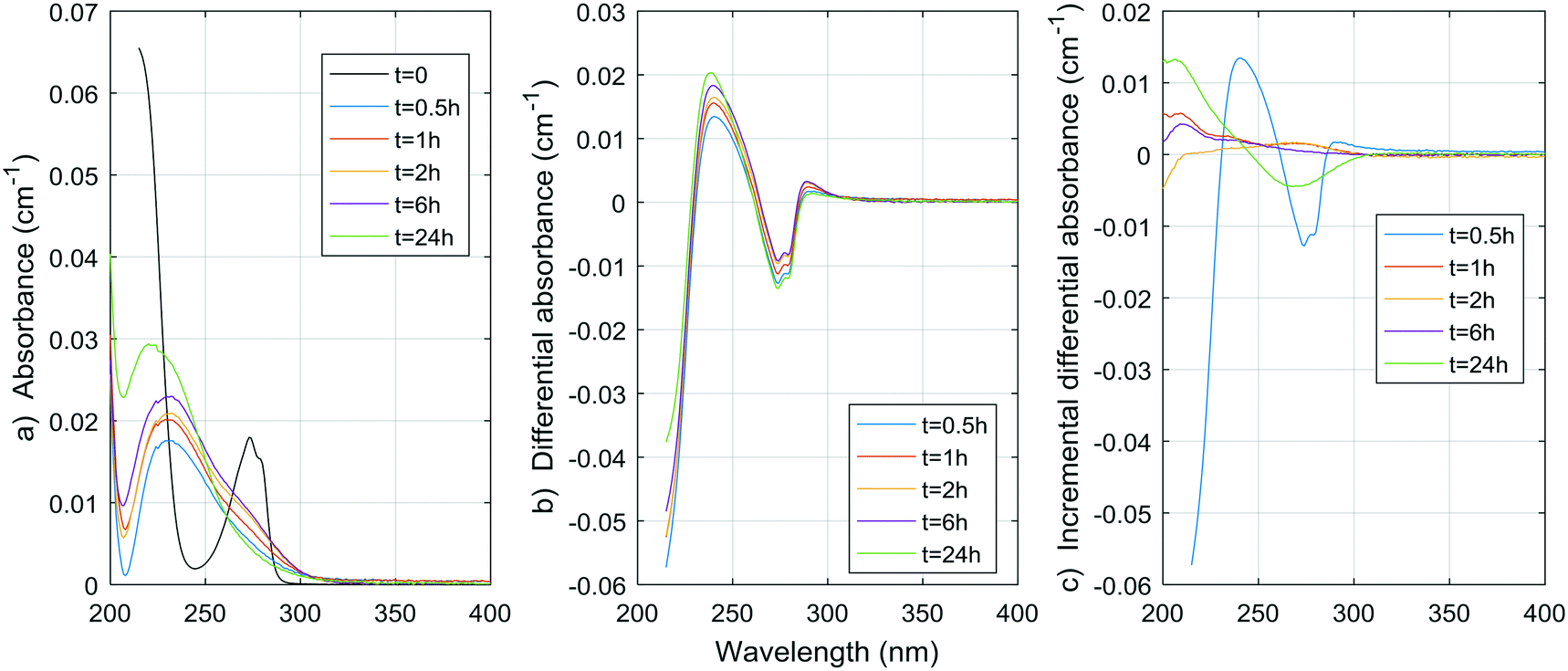 | ||
| Fig. 1 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a resorcinol aqueous solution. | ||
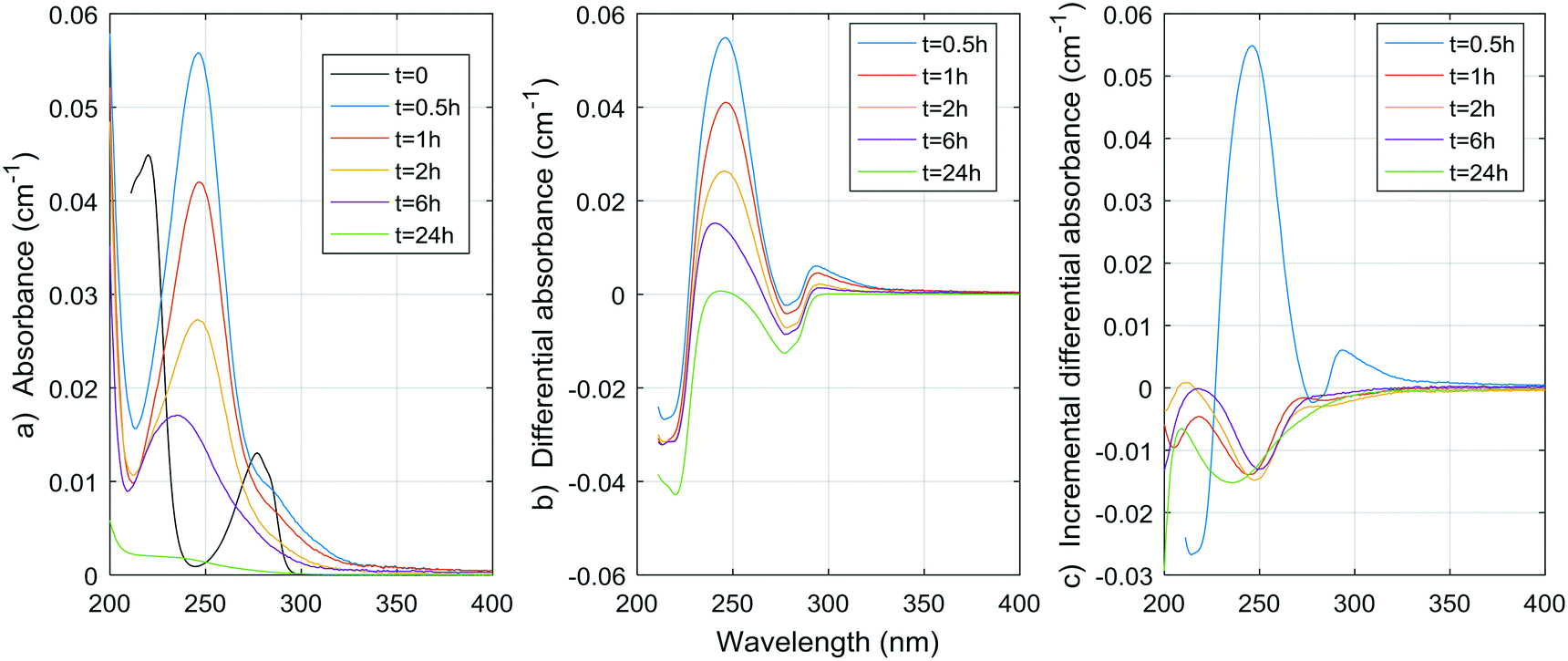 | ||
| Fig. 2 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a p-cresol aqueous solution. | ||
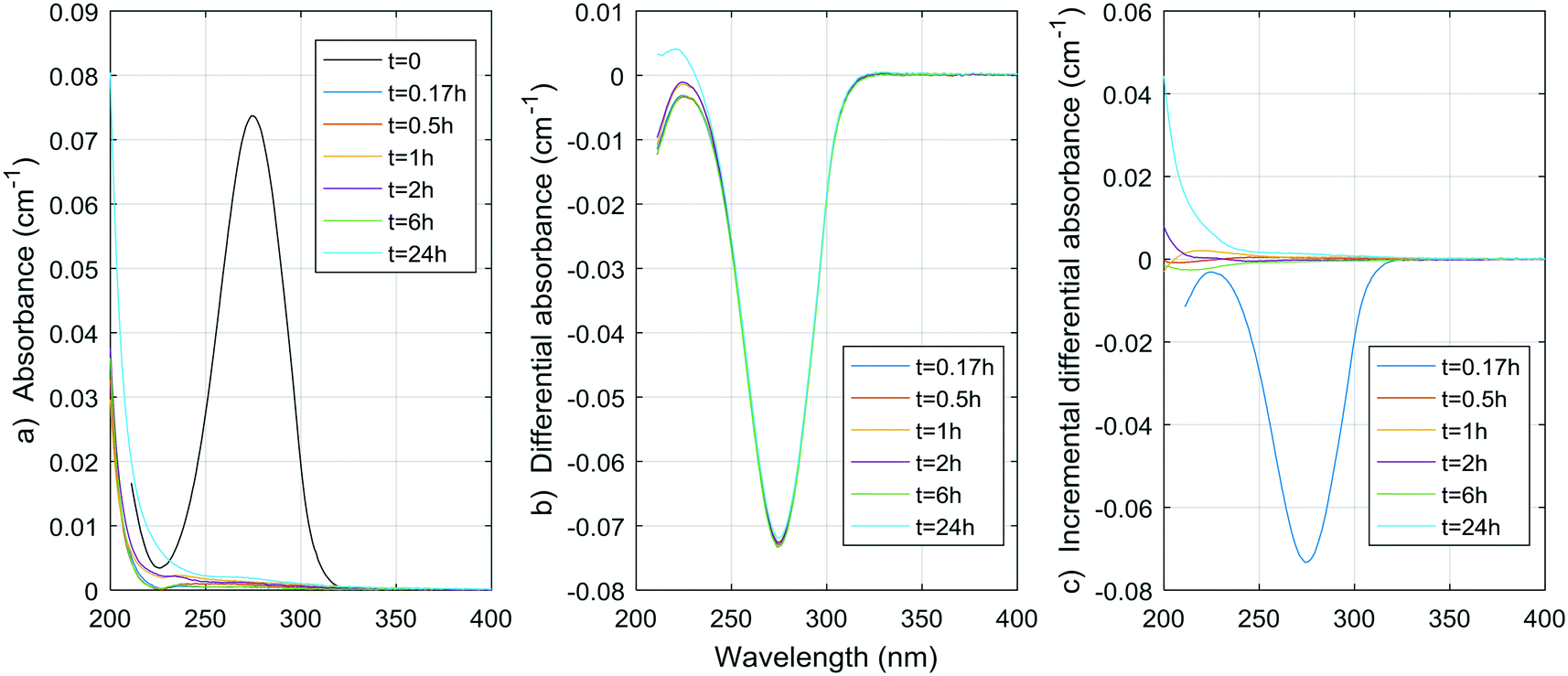 | ||
| Fig. 3 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of an acetylacetone aqueous solution. | ||
 | ||
| Fig. 4 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a citric acid aqueous solution. | ||
 | ||
| Fig. 5 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a malonic acid aqueous solution. | ||
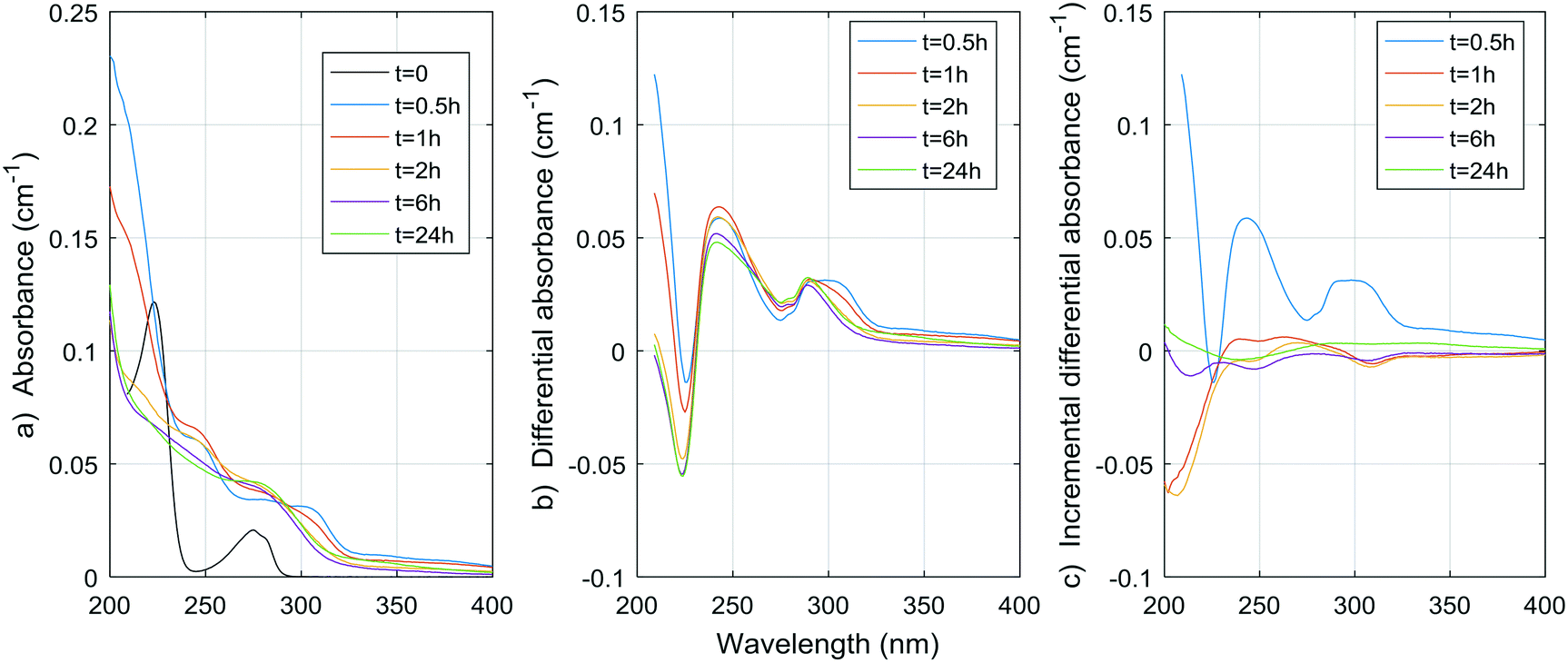 | ||
| Fig. 6 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a tyrosine aqueous solution. | ||
 | ||
| Fig. 7 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a histidine aqueous solution (Histidine 1). | ||
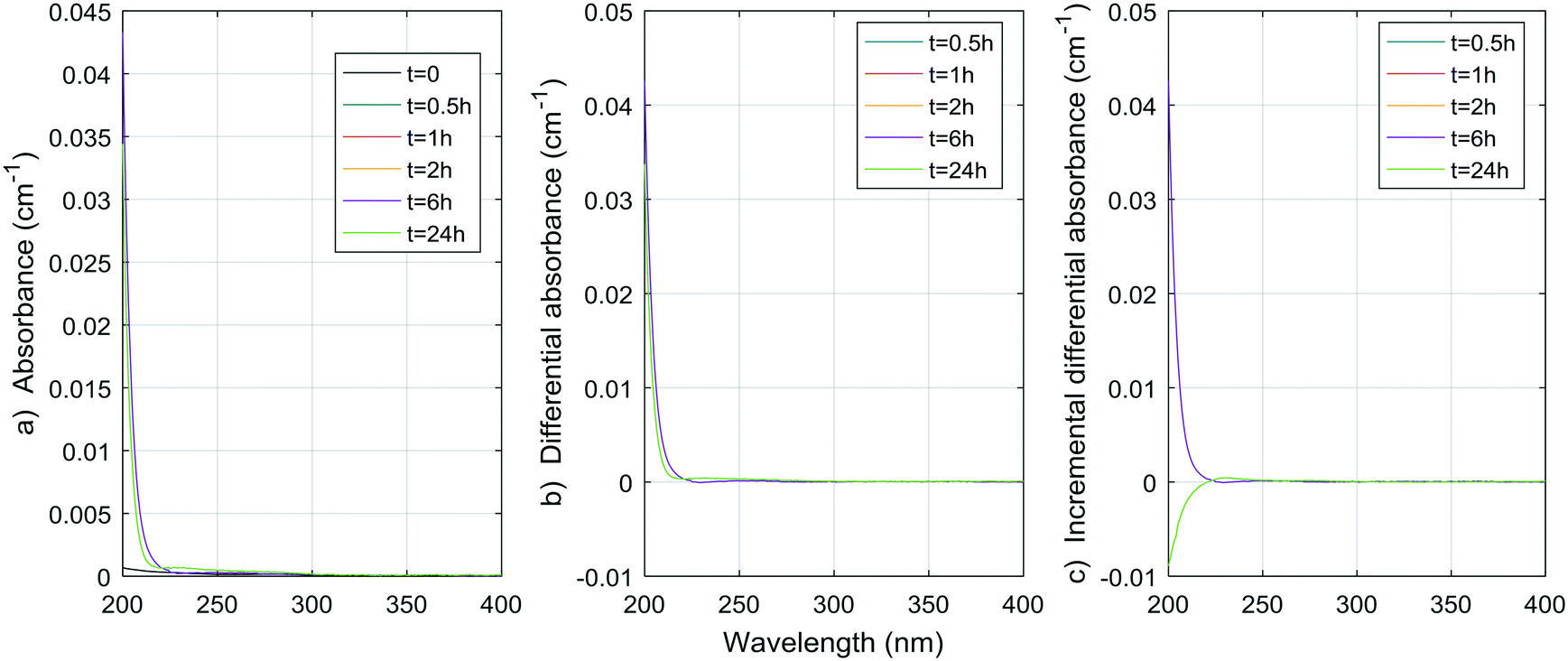 | ||
