
Catalyst-free ambient temperature synthesis of isoquinoline-fused benzimidazoles from 2-alkynylbenzaldehydes via alkyne hydroamination†
Manisha
Mishra
a,
Dylan
Twardy
b,
Clifford
Ellstrom
a,
Kraig A.
Wheeler
 c,
Roman
Dembinski
bd and
Béla
Török
*a
c,
Roman
Dembinski
bd and
Béla
Török
*a
aDepartment of Chemistry, University of Massachusetts Boston, 100 Morrissey Blvd., Boston, MA 02125, USA. E-mail: bela.torok@umb.edu
bDepartment of Chemistry, Oakland University, 146 Library Drive, Rochester, MI 48309-4479, USA
cDepartment of Chemistry, Whitworth University, 300 W. Hawthorne Rd., Spokane, WA 99251, USA
dCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Łódź, Poland
First published on 12th November 2018
Abstract
An efficient environmentally benign route for the synthesis of benzimidazo[2,1-a]isoquinoline has been developed by reacting 2-ethynylbenzaldehyde and related substituted alkynylbenzaldehydes with variously substituted ortho-phenylenediamines and aliphatic amines in ethanol. This method provides a convenient, room temperature, atom-economical, and catalyst-free access to diversely substituted isoquinoline fused benzimidazoles. Regioselectivity of the reaction, as referred to o-phenylenediamines, was confirmed by X-ray crystallography. The reaction was found to occur in three major steps (imine formations, cyclization, and aromatization) and a mechanism has been proposed.
Introduction
Among the multitude of nitrogen-containing heterocyclic compounds, isoquinolines1 and benzimidazoles2 exhibit a wide range of biological activities. A molecular scaffold that integrates both isoquinoline and benzimidazole subunits, such as fused benzimidazoisoquinoline rings, has an immense potential due to outstanding biological and pharmacological activities, such as anti-HIV-1, anticancer, antimicrobial, and antifungal properties.3 Therefore, a straightforward, simple, and furthermost environmentally benign route for the construction of benzimidazo[2,1-a]isoquinolines is of high interest to the fine chemicals and pharmaceutical industry.Numerous approaches towards the formation of benzimidazo[2,1-a]isoquinolines are available.4 An important protocol, described by Dyker et al. for constructing such fused rings,5 combines 2-ethynylbenzaldehyde 1 and ortho-phenylenediamine 2 in nitrobenzene, which serves as a solvent as well as an oxidizing agent. However the procedure (150 °C, 2 d) has recorded a low yield of the cyclized product. An alternative approach involves a microwave-accelerated tandem process in which a Sonogashira coupling, 5-endo-trig cyclization, oxidative aromatization, and 6-endo-dig cyclization takes place in one pot.6 The reaction, catalyzed by Pd(OAc)2 at elevated temperature (120 °C) in DMF in the presence of Bu4NOAc as a base, albeit quite competitive, features a stoichiometric amount of base, difficult to remove solvent, and high energy demand. Other methods for the synthesis of benzimidazo[2,1-a]isoquinolines via transition metal-catalyzed cross-coupling reactions have been reported in literature.7–10 However, most of these methods are carried out under conditions that involve the use of catalytic albeit often toxic metals at elevated temperature. These protocols also need pre-functionalization of precursors, and the removal of metal impurities to meet pharmaceutical standards is challenging.11 A metal free approach for the synthesis of benzimidazo[2,1-a]isoquinolines involving an intramolecular cyclization of N-arylisoquinoline-1-amine has also been reported. The reaction, catalyzed by a hypervalent iodine reagent proceeds in water under ambient condition in good yield.12 However, the method requires a highly pre-assembled starting material.
