 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultivariate analysis applied to complex biological medicines†
Timothy R.
Rudd
 *ab,
Lucio
Mauri
*c,
Maria
Marinozzi
c,
Eduardo
Stancanelli
c,
Edwin A.
Yates
b,
Annamaria
Naggi
c and
Marco
Guerrini
c
*ab,
Lucio
Mauri
*c,
Maria
Marinozzi
c,
Eduardo
Stancanelli
c,
Edwin A.
Yates
b,
Annamaria
Naggi
c and
Marco
Guerrini
c
aNational Institute for Biological Standards and Control (NIBSC), Blanche Lane, South Mimms, Potters Bar, Hertfordshire EN6 3QG, UK. E-mail: tim.rudd@nibsc.org; Tel: +44 (0)1707641120
bDepartment of Biochemistry, Biosciences Building, University of Liverpool, Crown Street, Liverpool, L69 7ZB, UK
cIstituto di Ricerche Chimiche e Biochimiche ‘G. Ronzoni’, Via G. Colombo 81, 20133 Milano, Italy
First published on 4th April 2019
Abstract
A biological medicine (or biologicals) is a term for a medicinal compound that is derived from a living organism. By their very nature, they are complex and often heterogeneous in structure, composition and biological activity. Some of the oldest pharmaceutical products are biologicals, for example insulin and heparin. The former is now produced recombinantly, with technology being at a point where this can be considered a defined chemical entity. This is not the case for the latter, however. Heparin is a heterogeneous polysaccharide that is extracted from the intestinal mucosa of animals, primarily porcine, although there is also a significant market for non-porcine heparin due to social and economical reasons. In 2008 heparin was adulterated with another sulfated polysaccharide. Unfortunately this event was disastrous and resulted in a global public health emergency. This was the impetuous to apply modern analytical techniques, principally NMR spectroscopy, and multivariate analyses to monitor heparin. Initially, traditional unsupervised multivariate analysis (principal component analysis (PCA)) was applied to the problem. This was able to distinguish animal heparins from each other, and could also separate adulterated heparin from what was considered bona fide heparin. Taught multivariate analysis functions by training the analysis to look for specific patterns within the dataset of interest. If this approach was to be applied to heparin, or any other biological medicine, it would have to be taught to find every possible alien signal. The opposite approach would be more efficient; defining the complex heterogeneous material by a library of bona fide spectra and then filtering test samples with these spectra to reveal alien features that are not consistent with the reference library. This is the basis of an approach termed spectral filtering, which has been applied to 1D and 2D-NMR spectra, and has been very successful in extracting the spectral features of adulterants in heparin, as well as being able to differentiate supposedly biosimilar products. In essence, the filtered spectrum is determined by subtracting the covariance matrix of the library spectra from the covariance matrix of the library spectra plus the test spectrum. These approaches are universal and could be applied to biological medicines such as vaccine polysaccharides and monoclonal antibodies.
Introduction
Biological medicines (or biologicals) are drugs that are derived from natural sources. They are, by definition, heterogeneous, which can be seen in both their composition and activity. Examples of biological medicines are vaccines, monoclonal antibodies and the family of heparin-based anticoagulants, the latter being amongst the most intrinsically diverse pharmaceutical products on the market.Many physico-chemical techniques are used to characterise biological medicines. These include HPLC techniques, mass spectrometry and nuclear magnetic resonance (NMR) spectroscopy. Each of these techniques have their own strengths, with NMR spectroscopy being, in the authors opinion, one of the most adaptable. The technique can be used to fingerprint, determine the structure (chemical and physical) and quantify the amount of material present. An event in 2008, the contamination of heparin with oversulfated chondroitin sulfate,1 further exemplified the usefulness of NMR spectroscopy, with the technique being used to determine the contaminant.2 Since then, the interest in using NMR spectroscopy to characterise biological medicines has increased even more. The technique is readily applied to the heparin active pharmaceutical product and there is currently great interest in applying NMR spectroscopy to peptide/protein based products, for example, to the qNMR analysis of small peptides,3 protamine sulfate4 (reversal of heparin administration), copaxone5 (glatiramer acetate, an immunomodulator used to treat multiple sclerosis) and monoclonal antibodies (immunotherapies for cancer and autoimmune diseases). These complex molecules are primarily fingerprinted using 1D and 2D-NMR spectroscopy.
The limitation of the manual spectral analysis of these biological medicines is the ability of the analyst to differentiate samples of interest when comparing complex 1D or 2D spectra, and the problem is further compounded when dealing with large datasets, where many samples are compared.
The solution to this is to use multivariate analysis, where complex datasets can be decomposed into a number of key trends that can be used to reconstruct the dataset, as well as where predictions about the sample(s) being analysed are made. These analyses fall into two camps, the first being untaught analysis, where the dataset is blindly analysed and the method differentiates the observations by correlations calculated between the variables. Examples of this type of analysis are principal component analysis or factor analysis. This type of analysis is very informative if the aim is to find the features within the dataset that discriminate the observations. The second type of analysis is taught or supervised analyses, and these are used where various parameters are known about an already existing dataset. This pre-existing dataset can then be used as a reference to compare a test sample against, allowing the parameter of interest to be determined. Analyses that fall into this category include partial least squares-discriminant analysis and orthogonal partial least squares analysis.
As previously mentioned, heparin is a biological medicine,6,7 principally being derived from the intestines of pigs, but it is also extracted from cows. Heparin has been long established as an anticoagulant drug, which prevents or slows blood clotting, and it is very important for patients undergoing surgery, dialysis and during recovery from surgical procedures. It functions by interacting with a number of proteins of the blood clotting cascade, notably, but not limited to, antithrombin and thrombin.8 It is composed of a linear, highly sulfated polysaccharide chain of varying lengths, from 2 to 40 kDa. The carbohydrate is formed of repeating disaccharide units of 1,4 linked α-L-iduronic or β-D-glucuronic acid, and α-D-glucosamine. The predominant substitution pattern comprises 2-O-sulfation of the iduronate residues and N- and 6-O-sulfation of the glucosamine residues. The α-D-glucosamine residue can also be O-sulfated at position 3, and this is important for the molecule’s antithrombotic properties.7 Currently there is no alternative for these applications. It has also been proposed that sheep or camelids could be useful sources of heparin, as well as possibly non-mammalian animals.9 Its diversity arises from manifold sources; the biosynthesis of heparin is complex involving many enzymes, the extraction method is initially mechanical in nature resulting in material of varying quality, and furthermore, once the mucosa has been extracted many steps of chemical purification, resin capture, precipitation and fractionation take place to produce a pure product, which is then bleached. This process produces a colourless and odourless material that is free from endotoxins, bacteria, mould, viruses and prions.9,10 The bleaching step can also chemically modify the underlying polysaccharide structure. This diversity means that heparin is a challenging material to analyse, and it was this property that provided the opportunity for heparin to be adulterated with oversulfated chondroitin sulfate.