| Fig. 8 a) Absorbance, b) ΔA and c) incremental ΔA spectra at increasing times during the chlorination of a methylcellulose aqueous solution. | ||
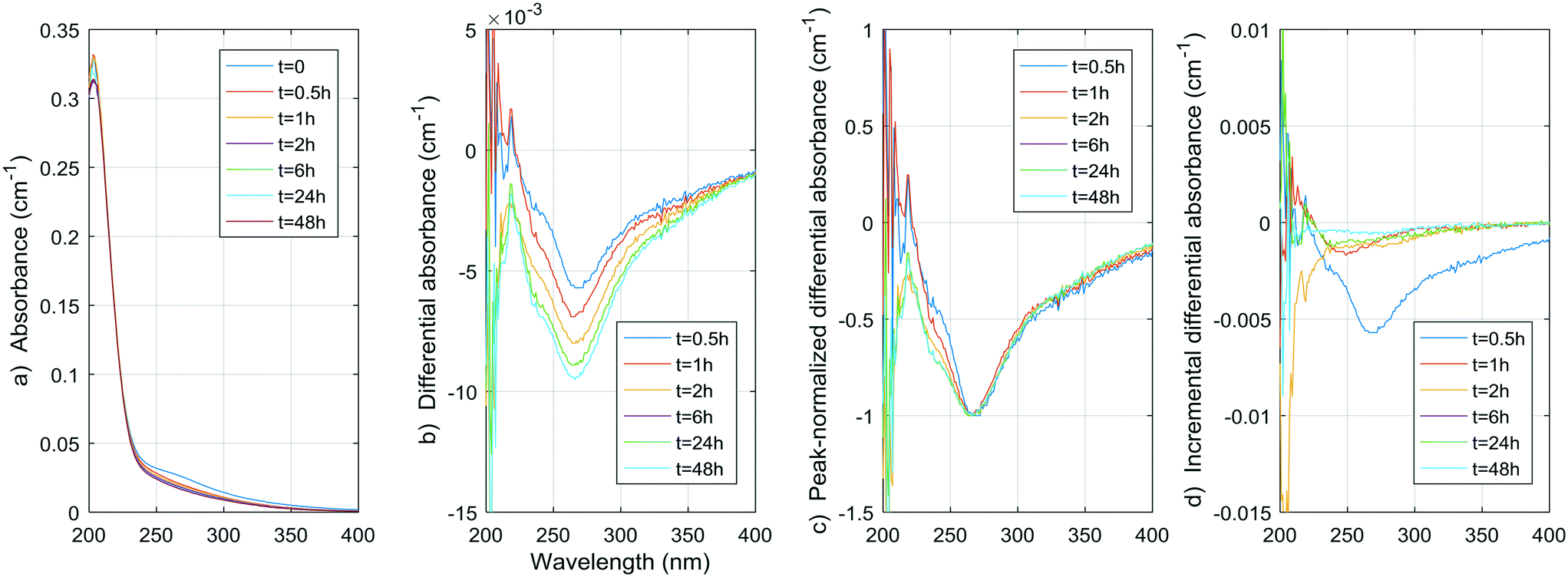 | ||
| Fig. 9 a) Absorbance, b) ΔA, c) peak normalized ΔA, and d) incremental ΔA spectra at increasing times during the chlorination of pretreated water (DOC = 1.96 mg C L−1, UV254 = 0.0305 cm−1, pH = 6.95, T = 4 °C, Cl2 dose = 3 mg L−1). | ||
In the pretreated water, the negative peak is at 268 nm, which is close to the negative peaks of the phenols. However, no absorbance increase is observed, probably because the lower concentration of phenolic moieties in NOM makes this phenomenon occur very rapidly, and because the increase is largely compensated by a decrease in absorbance due to the simultaneous cleavages of UV-absorbing moieties. Nonetheless, a continuous decrease in absorbance between 230 and 250 nm, which is larger than the