Our earlier efforts revealed that several traditional, commonly acid-catalyzed, high temperature protocols reported for the synthesis of heterocycles can, in fact, readily proceed at room temperature without the use of any catalyst and in certain cases even solvent-free, opening opportunities for more environmentally friendly assembly of important compounds.13 Based on these findings and our ongoing efforts towards the sustainable synthesis of nitrogen heterocycles,14 we examined the condensation reaction between o-phenylenediamines and 2-ethynylbenzaldehyde 1, which is an intriguing substrate for the synthesis of diverse polycyclic compounds. Continuing our sustainable synthesis development efforts,15 herein, we report a novel one pot catalyst and reagent free green strategy for the synthesis of benzimidazo[2,1-a]isoquinolines. This approach allows the synthesis of a diverse family of substituted derivatives of the target compounds in excellent yields with a simple reaction setup.
Results and discussion
To find the optimal reaction conditions, we selected the 2-ethynylbenzaldehyde 1 and o-phenylenediamine 2a as model compounds. The transformation conditions were investigated as outlined in Table 1.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Temperature [°C] | Time [min] | Yieldc [%] |
| a Reagents and conditions: 2-Ethynylbenzaldehyde 1 (0.5 mmol), diamine 2a (0.5 mmol), EtOH (2 mL), and K-10 (300 mg); MW microwave irradiation; CH conventional heating. b Ethanol was removed before the reaction. c Yield determined by GC. | ||||
| 1 | K-10b | 50MW | 15 | 71 |
| 2 | K-10b | 50MW | 30 | 81 |
| 3 | K-10b | 100MW | 15 | >99 |
| 4 | — | 50CH | 30 | 80 |
| 5 | — | 50CH | 60 | 98 |
| 6 | — | rt | 60 | 74 |
| 7 | — | rt | 120 | 98 |
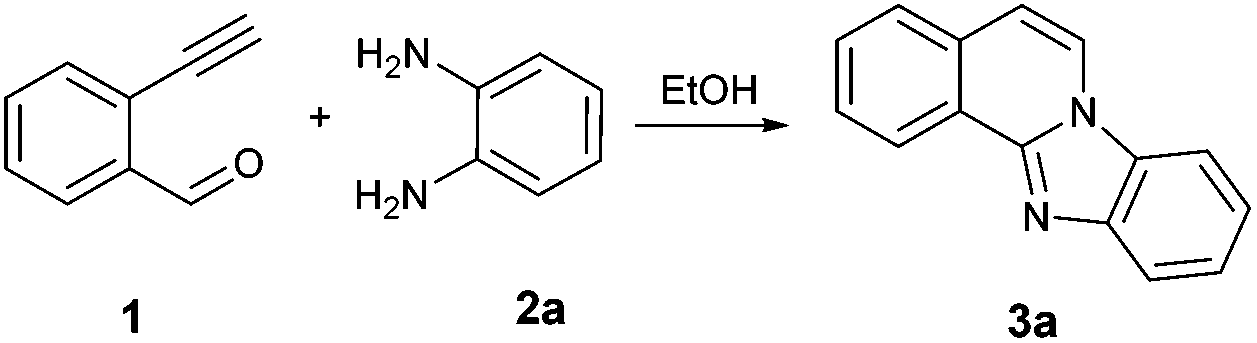
Initially the reaction was carried out using solvent-free conditions. The substrates were mixed in ethanol, and montmorillonite K-10, a well-known solid acid catalyst,16 was added, and the solvent was removed. The solid mixture was heated by microwave irradiation. Formation of the benzimidazo[2,1-a]isoquinoline 3a was observed in quantitative yield after a few steps of optimization (Table 1, entries 1–3). While K-10 has effectively catalyzed the cyclization reaction, a control experiment, the same reaction without catalyst was carried out. Interestingly, while the reaction occurred at a higher rate in the presence of K-10, it did not require the presence of a catalyst. Even at 50 °C, the reaction produced the desired tetracyclic product 3a with 80% yield within 30 min (Table 1, entry 4). The yield was excellent at 50 °C after 60 min stirring (98%, Table 1, entry 5). This result prompted us to explore the reaction in a catalyst-free setting even at room temperature (rt; Table 1, entries 6 and 7). Almost quantitative formation of the desired product at ambient temperature with no catalyst was achieved after 2 h (Table 1, entry 7). Even though the microwave-assisted K-10 catalyzed reaction afforded 100% yield of the cyclized compound (Table 1, entry 3), we decided to proceed with the synthesis under catalyst-free conditions at room temperature. Without need for catalyst separation and energy investment the reaction still gives nearly quantitative (98%) conversion to the desired product in 2 h, at conditions that we deem greener than the catalytic, microwave-assisted method. This is a second recent example of catalyst-free reactions formerly believed to proceed only in the presence of a promoter.17
Following, the scope of cyclization reaction with 2-ethynylbenzaldehyde 1 and diamines 2 was explored under the optimized reaction conditions (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time [h] | Yieldc [%] |
| a Reaction conditions: 2-Ethynylbenzaldehyde 1 (1 mmol) and diamines 2 (1 mmol) were dissolved in absolute ethanol (3 mL) and stirred at room temperature, unless otherwise noted. b Stirred at 50 °C. c GC yield. d Isolated yield. | ||||
| 1 |