NMR spectroscopy was used to identify the adulterant used to contaminate heparin,2 and it was quickly realised by the research groups working on the problem that manually analysing the data would be inefficient. Principal component analysis (PCA) has been readily used to analyse heparin and model adulterated heparin samples.11,12 Furthermore, taught analyses have been used to predict the amounts of known heparin contaminants (chondroitin sulfate and dermatan sulfate) present in test samples.13–16 The techniques can also be applied to the more complex crude heparin, that is composed of heparin as well as other glycosaminoglycans.17 Novel techniques were also applied, such as spectral filtering, to search for unknown contaminants in heparin.18–21 The aims of all of these analyses have all been directed to the quality control of heparin, with the goal of detecting heparin samples that contain contaminants, such as chondroitin sulfate/dermatan sulfate, or adulterants, such as oversulfated chondroitin sulfate.
This is not the case for the analysis described within this manuscript. Here, a combination of 2D-NMR spectroscopy and PCA will be used to differentiate heparin from different animal sources. Even though the biosynthesis of heparin in the different animal sources uses the same biosynthetic pathway, the materials have different chemical structures. Normally, the structural differences would be elucidated by enzymic digestion followed by either HPLC or HPLC-MS. The benefit of using a combination of NMR spectroscopy and multivariate analysis is that the sample pre-treatment is minimal; 2 steps of D2O exchange and lyophilisation and then final resuspension of the material in D2O or a deuterated buffer containing a chemical shift reference. The experiment used here is a standard HSQC experiment found in the Bruker library.
Historically, the researchers involved in the analysis of heparin were early adopters of NMR spectroscopy, with 1H and 13C spectra successfully being used to characterise the material. One dimensional-NMR measurements of complex materials suffer from many overlapping signals and this problem can be ameliorated by using 2D-NMR experiments. Heteronuclear Single Quantum Coherence (13C–1H HSQC) spectra are two-dimensional containing correlations between 13C atoms and the proton bound to them.
This dispersion in a second dimension means that the problem of overlapping signals is greatly diminished for heparin samples, although the problem is not eradicated entirely due to the heterogeneity of heparin.
The analysis contained within shows that the combination of 13C–1H HSQC NMR spectra and multivariate analysis (PCA) is able to differentiate heparin from different animal sources (porcine intestinal mucosa, bovine intestinal mucosa, ovine intestinal mucosa and bovine lung). Furthermore, if the relationships found within the data are examined, the spectral and therefore the chemical differences of the material can be revealed, thereby providing 2D-spectral fingerprints for the different heparins.
Methods
Materials
Heparin from porcine intestinal mucosa (PMH, 67 samples), bovine intestinal mucosa (BMH, 20 samples), ovine intestinal mucosa (OMH, 13 samples) and bovine lung (BLH, 6 samples) were sourced from different manufacturers. The PMH heparin represents samples from a number of different manufacturers, that have been sourced over many years. The material was lyophilised twice into D2O. After the final freeze-drying step, the material was resuspended in 600 μL of 20 mM phosphate buffer, which also contained 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TSP) as a chemical shift reference.NMR spectroscopy
The HSQC (13C–1H) spectra were measured on a Bruker AVANCE III 600 MHz spectrometer (Karlsruhe, Germany), equipped with a TCI 5 mm cryoprobe, using the Bruker hsqcetgpsisp2.2 (Phase-sensitive ge-2D HSQC using PEP and adiabatic pulses for inversion and refocusing with gradients in back-inept) pulse sequence. The experiments were recorded at 298 K using the following acquisition parameters: number of scans 12, number of dummy scans 16, relaxation delay 2.5 s, spectral width 8 ppm (F2) and 80 ppm (F1), transmitter offset 4.7 ppm (F2) and 80 ppm (F1), and 1JCH = 150 Hz.Multivariate analysis
The spectra were processed so that F2 was comprised of 8 k points and 2 k in the F1 dimension. Importantly, before converting the proprietary HSQC NMR spectra into numerical matrices, the offsets for every spectrum were set as the same values (F1 and F2). These values were for the first spectrum in the dataset, which had been calibrated correctly (TSP set to 1H and 13C equal to 0 ppm). The spectra were processed using Topspin software version 4.0.4 (Bruker BioSpin, Rheinstetten, Germany). Principal component analysis (PCA) of the HSQC NMR spectra was carried out using R (R: A Language and Environment for Statistical Computing22), and the 2D spectra were imported into R using the rNMR package.23 This involves reading the acquisition (parseAcqus) and processing (parseProcs) parameters, and the spectra are then converted into the sparky format (bruker2D), and then they are finally imported (ucsf2D) into R as a matrix. Before the spectra are analysed, they are aligned, normalised for area, and mean centred.24 PCA is then performed using the prcomp function. All the spectra were assigned the same offset values. Once the spectra were imported into R they were peak picked and then aligned to the signal due to I1(2OH)-A(6S), which can be found between 5.05–4.97 ppm 1H and 105.5–104.5 ppm 13C. This signal was chosen as it is insensitive to its environment, so it is not readily perturbed. The script to perform this task was written in-house. Due to the large size of the dataset involved in the analyses, cross-validation was very time consuming. To this end, a method was used that is an approximation of the leave-one-out cross-validation methods – the general cross-validation.25 This method is found in the R package FactoMineR,26 implemented by the function estim_ncp.Results and discussion
The aim of many multivariate analysis techniques is to reduce complex datasets to a number of key trends found within the dataset, that explain the variation within those data. This is the aim of techniques such as principal component analysis, single value decomposition, and factor analysis, to highlight three.Here, PCA27 is used to explore the 13C–1H HSQC NMR spectra of heparins from different animal sources. Using 13C–1H HSQC NMR spectra to analyse heparin has one major advantage over 1H NMR spectra. That is signal dispersion, which enables features to be assigned. Furthermore, the 13C–1H HSQC NMR experiment allows information to be gathered regarding the environment surrounding the 13C nuclei present in heparin in less time than a standard 1D-13C NMR experiment.