decrease at 268 nm, is seen on the peak-normalized ΔA spectra and incremental ΔA spectra of the pretreated water (Fig. 9c). This continuous change between 230 and 250 nm is possibly related to the gradual aromatic ring cleavage, as observed in the case of p-cresol. Hence, monitoring the ΔA in this range of wavelengths could present a way to monitor the ongoing reaction of chlorine with the aromatic moieties in raw and coagulated waters.
Hence, the rapid decline of acetylacetone implies a negative ΔA peak at 274 nm, which is very close to the observed peak in the pretreated water (268 nm) and the peak generally reported in the literature (272 nm). If we assume similar absorbance spectra to that of acetylacetone for moieties that release DCP and TCP, using the concentrations obtained during natural pretreated water chlorination (0.55 and 2.1 μg L−1, or 0.004 and 0.013 μmol L−1, respectively) or even chlorination in a full-scale treatment plant (1.6 and 4.1 μg L−1, or 0.012 and 0.025 μmol L−1, respectively8), a negative peak of no more than 0.000030 ± 0.000006 cm−1 for the pretreated water and 0.00007 ± 0.00001 cm−1 for the full-scale conditions (molar absorptivity of 1790 ± 10 M−1 cm−1) would be produced. This contribution to the ΔA spectrum of coagulated waters is likely no more than 1% of the total ΔA (1% of 0.01 cm−1) at wavelengths around 270 nm. As presented in Table S4,† the DCP yield per ΔA at 274 nm is indeed smaller for the pretreated water than for the acetylacetone by more than an order of magnitude. Given what we already know about the formation of TCP from acetylacetone (about 1 mol of TCP per mol of acetylacetone, see ESI†) and about the UV absorptivity of acetylacetone (1790 ± 10 M−1 cm−1 at 274 nm, see ESI†), we can fairly assume the TCP yield per ΔA at 274 nm would also be smaller for the pretreated water by about two orders of magnitude.