|
|
2 | 98 |
| 2 |

|

|
2 | >99 |
| 3 |
|

|
4b | 84d |
| 4 |
|
|
6 | 91 |
| 5 |
|

|
8b | 91 |
| 6 |
|
|
6b | 90 |
| 7 |
|
|
8b | 80d |
| 8 |
|

|
6b | 88d |
| 9 |

|

|
4 | 99 (88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12) :12) |
| 10 |
|

|
4 | 91 (60:40) |
| 11 |
|

|
6 | 90 (60:40) |
| 12 |
|

|
2 | 95 (65:35) |
| 13 |

|

|
6 | 98 (73:27) |
| 14 |

|
|
6b | 90 (65:35) |

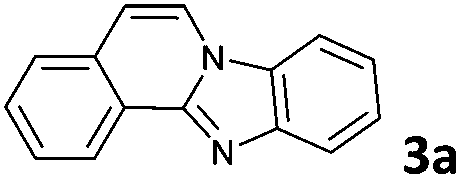
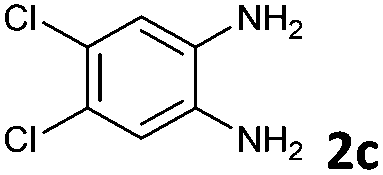
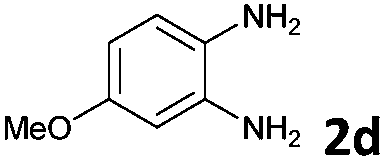
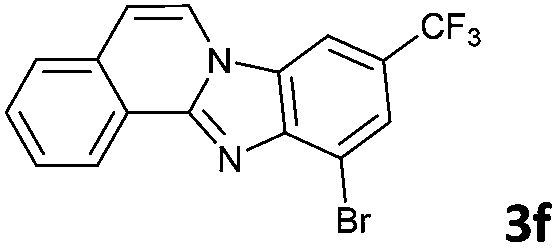
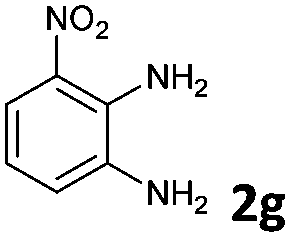
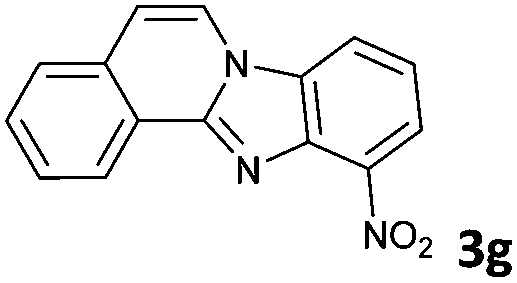
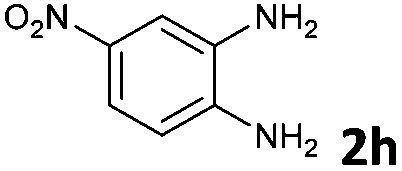
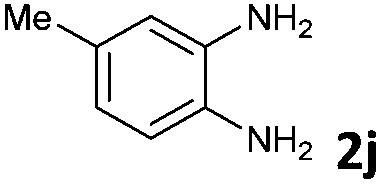
The reaction provided benzimidazo[2,1-a]isoquinolines 3a–n with good to excellent yields (80–99%), regardless of the electronic nature of the substituents of the o-phenylenediamines. Only in the case of strong electron withdrawing groups such as NO2 or CF3 (Table 2, entries 5–8), did the reaction require moderate heating (50 °C) to achieve completion. Symmetrical o-phenylenediamines (Table 2, entries 1–3), gave single isolated products as expected. The cyclization also showed exclusive regioselectivity when diamines with strong electron withdrawing groups were used (Table 2, entries 5–8). In addition, 4-methoxy-o-phenylenediamine 2d also yielded only one product. In other cases, when the reaction still occurred with exclusive chemoselectivity, the formation of regioisomers was observed (Table 2, entries 9–14).
After determining the substrate scope regarding substituted o-phenylenediamines, the effect of structure of the other starting materials, alkynylaldehydes, on the yield and selectivity of the approach was investigated. Several 2-alkynylbenzaldehydes 4 (e.g.4a,c,f) are commercially available. However, in order to further extend the scope, a few more alkynylaldehydes were synthesized following a literature procedure (see ESI, Scheme S1†).18 Characterization for all aldehydes 4 but 5-fluoro-4-methoxy-2-(phenylethynyl)benzaldehyde (4d) is available in the literature and referenced in the ESI.† The structures of alkynyl aldehydes used for the further scope extension are summarized in Chart 1.
 | ||
| Chart 1 Structure of substituted alkynylbenzaldehyde derivatives18 applied for further assessment of the reaction scope. | ||
Reactivity of the new alkynylbenzaldehydes was investigated with two o-phenylenediamines; the unsubstituted (2a) and 4,5-dimethyl (2b) derivative. The data are tabulated in Table 3.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 4 | 2 | Product | t (h) | Yieldb (%) |
| a Reaction conditions: Substituted 2-alkynylbenzaldehyde 4 (1 mmol) and diamine 2 (1 mmol) were dissolved in absolute ethanol (6 mL) and stirred at 50 °C. b Isolated yield. | |||||
| 1 |

|
2a |

|
6 | 88 |
| 2 |
|
2b |
|
6 | 63 |
| 3 |
|
2a |

|
6 | 82 |
| 4 |
|
2b |

|
6 | 92 |
| 5 |

|
2a |
|
6 | 80 |
| 6 |

|
2b |

|
6 | 85 |
| 7 |

|
2a |
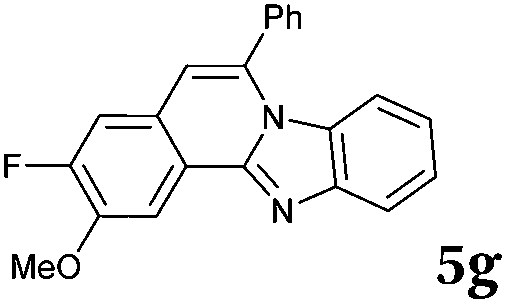
|
6 | 79 |
| 8 |

|
2b |

|
6 | 89 |
| 9 |
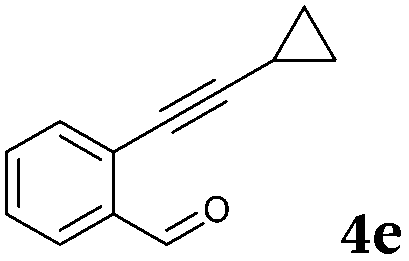
|
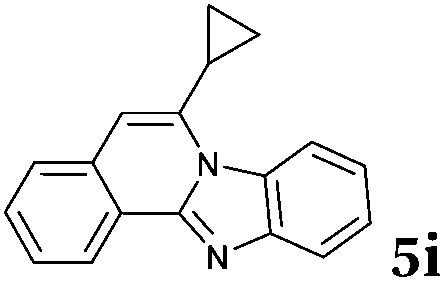
|
6 | 92 | |
| 10 |

|
2b |
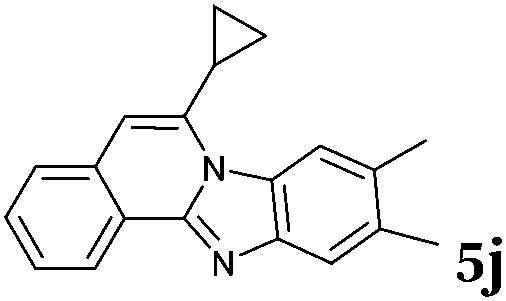
|
6 | 88 |
| 11 |

|
2a |

|
6 | 78 |
| 12 |

|
2b |

|
6 | 82 |
| 13 |
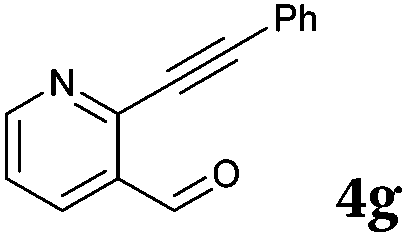
|
2a |
|
6 | 88 |
| 14 |
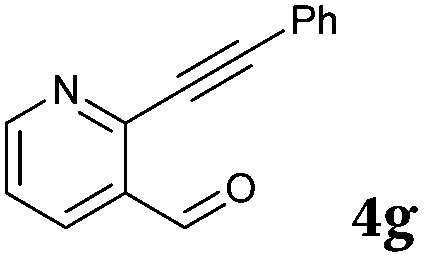
|
2b |
|
6 | 90 |