To avoid artefacts arising in the PCA, a number of steps have to be taken. Firstly, care has to be taken preparing the samples; samples were lyophilised into D2O to reduce the signal from water, furthermore the samples were reconstituted in a deuterated phosphate buffer, reducing any problems arising from the variations in pH. Secondly, the authors have noted that when preparing the data for analysis, the spectral offset (the furthest limits of the spectra in the F1 and F2 dimension) should be kept constant for the whole dataset. This may change if O1 (the centre of the direct dimension) is allowed to be determined for every experiment and even if the HSQC spectra are calibrated, they may still require internal alignment to avoid artefacts from ghost spectral shifts. The pre-treatment of the dataset that contained all of the HSQC spectra was simple. It was found that normalising the spectra for area and then mean-centring provided the best performance. Previously, the authors have found that when performing multivariate analysis of the 1D-NMR spectra of heparin, the additional normalisation of the data for area and mean centring, as well as Pareto scaling gave the best performance.11
PCA of PMH HSQC NMR spectra
Principal component analysis was performed on a dataset containing the 13C–1H HSQC spectra of 67 PMH samples. The analysis decomposed the dataset into 5 components, which explained 60.00% of the variance of the data; component 1 (36.52%), component 2 (8.23%), component 3 (6.22%), component 4 (5.04%) and component 5 (4.00%). Component 1 differentiated samples by the level of sulfation, epimerisation of the uronic acid and the linkage region (Fig. 1A). The linkage region is a tetrasaccharide at the non-reducing end of the carbohydrate that links the polysaccharide to a protein core; the sequence of this tetrasaccharide is GlcA-Gal-Gal-Xyl-serine.28 The features in red are due to the highly sulfated parts of the polysaccharide, and predominantly show signals from the major trisulfated disaccharide IdoA(2S)–GlcNS(6S). The blue features are due to regions in the heparin chain that contain low sulfation and the linkage region that links the polysaccharide to its protein core. Component 2 (Fig. 1B) is a little subtler. Samples are differentiated in this component by the varying levels of sulfation contained, not the stark differences found in component 1 (Fig. 1A). Interestingly, in both components 1 and 2 (Fig. 1A and B, respectively), signals are seen for the rare disaccharide IdoA(2OH)–GlcA(NH2), as well as signals due to chemical modifications arising in the chain from the manufacturing process, including epoxidation, in component 2. Component 3 (Fig. 1C) shows features specifically due to the linkage region, indicating that the rare GlcA(NH2) residue is correlated with the linkage region, and that this region of the chains adjacent to the linkage tetrasaccharide contains different levels of sulfation. Again, components 4 and 5 (Fig. 1D and E, respectively) differentiate the heparin samples by subtle features in the chain that contain varying levels of sulfation, uronic acid epimerisation and linkage region content. As with component 2, component 4 contains signals arising from chemical modifications in the chain, arising from the manufacturing process, and this time they are from the galacturonic acid residue.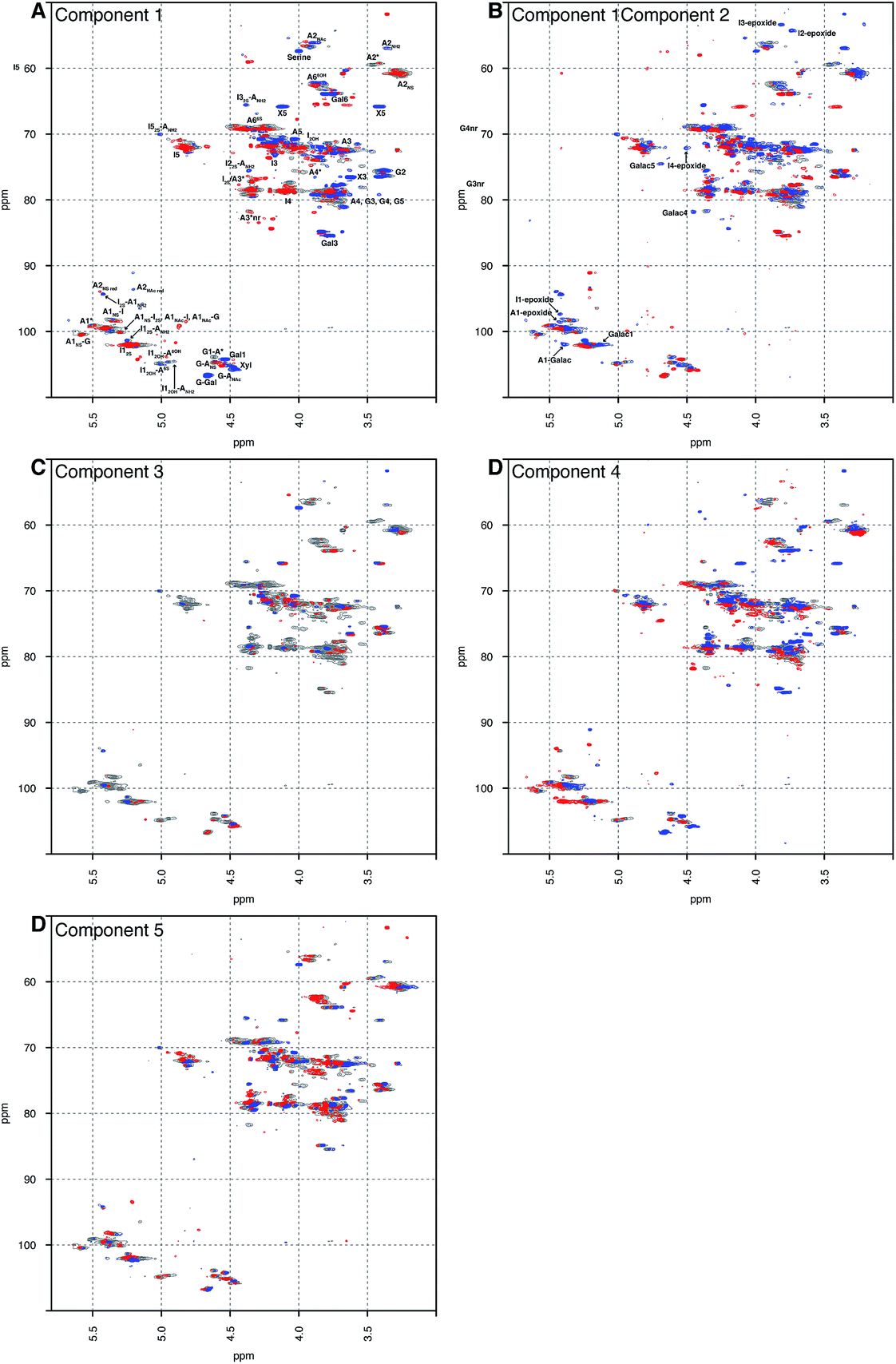 | ||