As a result, acetylacetone itself is likely not responsible for the change in UV spectra observed in natural and pretreated waters. However, it could be hypothesized that similar diketone moieties with different components R1 and R2 (R1–CH2–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–CH2–(CO)–CH2–R2) would split at the carbon atom at the center of the molecule. This would reduce the UV absorbance of the molecule in a way similar to that observed here, without releasing DCP or TCP (only molecules with protons (–H) at R1, R2, or both would yield DCP and TCP). All of these molecules could eventually yield TCM, DCAA and MCAA, although this remains to be proven.
O)–CH2–(CO)–CH2–R2) would split at the carbon atom at the center of the molecule. This would reduce the UV absorbance of the molecule in a way similar to that observed here, without releasing DCP or TCP (only molecules with protons (–H) at R1, R2, or both would yield DCP and TCP). All of these molecules could eventually yield TCM, DCAA and MCAA, although this remains to be proven.
Considering that carboxylic acids as a group are basically unaltered by conventional physicochemical treatment (coagulation–flocculation–settling–filtration), as shown by Jurado Sánchez et al.,33 a total of a few tens of micrograms per liter of carboxylic acids (approximately between 0.1 and 0.5 μmol L−1) can be found in treated waters to be chlorinated.33 The maximum yields of DBP for citric and malonic acids (0.014 mol TCM per mol citric acid, 0.010 mol TCAA per mol citric acid, 0.021 mol DCAA per mol malonic acid) could mean contributions of up to 0.007 μmol L−1 (0.8 μg L−1) for TCM, 0.005 μmol L−1 (0.8 μg L−1) for TCAA, and 0.0105 μmol L−1 (1.3 μg L−1) for DCAA, if we apply these maximum yields to carboxylic acid concentrations found in natural waters. It is important to note that citric and malonic acids have not been identified by Jurado Sánchez et al.,33 although they have been reported before in natural waters.34 These results suggest that, unless much higher DBP yields are observed during the chlorination of other aliphatic carboxylic acids, or much higher aliphatic carboxylic acid concentrations are found in natural waters, aliphatic carboxylic acids as a group does not contribute significantly to the total DBP yields observed in natural and treated waters. The results also suggest that aliphatic carboxylic acids do not contribute significantly to the UV absorbance and ΔA spectra of natural and treated waters either.
The spectrum of tyrosine is similar to those of resorcinol and p-cresol because of the aromatic side-chain of the molecule. As for histidine, most changes in the UV spectrum of tyrosine occurred after 0.5 h of reaction time upon chlorination. The increase in absorbance observed for both histidine and tyrosine are probably due to absorbance by the chloramine species formed. Mono-, di-, and tri-chloramines all exhibit increasing molar absorptivity with decreasing wavelengths between 200 and 300 nm. The resulting ΔA of both compounds tested here seem to be the result of a decrease proportional to the initial spectrum of the compound, plus an increase proportional to the concentration and species of chloramines formed.
Total amino acid concentrations in natural waters can reach more than 100 μg L−1,35 with concentrations varying from 0.12 to 2.5 μg-N L−1 for free amino acids and from 29 to 171 μg-N L−1 for total (free and combined) amino acids.31 Although tyrosine and histidine are not the most abundant species found, if we assume other amino acids have similar DBP yields at 24 h, then for concentrations of 100 μg L−1, THMs, DCAA, TCAA and DCAN yields in natural waters could reach 1.8 μg L−1, 0.9 μg L−1, 2.0 μg L−1, and 1.3 μg L−1, respectively. The contribution to THMs and HAAs is small, but 1.3 μg L−1 represents a significant percentage of DCAN concentrations measured in chlorinated waters, specifically 75% in the pretreated water studied here. Assuming a concentration of 100 μg per L of tyrosine and histidine, peak UV absorbance of tyrosine at 225 nm would be around 0.004 cm−1, and peak UV absorbance for histidine at 212 nm would also be around 0.004 cm−1. These are significant contributions to the ΔA spectrum of pretreated waters, for which values range from a few thousandths to a few hundredths cm−1. However, particulate forms of amino acids, such as large proteins, cell debris or living organisms can be present in pretreated waters,31,35 and not all HAN production is necessarily related to UV-absorbing forms of amino acids.
4 Discussion and research needs
4.1 Findings
The initial decrease of absorbance around 270 nm in natural waters is possibly due to a combination of phenolic and diketones moieties. Phenolic moieties have long been identified as contributors to the ΔA spectra,36–38 However, to our knowledge, this is the first time diketone moieties have been suggested to be important contributors to ΔA, although they have been previously identified as DBP precursors.17,39 Following the initial rapid decrease in absorbance, absorbance at wavelengths between 230 and 250 nm seem better suited to monitor the chlorine–phenol reaction than the often used wavelength of 272 nm. It is in this region that the ΔA spectra exhibit the highest changes after their initial decrease. This behaviour points towards the use of multi-wavelength DBP–ΔA relationships in order to take into account the contribution of the different precursors and intermediates, as suggested previously by Korshin36 and Yan et al.3 However, these previous studies suggested the use of ΔA at 272 nm and 350 nm. Using ΔA at 230–250 nm as well as at other wavelengths that are yet to be determined in multi-wavelength relationships should be attempted in future work.Based on the low DCP and TCP concentrations measured in the pretreated water, the DCP and TCP-releasing diketones probably account for only a fraction of all diketones. Other compounds in the present study have yielded DCP and TCP (p-cresol, citric and malonic acids), which further reduce the diketone contribution to DPC and TCP yields in real waters.