Although the above data clearly describe the versatility and the broad scope of the reaction for the synthesis of isoquinoline fused benzimidazoles we have attempted the reaction with aliphatic diamines such as 1,2-diaminoethane and 1,3-diaminopropane. Both diamino compounds appeared to readily react and provided the product with high selectivity resulting in the products 7a and 7b in excellent to moderate yields respectively (Scheme 1). It appears that similarly to o-phenylenediamines, the aliphatic diamines also formed products in which a secondary oxidation in the form of a loss of two H occurred. It is suggested that the extended number of already delocalized electrons (8 before oxidation) is a major driving force for this spontaneous oxidation to develop a ten electron containing delocalized system. Limited attempts to reveal the source of the oxidation have been carried out; however, the reduced species has not been identified.
 | ||
| Scheme 1 Application of the general procedure for the synthesis of 2,3-dihydroimidazo[2,1-a]isoquinoline (7a) and 3,4-dihydro-2H-pyrimido[2,1-a]isoquinoline (7b). | ||
Based on the explored substrate scope we can conclude that the reaction appears to be broadly applicable using a diverse group of compounds for both starting materials. Next the scale-up prospect of the reaction was examined. For this purpose, the reaction of 1 and 2a was carried out in a 5.2 g (40 mmol) scale. We have selected this scale as it is a benchmark in organic process research. During the scale up the general reaction conditions, such as temperature (rt) and reaction time was kept at the original levels, only the amount of starting materials were increased. It was observed that the reaction proceeded with the expected efficiency producing 3a in 92% yield, which is approximately the same as that of the much smaller scale reaction indicating that the reaction is potentially scalable.
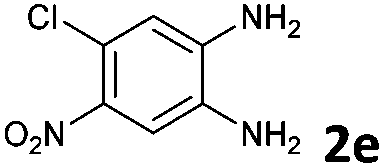
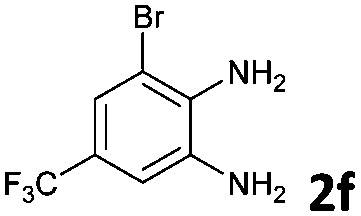
To understand the mechanism of the reaction, a controlled experiment was carried out with 2-ethynylbenzaldehyde 1 and amines 2e–g (Scheme 2). Nucleophiles, containing strongly electron withdrawing substituents, require elevated temperature to form the final cyclized products. Thus, the reaction was carried out at ambient temperature with the aim of isolating potential intermediates.
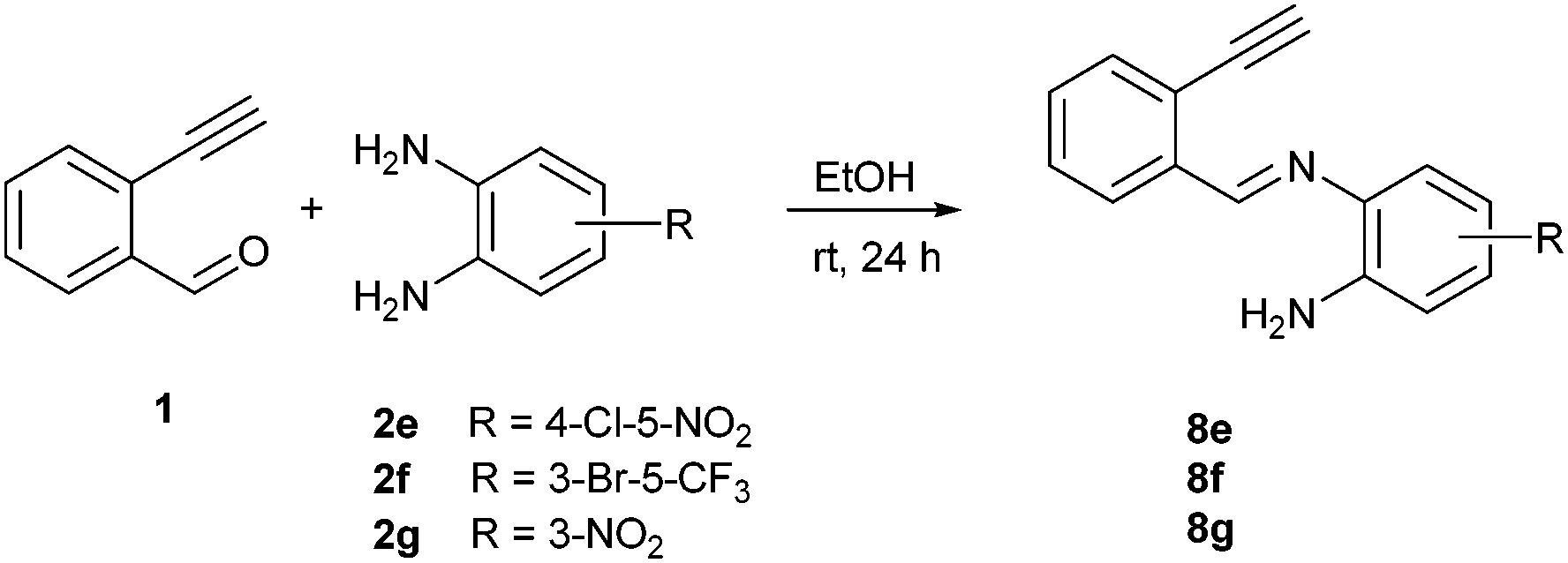 | ||
| Scheme 2 Non-catalytic reaction of ethynylbenzaldehyde (1) with selected o-phenylenediamines 2e,f,g at room temperature. | ||
Our previous experience regarding the reactions of alkynones with amines indicates that depending on the circumstances the first step of the reaction sequence may include the creation of the C–N bond at an alkyne unit13 or the formation of the imine involving a competing carbonyl group.19 As illustrated in Scheme 2, the formation of an intermediate imine 8 was observed at room temperature. The imines 8e,f,g were successfully isolated and characterized to confirm their structure. Their formation suggests that in the initial step the NH2 group reacts with the carbonyl group of the 2-ethynylbenzaldehyde 1. The imine intermediate subsequently undergoes ring closure via the addition of the second NH2 group to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. Based on the regioselectivity data (Table 2 and Scheme 2) we propose that the amino group which possess higher electron density would react with aldehyde group first during the formation of imine intermediate 8 (Scheme 3, first step). The 1H NMR of 8g shows a broad singlet at δ 6.81 ppm (2H integration) indicating the presence of –NH2 group engaged in hydrogen bonding with the ortho nitro group. The successive reaction between the imine formed and the second amino group leads to a dihydrobenzimidazole intermediate (I) that will undergo an N–H addition to the C