| Fig. 1 Principal component analysis of 67 PMH 13C–1H HSQC spectra. Prior analysis was performed: the spectra were aligned, normalised for area and mean centred. The analysis decomposed the dataset into 5 major components, and the figure shows the score plots of these 5 components, panels (A) to (E), respectively. The 5 components chosen here explain 60.00% of the variance contained within the dataset. The percentages of variance explained by each component are as follows: 36.52%, 8.23%, 6.22%, 5.40% and 4.00% of the variance, respectively. The scree and score plots can be found in the ESI.† I stands for iduronate, A for glucosamine, and nr indicates that the residue is at the nonreducing end of the molecule. The sub- and superscripts denote the position of sulfation (S) or acetylation (Ac), respectively. AN and IN refer to position N (either C atom or H atom depending on the context) of the glucosamine or iduronate residue, respectively. For example, I2S-A6SNS corresponds to the disaccharide 2-O sulfated iduronic acid linked to 6-O-sulfated N-sulfated glucosamine. A2* signifies position 2 of glucosamine, which is N-sulfated and O-sulfated at positions 6 and 3. IN-epoxide indicates that the iduronate has undergone epoxidation and galac indicates a galacturonic acid residue. Cross-validation of the dataset found that 11 components would explain the variance present in the PMH dataset (see Methods section). | ||
PCA of PMH HSQC NMR spectra compared or HSQC NMR spectra of BMH, BLH and OMH
Principal component analysis was then performed on the PMH HSQC NMR spectra dataset, comparing it to the spectra of BMH, BLH and OMH. Unsurprisingly, the analysis was able to differentiate the other types of heparin from PMH. The individual datasets for BMH, BLH and OMH were not analysed separately as they did not contain sufficient spectra to draw meaningful conclusions. This is a common error made by analysts when PCA is performed.The comparison of 20 BMH HSQC NMR spectra with the 67 PMH HSQC NMR spectra by PCA found two significant components, one major and one minor (component 1 62.30% and component 2 12.08%, Fig. 2A). The BMH samples are clearly differentiated from the PMH samples in component 1 (Fig. 2B and C). Bovine intestinal mucosal heparin has varying levels of O-sulfation at position 6 and this can clearly be seen in component 1 (Fig. 2C, blue features), as well as signals arising from GlcA-Glc(NAc), GlcA-Glc(NS) and GlcA(2S).
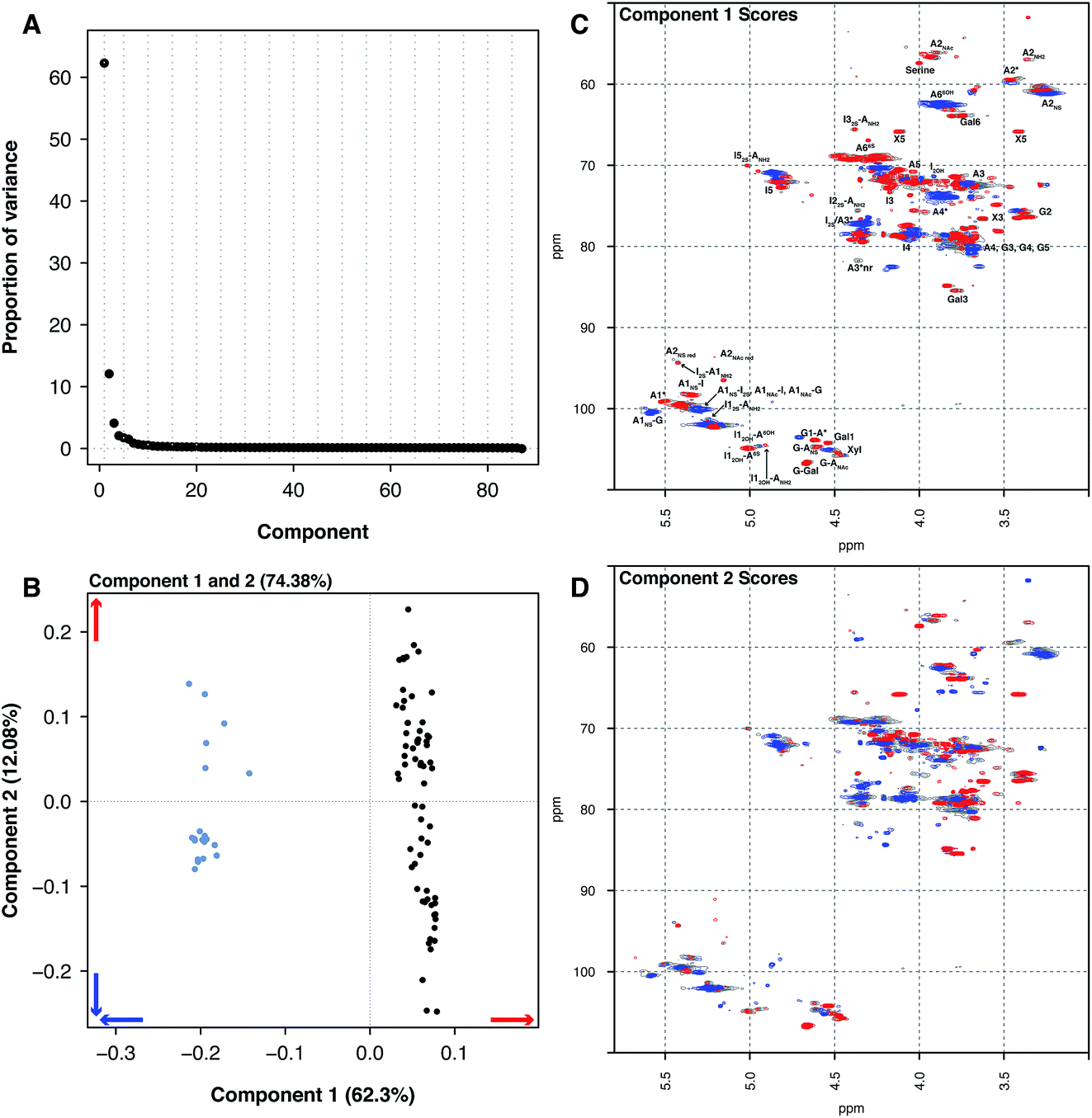 | ||
| Fig. 2 PMH 13C–1H HSQC spectra compared to BMH 13C–1H HSQC spectra. Principal component analysis of a dataset composed of 67 PMH and 20 BMH 13C–1H HSQC spectra. Prior to analysis, the spectra were aligned, normalised for area and mean centred. The analysis decomposed the dataset into 2 major components explaining 74.38% of the total variance. (A) Scree plot and (B) loading plot (BMH samples are light blue, while the PMH samples are black). The figure shows the score plots of components 1 (62.30%) (C) and 2 (12.08%) (D). Porcine intestinal mucosal heparin is differentiated from BMH by component 1 (B and C). The blue features observed in component 1 (C) are more prevalent in the BMH spectra and the red features are more prevalent in the PMH spectra. Cross-validation of the dataset found that 14 components would explain the variance present in the PMH–BMH dataset (see Methods section). | ||