The contribution of carboxylic acids to UV-visible absorbance and to THM and HAA yields of pretreated natural waters seem to be small, considering that their concentrations are only a few μg L−1 in natural and pretreated waters. If significant concentration variations (about two orders of magnitude) were observed in natural waters, they could explain some of the variation observed in DBP–ΔA relationships. However, this seems unlikely based on our results. Nonetheless, Beaulieu40 showed that the hydrophilic fraction of the residual organic matter found in distribution systems has high chlorine reactivity (0.56 mg Cl2 per mg C) and high HAA and THM formation potential (390 nmol THM per mg Cl2 consumed and 380 nmole HAA per mg Cl2 consumed) at pH 7 for a contact time of 24 h. If we assume that, like carboxylic acids, the hydrophilic fraction has low UV absorbance,40 variations of hydrophilic fractions between waters could explain the varying DBP–ΔA relationships found in the literature. Future research should therefore look into the UV absorbance spectra of the various NOM fractions and their contribution to the total ΔA spectrum of natural water.
The side chains of amino acids seem to be the most important contributor to their UV absorbance spectra. The UV absorbance of amino acids declines quickly upon chlorination, although an increase proportional to the formation of chloramines was observed. In drinking water treatment, low chloramine residuals are expected after breakpoint chlorination, so UV interference of chloramines should not be an issue (except when monochloramine is used as a disinfectant). Globally, it is therefore difficult to link the formation of HANs, the most important DBP group formed by amino acids, to any one particular feature of the pretreated water spectra. Nonetheless, previous studies9,10 suggest that DCAN-ΔA relationships can be obtained. Considering that HANs are toxicity drivers of the DBP mixture found in chlorinated waters,41–44 it is suggested that research efforts be directed toward understanding the underlying cause of HAN-ΔA relationships. The ability to estimate the concentrations of various DBPs simultaneously with the monitoring of ΔA, especially those that are considered toxicity drivers, would be a significant advantage of the technique over other methods of frequent, routine monitoring of DBP concentrations, such as online THM analysers, that focus on a single category of DBPs.
4.2 Limits of applicability
One limitation of this study is that the results and findings described in the previous sections do not constitute recommendations directly applicable to treatment plant operators who wish to routinely monitor ΔA, as these findings do not provide empirical relationships to predict DBP concentrations. On the other hand, and in accordance with the objective of the study, the results are useful for researchers, guiding them in their search for empirical DBP–ΔA relationships and providing interpretations for their empirical observations.Another limitation is that the compounds studied obviously do not mimic every possible compound or functional group found in natural waters. Nonetheless, they represent an important proportion of the functional groups that are considered DBP precursors in studies involving NOM surrogates.21 Moreover, we think the results discussed in this paper are representative of the moieties studied, and can be extrapolated to similar moieties found in natural waters. For example, because we selected the phenols as models of humic substances does not mean the results should be extrapolated to all and only to humic substances. Results from the chlorination of phenols are most likely representative of the chlorination of similar aromatic structures, even for small aromatic compounds that are usually not considered part of humic substances (e.g. aromatic carboxylic acids, aromatic amino acids).
Finally, none of the absorbances or ΔA spectra of the model compounds individually could be related to the entire absorbance and ΔA spectra of natural waters. In some cases, high DBP yields were obtained from compounds having low absorbance and minimal ΔA (e.g. DCAA from malonic acid), while in other cases there were high DBP yields and high absorbance and ΔA (e.g. TCM from resorcinol). Still other instances showed low DBP yields and high ΔA (TCM from p-cresol). However, some of the interesting ΔA features identified in the spectra of both model compounds and natural water are probably related to DBP concentrations. Moreover, the DBP formation and the absorbance changes are two phenomena initiated and controlled by the same driving force, namely the chlorine concentration. Therefore, in addition to the similar features between model compound spectra and natural water spectra identified here, DBP concentration and ΔA are at least empirically and indirectly related, hence supporting the possibility of DBP–ΔA relationships in natural waters.
5 Conclusions
The objective of this study was to provide a theoretical explanation for the empirical observations of ΔA in natural waters and their relationship to DBP formation, as discussed in previous literature.3,5–7,9,10,38 To our knowledge, this is the first time UV-visible absorbance spectra of some of these compounds (p-cresol, acetylacetone, tyrosine, histidine, citric and malonic acids) have been monitored during a chlorination reaction. The main findings from our experiments with model compounds are:• Model compounds representing the same NOM fractions (e.g. resorcinol and p-cresol) can have different DBP–ΔA relationships.
• The initial decrease in UV absorbance of natural and pretreated waters, which peaks around 270 nm, is likely due to the chlorination of phenolic and diketones moieties, although not necessarily associated with the release of THMs and HAAs.
• Subsequent gradual changes in absorbance of the pretreated water resembles the gradual decline observed during the chlorination of p-cresol, with a peak around 250 nm.
• Based on this result, monitoring ΔA at wavelengths between 230–250 nm in the pretreated water as a means to follow the DBP formation reaction seems more relevant than the often cited 272 nm.
• The contribution of carboxylic acids and amino acids to the total THM and HAA yields of pretreated water is likely small. Nonetheless, amino acids seem to contribute to a significant proportion of the total HAN measured.