N double bond. Based on the regioselectivity data (Table 2 and Scheme 2) we propose that the amino group which possess higher electron density would react with aldehyde group first during the formation of imine intermediate 8 (Scheme 3, first step). The 1H NMR of 8g shows a broad singlet at δ 6.81 ppm (2H integration) indicating the presence of –NH2 group engaged in hydrogen bonding with the ortho nitro group. The successive reaction between the imine formed and the second amino group leads to a dihydrobenzimidazole intermediate (I) that will undergo an N–H addition to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond forming the final cyclic structure (III). Considering the significant driving force for intermediate III to form a conjugated four ring-system, it undergoes an oxidation to form the conjugated aromatic product 3. While prior literature suggest that the aromatization may occur earlier, immediately after the formation of I,8 we suggest that even if the oxidation would be regioselective the potential tautomers of benzimidazole would likely give equal probability to both amino groups to react with the CC triple bond. In addition, the aromatic NH group will possess significantly diminished reactivity. This, however, could not explain the facile progress of the reaction and the often excellent regioselectivity observed. Considering the dihydroimidazole intermediate (I) the electronic orientation effect of the substituents on the phenylenediamine ring would ensure the regioselective ring closure in many cases, just as it ensures the selective reaction of the partially activated NH2 in the imine formation. The proposed mechanistic outline is summarized in Scheme 3.
C triple bond forming the final cyclic structure (III). Considering the significant driving force for intermediate III to form a conjugated four ring-system, it undergoes an oxidation to form the conjugated aromatic product 3. While prior literature suggest that the aromatization may occur earlier, immediately after the formation of I,8 we suggest that even if the oxidation would be regioselective the potential tautomers of benzimidazole would likely give equal probability to both amino groups to react with the CC triple bond. In addition, the aromatic NH group will possess significantly diminished reactivity. This, however, could not explain the facile progress of the reaction and the often excellent regioselectivity observed. Considering the dihydroimidazole intermediate (I) the electronic orientation effect of the substituents on the phenylenediamine ring would ensure the regioselective ring closure in many cases, just as it ensures the selective reaction of the partially activated NH2 in the imine formation. The proposed mechanistic outline is summarized in Scheme 3.
 | ||
| Scheme 3 Mechanistic outline for the formation of benzimidazo[2,1-a]isoquinolines 3. | ||
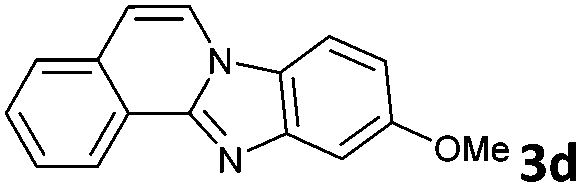
The above proposed mechanism is further supported by the X-ray structure analysis of the product from the reaction between 2-ethynylbenzaldehyde 1 and amine 2d that formed with exclusive regioselectivity. X-ray crystallography confirmed the structure of 10-methoxybenzimidazo[2,1-a]isoquinoline (3d), supporting the proposed regiochemistry of the reaction as referred to o-phenylenediamine, as well as the full aromatization of the five-membered (double-side fused imidazole) ring (Fig. 1).20 The X-ray structure confirms that the initial step occurs by the condensation of the amino group, which is in a para position to the methoxy group, and possess higher electron density than the meta amino group due to the +M effect of OMe group. Corresponding influence was observed for o-phenylenediamines 2e–g that possessed strongly electron-withdrawing substituents (NO2 or CF3). In these cases it was observed that the least deactivated meta-NH2 reacted first.
 | ||
| Fig. 1 An ORTEP view of selected crystallographically independent molecule (A) of 10-methoxybenzo[4,5]imidazo[2,1-a]isoquinoline 3d, illustrating atom labeling scheme and thermal ellipsoids (50% probability level). Selected interatomic distances for molecules A/B (Å): N1–C6 1.393(4)/1.390(4), N1–C7 1.397(4)/1.395(4), N1–C14 1.397(4)/1.392(4), N2–C7 1.333(4)/1.331(4), N2–C15 1.393(4)/1.400(4). | ||
Although the formation of 3d occurred with exclusive selectivity, in certain cases a mixture of regioisomers were obtained. Some of the product mixtures were analyzed in more depth. Product 3l has been isolated in high yields (95%), however, the formation of two regioisomers in a ratio of 65:35 was observed. After the successful isolation of the major isomer, X-ray crystallography confirmed the structure of 8,9-dimethylbenzimidazo[2,1-a]isoquinoline 3l,20 which describes the regiochemistry of the reaction as referred to the o-phenylenediamine, as well as full aromatization of the five-membered (double-side fused imidazole) ring (Fig. 2).
 | ||