While the PMH samples analysed have higher levels of the standard disaccharide IdoA(2S)–GlcNS(6S), as well as containing more of the linkage region (GlcA-Gal-Gal-Xyl-serine), signals also arose from the trisulfate glucosamine (Glc(3S,6S,NS)) which is important for the antithrombotic activity of the molecules and disulfate iduronic acid linked to 6-O-sulfated glucosamine (IdoA(2OH)–Glc(6S)). Component 2 differentiated samples based on their overall sulfation level (Fig. 2D), separating both PMH and BMH.
Another source of pharmaceutical heparin that is being considered is sheep. Many regions of the world consume large amounts of lamb or mutton, and therefore a significant amount of ovine mucosa is available. As with the BMH material, OMH is distinct from PMH and PCA of the HSQC NMR spectra can differentiate PMH from OMH. Two significant components are found by PCA, similarly with 1 major and 1 minor component (component 1 52.4% and component 2 9.14%, these two components explain 61.54% of the variance found in the dataset, Fig. 3A). The OMH and PMH samples are differentiated by component 1 (Fig. 3C). The blue features observed in the score plot for component 1 are those that are more prevalent in OMH. The OMH samples have a different amount of the standard IdoA(2S)–GlcA(NS,6S) disaccharide to that seen in PMH. Interestingly, signals due to the trisulfated glucosamine (Glc(3S,6S,NS)) indicate that the antithrombin binding site found in OMH is distinct to that found in PMH. These are signals for positions 1 and 2 of Glc(3S,6S,NS), as well as position 3 of Glc(3S,6S,NS) located at the non-reducing end of the polysaccharide. As can be seen from the loading plot, the samples from OMH and PMH are not completely orthogonal, so the major variation that differentiates OMH from PMH also arises within the PMH samples. The red features in component 1 (Fig. 3C) are those found more prevalently in the PMH samples and contain signals due to the less sulfate residues, GlcA containing disaccharides and the linkage region. These observations suggest that the OMH samples analysed here have a more homogeneous sequence than the PMH samples. Component 2 disperses the PMH samples (Fig. 3D), with the PMH samples containing varying amounts of the component. As can be seen from the loading plot for the analysis (Fig. 3B), the OMH samples only contain the positive features of component 2, which contains signals from the linkage region, as well as signals for the standard IdoA(2S)–GlcA(NS,6S) disaccharide and the trisulfated glucosamine (Glc(3S,6S,NS)). This suggests that the non-reducing end of the OMH samples is, on the whole, more sulfated than the same region found in the PMH samples and, potentially, it also contains a possibly distinct antithrombin binding site.