The hypothesis behind the interpretation of the results is that the model compounds are representative of the various NOM fractions mentioned in this study. Future research should seek to test this hypothesis by reproducing this experiment using NOM fractions (humic acids, fulvic acids, hydrophilic fractions, etc.) instead of model compounds, and to identify what type of DBP-precursor/moieties are present in each fraction based on ΔA and DBP yields. Finally, the issue of generalizing DBP–ΔA relationships using another wavelength around 250 nm or using multiple wavelengths should be addressed in studies involving natural and treated waters.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors want to thank all the staff of Université Laval Industrial Research Chair in Monitoring and Management of Drinking Water Quality from Source to Consumer. Thanks to Sabrina Simard, head of the lab, and to undergraduate students, Pamela Ouellet, Jessica Beaupré, Vincent Boutet, Antoine Grondin and Émilie Leclerc for their valuable help extracting and analysing DBPs. A special thank you to undergraduate student Gabriel Caron, whose lab work was essential for the chlorination experiments and UV measurements. We also wish to recognize the help of Michel Bisping, lab technician at the Department of Civil and Water Engineering, with the manipulations of the compounds. The authors recognize the financial support of the National Science and Engineering Research Council of Canada (NSERC) through the Alexander Graham Bell Canada Graduate Scholarship-Doctoral program. The role of NSERC was limited to the funding of the study.References
- G. V. Korshin, M. M. Benjamin, O. Hemingway and W. W. Wu, Development of Differential UV Spectroscopy for DBP Monitoring, AWWA Research Foundation, Denver, CO, 2002 Search PubMed.
- P. Roccaro, F. G. A. Vagliasindi and G. V. Korshin, Comparison of the Performance of Spectroscopic Indices Developed to Quantify the Halogenation of Natural Organic Matter at Varying Chlorine Concentrations, Reaction Times and Temperatures, in Disinfection By-Products in Drinking Water, American Chemical Society, 2008, vol. 995, pp. 198–212 Search PubMed.
- M. Yan, G. V. Korshin and H.-S. Chang, Examination of disinfection by-product (DBP) formation in source waters: A study using log-transformed differential spectra, Water Res., 2014, 50, 179–188 CrossRef CAS PubMed.
- G. V. Korshin and H.-S. Chang, Spectroscopic Studies of the Roles of Distinct Chromophores in NOM Chlorination and DBP Formation, in Disinfection By-Products in Drinking Water, American Chemical Society, 2008, vol. 995, pp. 158–171 Search PubMed.
- C.-W. Li, G. V. Korshin and M. M. Benjamin, Monitoring DBP formation with differential UV spectroscopy, J. - Am. Water Works Assoc., 1998, 90, 88–100 CrossRef CAS.
- P. Roccaro and F. G. A. Vagliasindi, Differential vs. absolute UV absorbance approaches in studying NOM reactivity in DBPs formation: Comparison and applicability, Water Res., 2009, 43, 744–750 CrossRef CAS.
- V. Uyak and K. D. Demirbas, Formation of Disinfection Byproducts (DBPs) in Surface Water Sources: Differential Ultraviolet (UV) Absorbance Approach, Environ. Forensics, 2014, 15, 52–65 CrossRef.
- N. Beauchamp, O. Laflamme, S. Simard, C. Dorea, G. Pelletier, C. Bouchard and M. Rodriguez, Relationships between DBP concentrations and differential UV absorbance in full-scale conditions, Water Res., 2018, 131, 110–121 CrossRef CAS PubMed.
- G. V. Korshin, W. W. Wu, M. M. Benjamin and O. Hemingway, Correlations between differential absorbance and the formation of individual DBPs, Water Res., 2002, 36, 3273–3282 CrossRef CAS.
- P. Roccaro, H.-S. Chang, F. G. A. Vagliasindi and G. V. Korshin, Differential absorbance study of effects of temperature on chlorine consumption and formation of disinfection by-products in chlorinated water, Water Res., 2008, 42, 1879–1888 CrossRef CAS.
- N. Beauchamp, C. Dorea, C. Bouchard and M. Rodriguez, Use of differential absorbance to estimate concentrations of chlorinated disinfection by-product in drinking water: Critical review and research needs, Crit. Rev. Environ. Sci. Technol., 2018, 48, 210–241 CrossRef CAS.
- K. Özdemir, İ. Toröz and V. Uyak, Assessment of Trihalomethane Formation in Chlorinated Raw Waters with Differential UV Spectroscopy Approach, Sci. World J., 2013, 2013, 890854 Search PubMed.
- M. Yan, D. Dryer and G. V. Korshin, Spectroscopic characterization of changes of DOM deprotonation–protonation properties in water treatment processes, Chemosphere, 2016, 148, 426–435 CrossRef CAS PubMed.
- M. Yan, G. V. Korshin, F. Claret, J.-P. Croué, M. Fabbricino, H. Gallard, T. Schäfer and M. F. Benedetti, Effects of charging on the chromophores of dissolved organic matter from the Rio Negro basin, Water Res., 2014, 59, 154–164 CrossRef CAS PubMed.
- S. J. Traina, J. Novak and N. E. Smeck, An Ultraviolet Absorbance Method of Estimating the Percent Aromatic Carbon Content of Humic Acids, J. Environ. Qual., 1990, 19, 151–153 CrossRef CAS.
- J. de Laat, N. Merlet and M. Dore, Chloration de composés organiques: demande en chlore et réactivite vis-a-vis de la formation des trihalométhanes. Incidence de l'azote ammoniacal, Water Res., 1982, 16, 1437–1450 CrossRef CAS.
- M. Deborde and U. von Gunten, Reactions of chlorine with inorganic and organic compounds during water treatment—Kinetics and mechanisms: A critical review, Water Res., 2008, 42, 13–51 CrossRef CAS.
- H. Gallard and U. von Gunten, Chlorination of Phenols: Kinetics and Formation of Chloroform, Environ. Sci. Technol., 2002, 36, 884–890 CrossRef CAS.
- D. L. Norwood, J. D. Johnson, R. F. Christman, J. R. Hass and M. J. Bobenrieth, Reactions of chlorine with selected aromatic models of aquatic humic material, Environ. Sci. Technol., 1980, 14, 187–190 CrossRef CAS.
- D. A. Reckhow, P. C. Singer and R. L. Malcolm, Chlorination of humic materials: byproduct formation and chemical interpretations, Environ. Sci. Technol., 1990, 24, 1655–1664 CrossRef CAS.