| Fig. 2 An ORTEP view of selected crystallographically independent molecule of 3l, illustrating atom labeling scheme and thermal ellipsoids (50% probability level). Selected interatomic distances for molecules A/B (Å): N1–C6 1.398(4)/1.406(4), N1–C7 1.409(4)/1.419(4), N1–C14 1.398(4)/1.391(4), N2–C7 1.345(4)/1.333(4), N2–C15 1.373(4)/1.364(4). | ||
The structure of the major regioisomer of 3l shown in Fig. 2 supports the mechanistic conclusion described above regarding that the amino group with higher electron density will react first with the carbonyl group of the alkynone.
Based on the above presented data we can highlight the advantages of the target reaction using common green metrics.21,22 Among the theoretical factors the atom economy (AE) of the reaction is AE = 92–95% depending on the molecular weight of the starting materials, very close to the maximum possible 100%. As the AE is a theoretical (i.e. calculated) descriptor using the reaction mass efficiency (RME), that considers actual isolated yields, better describes the efficacy of the reaction. The reaction can be characterized by RME = 87–89%. As the isolated yields are mostly above 90% the RME values are only a little lower than the AE values indicating that the reaction is performing near to its very high possible theoretical efficiency. The reaction yields water as the only byproduct and the solvent ethanol is recyclable after the reaction thus the E-factor (waste to product ratio) is approximately 0.054–0.084 which indicates negligibly small waste production during the reaction. Certainly the amount of waste in itself does not describe the harm a process can cause, the nature of the waste such as toxicity, or environmental friendliness should also be considered. Based on the non-toxic and thus environmentally benign waste (water) we can use 1 as environmental unfriendliness quotient (Q) (vs. e.g. chromium oxide with over Q = 1000) thus the environmental quotient22 of the reaction is EQ = 0.084–0.054. Given that the reaction occurs at room temperature and it does not require heating or cooling, therefore, the E-scale, that describes the energy efficiency of a reaction, is also excellent for the described process. Finally, the reaction uses ethanol as a solvent and according to a recent categorization of solvents23 ethanol is the second on this list only n-propanol passes it in terms of environmental friendliness. However, considering that the ranking process does not involve the sustainable character of the solvent, in fact ethanol should rank higher than propanol as it is a renewable solvent, in contrary to propanol.
Based on the above analysis, the described target process is far superior in environmental metrics to most chemical reactions, certainly to all that has been published for the synthesis of these products. It is worth mentioning that during the revision process of our work a paper was published24 describing the synthesis of compound 5i that was not yet known. For comparison, that work describes about 30% yield for the product as compared to 92% in the present work. This clearly shows that this process is not only green but also highly effective.
Conclusions
In conclusion, an environmental-friendly, catalyst-free method has been developed for the rapid synthesis of biologically important isoquinoline-fused-benzimidazole compounds. The method, which includes formal hydroamination of an alkyne, appears to possess a broad substituent tolerance with respect to the different electronic properties of the starting materials. The novelty and sustainable nature of the method can be found in its operational simplicity; (i) without use of any catalyst there is no need for catalyst separation; (ii) the reaction occurs at room temperature with no extra energy investment required; (iii) a renewable solvent, such as ethanol can be used; (iv) the cyclized products form with exclusive chemoselectivity and nearly quantitative yields; (v) high atom economy – one mole of water loss; (vi) there are no toxic by-products; (vii) and finally, the reactants are inexpensive and commercially available.Experimental section