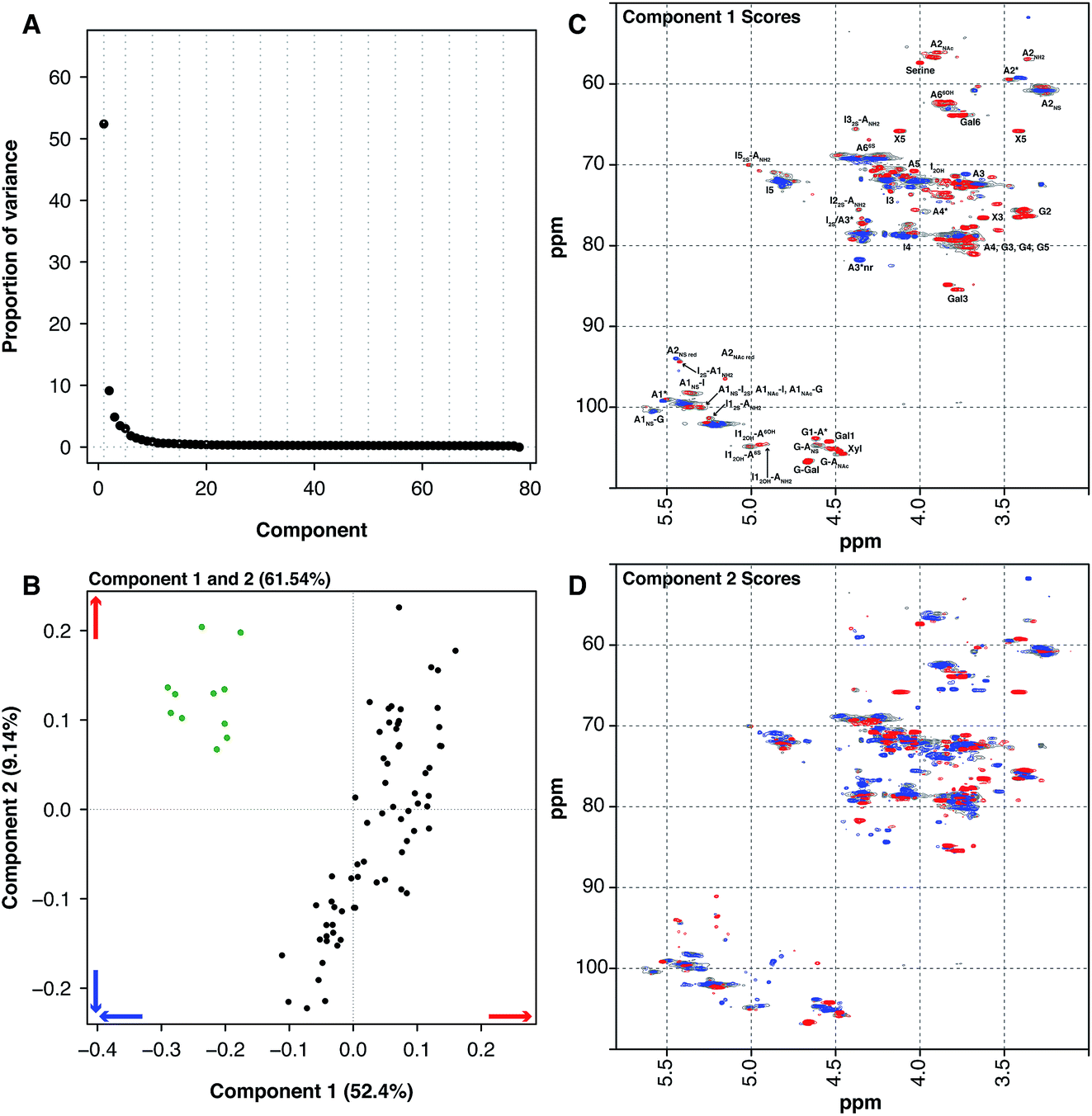 | ||
| Fig. 3 PMH 13C–1H HSQC spectra compared to OMH 13C–1H HSQC spectra. Principal component analysis of a dataset composed of 67 PMH and 13 OMH 13C–1H HSQC spectra. Before the analysis was performed, the spectra were aligned, normalised for area and mean centred. The analysis decomposed the dataset into 2 major components explaining 61.54% of the total variance. (A) Scree plot and (B) loading plot (OMH samples are green, while the PMH samples are black). The figure shows the score plots of components 1 (52.40%) (C) and 2 (9.14%) (D). Porcine intestinal mucosal heparin is differentiated from OMH by component 1 (B and C). The blue features observed in component 1 (C) are more prevalent in the OMH spectra and the red features are more prevalent in the PMH spectra. Cross-validation of the dataset found that 14 components would explain the variance present in the PMH–OMH dataset (see Methods section). | ||
Historically, heparin was sourced from both cows and pigs, however the emergence of bovine spongiform encephalopathy (BSE) put an end to the use of bovine heparin in most of the world, due to safety concerns. When heparin was widely sourced from cows, the material was extracted from both the intestinal mucosa and lungs. The 6 BLH samples analysed here are distinct from the 67 PMH samples. PCA of the dataset containing the BLH and PMH HSQC NMR spectra isolated 2 significant components, 1 major and 1 minor (component 1 68.87% and component 2 6.43%, these two components explain 75.30% of the variance found in the dataset) (Fig. 4A). The BLH samples have a very homogenous structure, being enriched in the standard IdoA(2S)–GlcA(NS,6S) disaccharide, which is evident in component 1 (Fig. 4C). The PMH samples were dispersed by component 2 (Fig. 4B). The blue signals seen in the score plot for component 2 are the features that separate the PMH samples (Fig. 4D). The PMH samples contain varying levels of the signals originating from the trisulfated glucosamine (Glc(3S,6S,NS)) residue, positions 1, 2 and 4 of Glc(3S,6S,NS), and position 1 of GlcA attached to Glc(3S,6S,NS), indicating that the antithrombin site within PMH is different to that seen in BLH. The BLH samples only contain the red features observed in component 2, the minor signals (Fig. 4C) corresponding with the major repeating disaccharide observed in component 1 (Fig. 4D).
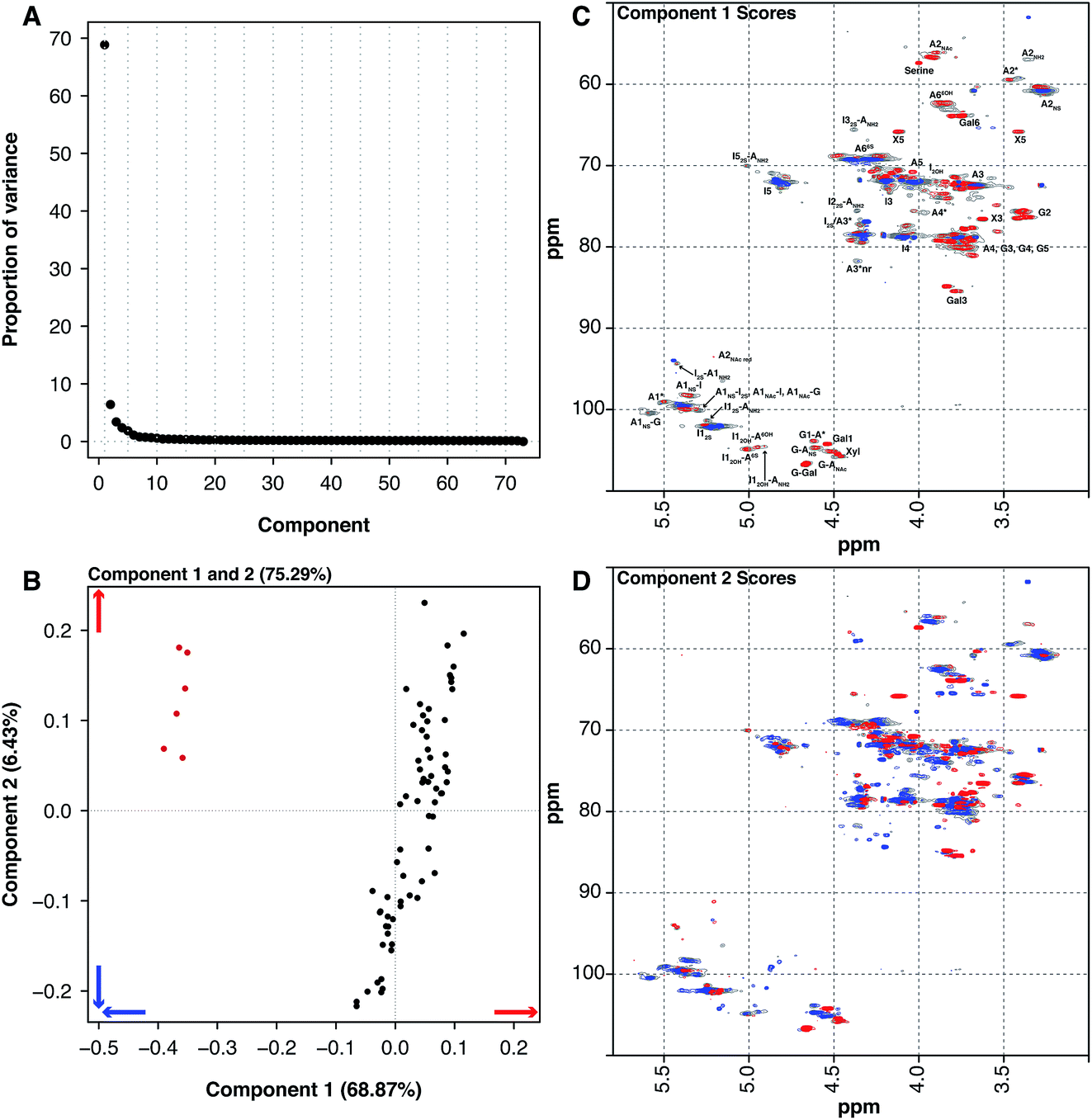 | ||
| Fig. 4 PMH 13C–1H HSQC spectra compared to BLH 13C–1H HSQC spectra. Principal component analysis of a dataset composed of 67 PMH and 6 BLH 13C–1H HSQC spectra. Before the analysis was performed, the spectra were aligned, normalised for area and mean centred. The analysis decomposed the dataset into 2 components (1 major and 1 nominal minor component), explaining 75.3% of the total variance. (A) Scree plot and (B) loading plot (BLH samples are red, while the PMH samples are black). The figure shows the score plots of components 1 (68.87%) (C) and 2 (6.43%) (D). Porcine intestinal mucosal heparin is differentiated from BLH by component 1 (B and C). The blue features observed in component 1 (C) are more prevalent in the BLH spectra and the red features are more prevalent in the PMH spectra. Cross-validation of the dataset found that 14 components would explain the variance present in the PMH–BLH dataset (see Methods section). | ||