- T. Bond, E. H. Goslan, S. A. Parsons and B. Jefferson, A critical review of trihalomethane and haloacetic acid formation from natural organic matter surrogates, Environ. Technol. Rev., 2012, 1, 93–113 CrossRef CAS.
- J. Leenheer and E. Huffman, Classification of organic solutes in water by using macroreticular resins, J. Res. U.S. Geol. Surv., 1976, 4, 737–751 CAS.
- J. A. Leenheer and J.-P. Croué, Peer Reviewed: Characterizing Aquatic Dissolved Organic Matter, Environ. Sci. Technol., 2003, 37, 18A–26A CrossRef CAS.
- S. Platikanov, R. Tauler, P. M. S. M. Rodrigues, M. C. G. Antunes, D. Pereira and J. C. G. Esteves da Silva, Factorial analysis of the trihalomethane formation in the reaction of colloidal, hydrophobic, and transphilic fractions of DOM with free chlorine, Environ. Sci. Pollut. Res., 2010, 17, 1389–1400 CrossRef CAS PubMed.
- X. Yang, C. Shang, W. Lee, P. Westerhoff and C. Fan, Correlations between organic matter properties and DBP formation during chloramination, Water Res., 2008, 42, 2329–2339 CrossRef CAS PubMed.
- D. Chaza, E. Mazen, S. Georg and D. Henri, Chlorine-Atom Transfer Reactions between Chloramine (=Chloramide) and Piperidine: Kinetic Reactivity and Characterization in a Raschig Medium, Helv. Chim. Acta, 2009, 92, 98–111 CrossRef.
- M. Ferriol and J. Gazet, Ultraviolet absorption spectra of chloramines and chlorine in carbon tetrachloride, Anal. Chim. Acta, 1988, 209, 321–325 CrossRef CAS.
- J. Kleinberg, M. Tecotzky and L. F. Audrieth, Absorption Spectrum of Aqueous Monochloramine Solutions, Anal. Chem., 1954, 26, 1388–1389 CrossRef CAS.
- R. A. Larson and A. L. Rockwell, Chloroform and chlorophenol production by decarboxylation of natural acids during aqueous chlorination, Environ. Sci. Technol., 1979, 13, 325–329 CrossRef CAS.
- R. A. Larson and A. L. Rockwell, Citric acid: Potential precursor of chloroform in water chlorination, Naturwissenschaften, 1978, 65, 490 CrossRef CAS.
- T. Bond, M. R. Templeton and N. Graham, Precursors of nitrogenous disinfection by-products in drinking water––A critical review and analysis, J. Hazard. Mater., 2012, 235–236, 1–16 CrossRef CAS.
- L. Hureiki, J. P. Croué and B. Legube, Chlorination studies of free and combined amino acids, Water Res., 1994, 28, 2521–2531 CrossRef CAS.
- B. Jurado-Sánchez, E. Ballesteros and M. Gallego, Occurrence of carboxylic acids in different steps of two drinking-water treatment plants using different disinfectants, Water Res., 2014, 51, 186–197 CrossRef.
- E. M. Thurman, Organic geochemistry of natural waters, M. Nijhoff, Dordrecht, 1985 Search PubMed.
- R. Chinn and S. E. Barrett, Occurrence of Amino Acids in Two Drinking Water Sources, in Natural Organic Matter and Disinfection By-Products, American Chemical Society, 2000, vol. 761, pp. 96–108 Search PubMed.
- G. V. Korshin, M. M. Benjamin, H.-S. Chang and H. Gallard, Examination of NOM Chlorination Reactions by Conventional and Stop-Flow Differential Absorbance Spectroscopy, Environ. Sci. Technol., 2007, 41, 2776–2781 CrossRef CAS.
- G. V. Korshin, M. M. Benjamin and C.-W. Li, Use of Differential Spectroscopy to Evaluate the Structure and Reactivity of Humics, Water Sci. Technol., 1999, 40, 9 CrossRef CAS.
- G. V. Korshin, C.-W. Li and M. M. Benjamin, Use of UV Spectroscopy To Study Chlorination of Natural Organic Matter, in Water Disinfection and Natural Organic Matter, American Chemical Society, 1996, vol. 649, pp. 182–195 Search PubMed.
- J. C. Morris, The chemistry of aqueous chlorine in relation to water chlorination, in Water chlorination: environmental impact and health effects, Ann Arbor Science Publishers, Ann Arbor, Michigan, 1978, vol. 1 Search PubMed.
- C. Beaulieu, Caractérisation spatio-temporelle de la réactivité des précurseurs de sous-produits de la désinfection en réseau de distribution d'eau potable, PhD Thesis, Université Laval, 2010 Search PubMed.
- E. D. Wagner and M. J. Plewa, CHO cell cytotoxicity and genotoxicity analyses of disinfection by-products: An updated review, J. Environ. Sci., 2017, 58, 64–76 CrossRef.
- M. J. Plewa, E. D. Wagner and S. D. Richardson, TIC-Tox: A preliminary discussion on identifying the forcing agents of DBP-mediated toxicity of disinfected water, J. Environ. Sci., 2017, 58, 208–216 CrossRef PubMed.
- Y. Kargalioglu, B. J. McMillan, R. A. Minear and M. J. Plewa, A New Assessment of the Cytotoxicity and Genotoxicity of Drinking Water Disinfection By-Products, in Natural Organic Matter and Disinfection By-Products, American Chemical Society, 2000, vol. 761, pp. 16–27 Search PubMed.
- S. D. Richardson, M. J. Plewa, E. D. Wagner, R. Schoeny and D. M. DeMarini, Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research, Mutat. Res., Rev. Mutat. Res., 2007, 636, 178–242 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ew00662h |
| This journal is © The Royal Society of Chemistry 2019 |