All solvents, phenylenediamines, 2-ethynylbenzaldehyde (1) and 2-((trimethylsilyl)ethynyl)benzaldehyde (4f) were commercially available from Sigma-Aldrich (currently MilliporeSigma) and used without further purification.The 1H and 13C NMR spectra were recorded on a 400 MHz Agilent MR400DD2 spectrometer in CDCl3, (CD3)2SO, using the residual solvent signal for reference. The mass spectrometric identification of the products has been carried out by an Agilent 6850 gas chromatograph-5973 mass spectrometer system (70 eV electron impact ionization) using a 30 m long DB-5 type column (J&W Scientific). LC-MS was recorded in Agilent 2100 system with a C18 (5.0 μm, 6.0 mm × 50 m) LC column. FT-IR spectra were recorded using neat, dry samples by a Thermo Fisher Nicolet 380 FT-IR equipped with Smart Orbit in ATR mode.
General procedure for synthesis of benzimidazo[2,1-a]isoquinolines 3 and dihydroimidazo- and dihydro-2H-pyrimido[2,1-a]isoquinolines 7
2-Ethynylbenzaldehyde 1 (1 mmol) and o-phenylenediamine 2 or alkyldiamines 6 (1 mmol) were dissolved in ethanol (3–5 mL) and stirred at rt. The reaction was monitored by TLC. After the reaction was completed, the reaction mixture was filtered over silica gel 2 cm height pad and the solvent was removed in vacuo. The spectral characterization of the new compounds is described below. The spectral data of compounds 3a and 3b were in agreement with those reported in literature.10General procedure for synthesis of benzimidazo[2,1-a]isoquinolines 5
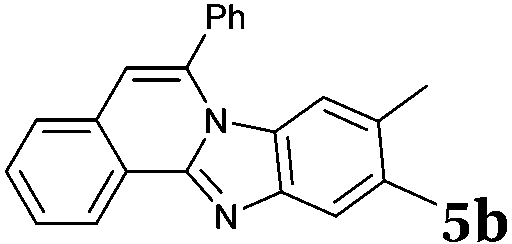
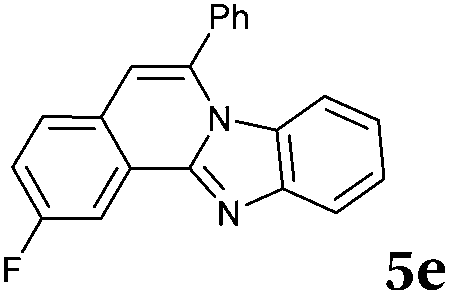
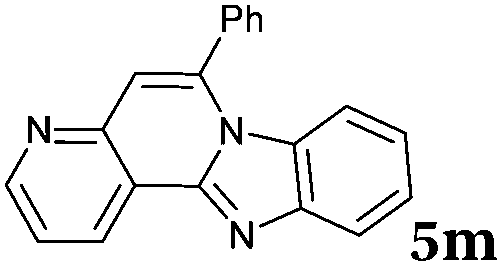
Similar procedure that was used for the synthesis of compounds 3a–3n was used. 2-Alkynylbenzaldehyde 4 (1 mmol) and diamines 2a or 2b, respectively (1 mmol) were dissolved in ethanol (6 mL) and stirred at 50 °C. To obtain selective cyclization the trimethylsilyl derivatives were reacted under more dilute conditions using 15 mL ethanol. The reaction was monitored by TLC. After the reaction was completed, the reaction mixture was filtered over silica gel 2 cm height pad and the solvent was removed in vacuum. The spectral data of 6-phenylbenzo[4,5]imidazo[2,1-a]isoquinoline (5a),25 9,10-dimethyl-6-phenylbenzo[4,5]imidazo[2,1-a]isoquinoline (5b),6,25 2-fluoro-6-phenylbenzo[4,5]imidazo[2,1-a]iso-quinoline (5e),6 and 6-phenylbenzo[4,5]imidazo[2,1-f][1,6]-naphthyridine (5m)6 were in agreement with those reported in literature.Synthesis of [2,1-a]isoquinolines 7
The same procedure that was used for the synthesis of compounds 3a–3n was applied.General procedure for preparation of imines 8
2-Ethynylbenzaldehyde 1 (0.5 mmol) and o-phenylenediamine 2 (0.5 mmol) were dissolved in absolute EtOH (5 mL) and stirred at room temperature for 24 h. The solid precipitate was filtered off and characterized by NMR spectroscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by University of Massachusetts Boston. Donors of the American Chemical Society Petroleum Research Fund (PRF #52858) and the NSF awards (CHE-0821487, CHE-0722547, and CHE-1048719) are also acknowledged.References
- J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; K. Frisch, A. Linda, S. Sabby and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 6058 CrossRef PubMed; A. Morrell, M. Placzek, S. Parmley, B. Grella, S. Antony, Y. Pommier and M. Cushman, J. Med. Chem., 2007, 50, 4388 CrossRef PubMed; J. S. Yadav, B. V. S. Reddy, N. N. Yadav, M. K. Gupta and B. Sridhar, J. Org. Chem., 2008, 73, 6857 CrossRef PubMed.
- P. Singla, V. Luxami and K. Paul, RSC Adv., 2014, 4, 12422 RSC; Y. Wan, C. Wallinder, B. Plouffe, H. Beaudry, A. K. Mahalingam, X. Wu, B. Johansson, M. Holm, M. Botoros, A. Karlen, A. Pettersson, F. Nyberg, L. Fandriks, N. Gallo-Payet, A. Hallberg and M. Alterman, J. Med. Chem., 2004, 47, 5995 CrossRef CAS PubMed; X. Song, B. S. Vig, P. L. Lorenzi, J. C. Darch, L. B. Townsend and G. L. Amidon, J. Med. Chem., 2005, 48, 1274 CrossRef PubMed.
- H.-C. Ouyang, R.-Y. Tang, P. Zhong, X.-G. Zhang and J.-H. Li, J. Org. Chem., 2011, 76, 223 CrossRef CAS PubMed; S. M. Rida, S. A. M. El-Hawash, H. T. Y. Fahmy, A. A. Hazzaa and M. M. M. Ei-Meligy, Arch. Pharmacal Res., 2006, 29, 826 CrossRef; L. W. Deady, P. M. Loria and T. Rodermann, Aust. J. Chem., 1998, 51, 941 CrossRef; R. L. Weinkauf, A. Y. Chen, C. Yu, L. Liu, L. Barrows and E. J. LaVoie, Bioorg. Med. Chem., 1994, 2, 781 CrossRef PubMed; P. Molina, E. Aller, A. Lorenzo, C. Foces and A. L. L. Saiz, Tetrahedron, 1996, 52, 13671 CrossRef.