The pairwise approach here allows the differences between PMH and BMH, OMH or BLH to be investigated. This analysis can be expanded to look at global differences between the heparins from 4 difference sources. The ESI† contains the PCA of all the heparin HSQC spectra; components 1 and 2 differentiate the four heparin. Component 1 differentiates PMH and OMH from BLH and BMH, and component 2 differentiates the heparin from the bovine sources.
Conclusions
Multivariate analysis techniques provide a powerful toolbox that can be used to analyse the most complicated mixtures. The material of interest in this paper, heparin, is a highly heterogeneous polysaccharide comprising of chains of varying length, charge and substitution pattern. The application of PCA to the 13C–1H HSQC NMR spectra of heparin allowed heparin from different animal sources and organs to be differentiated. Furthermore, the analysis extracted spectral signatures that are specific to the 4 heparin types (porcine intestinal mucosa, bovine intestinal mucosa, ovine intestinal mucosa and bovine lung). While the 13C–1H HSQC NMR spectrum of heparin provides a great deal of information, the analysis performed here is mainly qualitative, although through the integration of a number of signals in the 13C–1H HSQC NMR spectra of heparin samples, the average disaccharide composition of the polysaccharide can be determined.29,30 These approaches are much quicker than other traditional methods, such as digestion followed by HPLC or HPLC–MS, and require much less preparation time.Such approaches are highly valuable to the quality control of the heparin pharmaceutical product; the NMR experiment, spectral processing and subsequent multivariate analysis could all be performed within one working day, with the only barrier being the exchange of the sample into D2O. This exchange could be circumvented, and the measurement performed in 90% H2O/10% D2O. The only drawback would be that the water signal may obscure signals of interest and further complications could be caused by the presence of signals from exchange protons.31
The HSQC spectra provide information regarding the average electronic environment surrounding the hydrogen and carbon nuclei present in the carbohydrate. One important piece of data that is lacking is information regarding the sequence/substitution pattern found within the carbohydrate. To provide such information it might be necessary to perform different NMR experiments, possibly analysing datasets of TOCSY or NOESY experiments, or by analysing datasets that contain different experiment types, for example HSQC and TOCSY spectra. The only limitation would be time, since both high quality NOESY and TOCSY spectra take much more time to record than the equivalent HSQC spectrum.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the contribution of the late Professor Benito Casu, without whom, these developments would not have been possible.Notes and references
- T. K. Kishimoto, K. Viswanathan, T. Ganguly, S. Elankumaran, S. Smith, K. Pelzer, J. C. Lansing, N. Sriranganathan, G. Zhao, Z. Galcheva-Gargova, A. Al-Hakim, G. S. Bailey, B. Fraser, S. Roy, T. Rogers-Cotrone, L. Buhse, M. Whary, J. Fox, M. Nasr, G. J. Dal Pan, Z. Shriver, R. S. Langer, G. Venkataraman, K. F. Austen, J. Woodcock and R. Sasisekharan, N. Engl. J. Med., 2008, 358, 2457–2467 CrossRef CAS PubMed.
- M. Guerrini, D. Beccati, Z. Shriver, A. Naggi, K. Viswanathan, A. Bisio, I. Capila, J. C. Lansing, S. Guglieri, B. Fraser, A. Al-Hakim, N. S. Gunay, Z. Zhang, L. Robinson, L. Buhse, M. Nasr, J. Woodcock, R. Langer, G. Venkataraman, R. J. Linhardt, B. Casu, G. Torri and R. Sasisekharan, Nat. Biotechnol., 2008, 26, 669–675 CrossRef CAS PubMed.
- C. Li, S. Bhavaraju, M. P. Thibeault, J. Melanson, A. Blomgren, T. Rundlof, E. Kilpatrick, C. J. Swann, T. Rudd, Y. Aubin, K. Grant, M. Butt, W. Shum, T. Kerim, W. Sherwin, Y. Nakagawa, S. Pavon, S. Arrastia, T. Weel, A. Pola, D. Chalasani, S. Walfish and F. Atouf, J. Pharm. Biomed. Anal., 2019, 166, 105–112 CrossRef CAS PubMed.
- A. C. Gucinski, M. T. Boyne II and D. A. Keire, Anal. Bioanal. Chem., 2015, 407, 749–759 CrossRef CAS PubMed.
- S. Rogstad, E. Pang, C. Sommers, M. Hu, X. Jiang, D. A. Keire and M. T. Boyne II, Anal. Bioanal. Chem., 2015, 407, 8647–8659 CrossRef CAS PubMed.
- T. W. Barrowcliffe, Handb. Exp. Pharmacol., 2012, 207, 3–22, DOI:10.1007/978-3-642-23056-1_1.
- D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC.
- B. Mulloy, J. Hogwood, E. Gray, R. Lever and C. P. Page, Pharmacol. Rev., 2016, 68, 76–141 CrossRef PubMed.
- J. Y. van der Meer, E. Kellenbach and L. J. van den Bos, Molecules, 2017, 22, 1025 CrossRef PubMed.
- R. J. Linhardt and N. S. Gunay, Semin. Thromb. Hemostasis, 1999, 25(suppl. 3), 5–16 CAS.
- T. R. Rudd, D. Gaudesi, M. A. Skidmore, M. Ferro, M. Guerrini, B. Mulloy, G. Torri and E. A. Yates, Analyst, 2011, 136, 1380–1389 RSC.
- T. R. Rudd, M. A. Skidmore, S. E. Guimond, C. Cosentino, G. Torri, D. G. Fernig, R. M. Lauder, M. Guerrini and E. A. Yates, Glycobiology, 2009, 19, 52–67 CrossRef CAS PubMed.
- Q. Zang, D. A. Keire, L. F. Buhse, R. D. Wood, D. P. Mital, S. Haque, S. Srinivasan, C. M. Moore, M. Nasr, A. Al-Hakim, M. L. Trehy and W. J. Welsh, Anal. Bioanal. Chem., 2011, 401, 939–955 CrossRef CAS PubMed.
- Q. Zang, D. A. Keire, R. D. Wood, L. F. Buhse, C. M. Moore, M. Nasr, A. Al-Hakim, M. L. Trehy and W. J. Welsh, J. Pharm. Biomed. Anal., 2011, 54, 1020–1029 CrossRef CAS PubMed.
- Q. Zang, D. A. Keire, R. D. Wood, L. F. Buhse, C. M. Moore, M. Nasr, A. Al-Hakim, M. L. Trehy and W. J. Welsh, Anal. Bioanal. Chem., 2011, 399, 635–649 CrossRef CAS PubMed.
- Q. Zang, D. A. Keire, R. D. Wood, L. F. Buhse, C. M. Moore, M. Nasr, A. Al-Hakim, M. L. Trehy and W. J. Welsh, Anal. Chem., 2011, 83, 1030–1039 CrossRef CAS PubMed.
- L. Mauri, M. Marinozzi, G. Mazzini, R. E. Kolinski, M. Karfunkle, D. A. Keire and M. Guerrini, Molecules, 2017, 22, 1146 CrossRef PubMed.
- M. Guerrini, T. R. Rudd, L. Mauri, E. Macchi, J. Fareed, E. A. Yates, A. Naggi and G. Torri, Anal. Chem., 2015, 87, 8275–8283 CrossRef CAS PubMed.
- T. R. Rudd, D. Gaudesi, M. A. Lima, M. A. Skidmore, B. Mulloy, G. Torri, H. B. Nader, M. Guerrini and E. A. Yates, Analyst, 2011, 136, 1390–1398 RSC.
- T. R. Rudd, E. Macchi, L. Muzi, M. Ferro, D. Gaudesi, G. Torri, B. Casu, M. Guerrini and E. A. Yates, Anal. Chem., 2013, 85, 7487–7493 CrossRef CAS PubMed.
- T. R. Rudd, E. A. Yates and M. Guerrini, New Methods for the Analysis of Heterogeneous Polysaccharides – Lessons Learned from the Heparin Crisis, in New Developments in NMR, 2017, pp. 305–334 Search PubMed.
- R Core Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2018, http://https://www.R-project.org/ Search PubMed.
- I. A. Lewis, S. C. Schommer and J. L. Markley, Magn. Reson. Chem., 2009, 47(suppl. 1), S123–S126 CrossRef CAS PubMed.
- R. A. van den Berg, H. C. Hoefsloot, J. A. Westerhuis, A. K. Smilde and M. J. van der Werf, BMC Genomics, 2006, 7, 142 CrossRef PubMed.
- J. Josse and F. Husson, Comput. Stat. Data Anal., 2012, 56, 1869–1879 CrossRef.
- S. Lê, J. Josse and F. Husson, J. Stat. Softw., 2008, 1(1) DOI:10.18637/jss.v025.i01.
- I. T. Jolliffe, Principal component analysis, Springer-Verlag, New York, 2002 Search PubMed.
- M. Iacomini, B. Casu, M. Guerrini, A. Naggi, A. Pirola and G. Torri, Anal. Biochem., 1999, 274, 50–58 CrossRef CAS PubMed.
- M. Guerrini, A. Bisio and G. Torri, Semin. Thromb. Hemostasis, 2001, 27, 473–482 CrossRef CAS PubMed.
- L. Mauri, G. Boccardi, G. Torri, M. Karfunkle, E. Macchi, L. Muzi, D. Keire and M. Guerrini, J. Pharm. Biomed. Anal., 2017, 136, 92–105 CrossRef CAS PubMed.
- C. N. Beecher and C. K. Larive, Anal. Chem., 2015, 87, 6842–6848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fd00009g |
| This journal is © The Royal Society of Chemistry 2019 |