- R. Zhu, Y. Wang, J. Liu, Q. Wang and J. Huang, Synthesis, 2017, 49, 1335 CrossRef CAS; V. D. Gvozdev, K. N. Shavrin, E. G. Baskir, M. P. Egorov and O. M. Nefedov, Mendeleev Commun., 2017, 27, 231 CrossRef; A. L. Bagdasarian, H. H. Nguyen, T. A. Palazzo, J. C. Fettinger, M. J. Haddadin and M. J. Kurth, J. Org. Chem., 2016, 81, 3924 CrossRef PubMed; E. Moriarty and F. Aldabbagh, Tetrahedron Lett., 2009, 50, 5251 CrossRef; L. Chen, X. Zhang, B. Chen, B. Li and Y. Li, Chem. Heterocycl. Compd., 2017, 53, 618 CrossRef; W.-Q. Miao, J.-Q. Liu and X.-S. Wang, Org. Biomol. Chem., 2017, 15, 5325 RSC; J. Jie, H. Li, S. Wu, Q. Chai, H. Wang and X. Yang, RSC Adv., 2017, 7, 20548 RSC; S. Dhiman, K. Pericherla, N. K. Nandwana, D. Kumar and A. Kumar, J. Org. Chem., 2014, 79, 7399 CrossRef PubMed; B. W. Yang, P. D. Q. Dao, N. S. Yoon and C. S. Cho, J. Organomet. Chem., 2017, 851, 136 CrossRef; M.-Y. Gang, J.-Q. Liu and X.-S. Wang, Tetrahedron, 2017, 73, 4698 CrossRef; Y. Ohta, Y. Kubota, T. Watabe, H. Chiba, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 6299 CrossRef PubMed.
- G. Dyker, W. Stirner and G. Henkel, Eur. J. Org. Chem., 2000, 1433 CrossRef CAS.
- N. Okamoto, K. Sakurai, M. Ishikura, K. Takeda and R. Yanada, Tetrahedron Lett., 2009, 50, 4167 CrossRef CAS.
- S. Nandi, S. Samanta, S. Jana and J. K. Ray, Tetrahedron Lett., 2010, 51, 5294 CrossRef CAS.
- V. Rustagi, R. Tiwari and A. K. Verma, Eur. J. Org. Chem., 2012, 4590 CrossRef CAS.
- N. Kavitha, G. Sukumar, V. P. Kumar, P. S. Mainkar and S. Chandrasekhar, Tetrahedron Lett., 2013, 54, 4198 CrossRef CAS.
- J. Peng, G. Shang, C. Chen, Z. Miao and B. Li, J. Org. Chem., 2013, 78, 1242 CrossRef CAS PubMed; M. Sun, H. Wu, J. Zheng and W. Bao, Adv. Synth. Catal., 2012, 354, 835 CrossRef; V. P. Reddy, T. Iwasaki and N. Kambe, Org. Biomol. Chem., 2013, 11, 2249 RSC.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS; V. W. Rosso, D. A. Lust, P. J. Bernot, J. A. Grosso, S. P. Modi, A. Rusowicz, T. C. Sedergran, J. H. Simpson, S. K. Srivastava, M. J. Humora and N. G. Anderson, Org. Process Res. Dev., 1997, 1, 311 CrossRef.
- D. N. Rao, Sk. Rasheed, R. A. Vishwakarma and P. Das, RSC Adv., 2014, 4, 25600 RSC.
- A. Solan, B. Nisanci, M. Belcher, J. Young, C. Schaefer, K. A. Wheeler, B. Török and R. Dembinski, Green Chem., 2014, 16, 1120 RSC; J. Young, C. Schäfer, A. Solan, A. Baldrica, M. Belcher, B. Nişanci, K. A. Wheeler, E. Trivedi, B. Török and R. Dembinski, RSC Adv., 2016, 6, 107081 RSC.
- A. Kokel and B. Török, Green Chem., 2017, 19, 2515 RSC; A. Kulkarni, W.-H. Zhou and B. Török, Org. Lett., 2011, 13, 5124 CrossRef CAS PubMed; A. Kulkarni and B. Török, Green Chem., 2010, 12, 875 RSC; P. Wyrębek, A. Sniady, N. Bewick, Y. Li, A. Mikus, K. A. Wheeler and R. Dembinski, Tetrahedron, 2009, 65, 1268 CrossRef; S. M. Landge and B. Török, Catal. Lett., 2008, 122, 338 CrossRef.
- G. Pandey and B. Török, Green Chem., 2017, 19, 5390 RSC; A. Kokel, C. Schäfer and B. Török, Green Chem., 2017, 19, 3729 RSC; C. Schäfer, C. J. Ellstrom, H. Cho and B. Török, Green Chem., 2017, 19, 1230 RSC; H. Cho, F. Török and B. Török, Green Chem., 2014, 16, 3623 RSC; A. Daştan, A. Kulkarni and B. Török, Green Chem., 2012, 14, 17 RSC.
- S. M. Landge and B. Török, Catal. Lett., 2008, 122, 338 CrossRef CAS; S. M. Landge, A. Schmidt, V. Outerbridge and B. Török, Synlett, 2007, 1600 Search PubMed; B. Török, G. London and M. Bartok, Synlett, 2000, 631 Search PubMed.
- H. Cho, R. Madden, B. Nisanci and B. Török, Green Chem., 2015, 17, 1088 RSC.
- S. Zhu, H. Huang, Z. Zhang, T. Ma and H. Jiang, J. Org. Chem., 2014, 79, 6113 CrossRef CAS PubMed.
- D. A. Borkin, M. Puscau, A. Carlson, K. A. Wheeler, B. Török and R. Dembinski, Org. Biomol. Chem., 2012, 10, 4505 RSC.
- Crystals of 3d and 3l (the second one did not diffract well) were grown from the dichloromethane/hexanes and ethyl acetate-dichloromethane/hexanes, respectively, by diffusion. CCDC 1547129 and 1811816 contains the supplementary crystallographic data for this paper.†.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- B. Török and T. Dransfield, Green Chemistry: Historical Perspectives and Basic Concepts, in Green Chemistry: An Inclusive Approach, Elsevier, Waltham, Oxford, 2017, ch. 1, pp. 6–7 Search PubMed.
- M. Tobiszewski, J. Namieśnika and F. Pena-Pereira, Green Chem., 2017, 19, 1034 RSC.
- During the course of this work the compound was reported. X. Liu, G. Deng and Y. Liang, Tetrahedron Lett., 2018, 59, 2844 CrossRef CAS.
- K. R. Roesch and R. C. Larock, J. Org. Chem., 2002, 67, 86 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new products. CCDC 1547129 and 1811816. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8gc02520g |
| This journal is © The Royal Society of Chemistry 2019 |