
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional clustering of B cell receptors using sequence and structural features†
Zichang
Xu‡
 a,
Songling
Li‡
ab,
John
Rozewicki‡
ab,
Kazuo
Yamashita§
a,
Shunsuke
Teraguchi
ab,
Takeshi
Inoue
b,
Ryo
Shinnakasu
b,
Sarah
Leach
b,
Tomohiro
Kurosaki
b and
Daron M.
Standley
*ab
a,
Songling
Li‡
ab,
John
Rozewicki‡
ab,
Kazuo
Yamashita§
a,
Shunsuke
Teraguchi
ab,
Takeshi
Inoue
b,
Ryo
Shinnakasu
b,
Sarah
Leach
b,
Tomohiro
Kurosaki
b and
Daron M.
Standley
*ab
aResearch Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: standley@biken.osaka-u.ac.jp
bImmunology Frontier Research Center, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 24th July 2019
Abstract
The repertoires of B cell receptors (BCRs), which can be captured by single cell-resolution sequencing technologies, contain a personal history of a donor's antigen exposure. One of the current challenges in analyzing such BCR sequence data is to assign sequences to groups with similar antigen and epitope binding specificity. This is a non-trivial task given the paucity of experimentally-determined antibody–antigen structures and the fact that different gene combinations in B cells can lead to receptors that target the same antigen and epitope. Here, we describe a method for clustering BCRs based on sequence and predicted structural features in order to predict groups with similar antigen and epitope binding specificity. We show that all known experimentally-determined structures of antibody–antigen complexes can be clustered accurately (AUC 0.981) and that use of predicted structural features improved the accuracy of the epitope classification. We next show that an independent and non-redundant set of 104 anti-HIV antibody sequences could be clustered corresponding to manually-assigned epitopes with a specificity of 99.7% and a sensitivity of 61.93%, with the imbalance in sensitivity due almost entirely to one group of antibodies—those that target the gp120 V3 loop, which do not form a single, well-defined cluster. We next examined a diverse set of anti-hemagglutinin BCR sequences from humans and mice. We observed clusters that included human or mouse sequences with anti-hemagglutinin antibodies of known structure. We also observed clusters that included both human and mouse sequences. Importantly, to the extent that the epitopes have been experimentally characterized, none of the observed clusters erroneously grouped different hemagglutinin binding regions. Taken together, these results demonstrate that the proposed clustering method provides high-throughput prediction of BCRs with common binding specificity across clonal lineages, donors and even species.
Design, System, ApplicationComputational identification of B cell receptor (BCR) antigen and epitope specificity is currently an open problem. Previous work in this area has utilized antibody-epitope docking in order to predict antibody-specific epitopes. However, such methods are currently very computationally intensive and have low precision. Here, we employ a complimentary strategy: instead of attempting to dock an antibody to an antigen, we ask “are two antibodies likely to target the same antigen and epitope?” Our approach to answering this question utilizes a similarity function optimized for classifying known antibodies according to their antigen and epitope specificity. We demonstrate robust and highly specific clusters built from models using our companion server, Repertoire Builder. The resulting clusters can simplify downstream analysis for researchers working on large-scale BCR repertoire datasets from multiple lineages, donors or even species. Such clustering may also be useful for identifying disease-specific BCRs. Such BCRs may, in turn, be useful as disease biomarkers or, in some cases, as novel antibody-based therapeutics. |
Introduction
In humans, a diverse B cell receptor (BCR) repertoire is generated by rearrangement of receptor gene segments, followed by deletion or expansion of BCR lineages in response to self- or non-self-antigens. The observed repertoire of circulating BCRs in a given donor constitutes a record of past and present antigen exposure. Because infection, cell damage or genetic abnormalities can elicit antigen-driven expansion of specific B cell lineages, BCRs represent a potentially sensitive and abundant class of disease biomarkers.1 Moreover, due to their high stability, affinity and specificity, the soluble forms of BCRs (antibodies) can be engineered into antigen-targeted therapeutics.2 The depth and coverage of lymphocyte receptor sequencing has undergone a major inflection in recent years due to technological advances in cell isolation and molecular barcoding.3 There is thus a strong motivation to develop computational methods that can leverage sequence and structural data in order to predict the targeted antigen and epitope of a given BCR. Such methods would be highly beneficial for defining BCRs with shared binding specificity across donors, and may contribute to the development of diagnostic or therapeutic antibodies.To date, much of the work on BCR sequence analysis has focused on sequence variations within lineages (i.e., B cells that have descended from a common ancestor).4 Lineage analysis is an effective means of identifying dominant clones and somatic hypermutation (SHM) events, which can correlate with antigen affinity and thus provide clues to the characteristics of the epitope and binding residues on the BCR.5 Much less is known about how different lineages are functionally related. Structural and sequence similarity in BCRs arising from different clonal lineages that target common antigens and epitopes have been observed in anti-HIV and anti-influenza antibodies.6,7 In these studies, structural analysis was carried out using X-ray crystallography. However, because crystallographic analysis of antibody–antigen complexes typically requires months of human effort at a high financial cost, this approach cannot be expected to scale with emerging high-throughput paired (heavy–light chain) sequencing methods, which currently yield thousands or more sequences in a single experiment. In contrast, BCR structural modeling methods can scale well with large sequence datasets. For example, Marcatili et al. examined clusters of structural models of BCR sequences from chronic lymphocytic leukemia patients using the PIGS modeling software;8 DeKosky et al. carried out a large-scale study of naive and antigen-experienced BCRs using the Rosetta Antibody software;9 Kovaltsuk et al. developed an online resource to store 3D models of numerous BCRs, including several datasets acquired post-vaccination, using ABodyBuilder;10 more recently, Raybould et al. developed a Therapeutic Antibody Profiling score derived from sequence and 3D models built using ABodyBuilder.11 In this issue, we describe our own software, Repertoire Builder (https://sysimm.org/rep_builder/), which can model BCRs efficiently and accurately.31 3D modeling may, therefore, enable observation of structural convergence in BCRs, just as it has been used to infer distant homology in structural genomics studies.12
Several groups have investigated the use of BCR modeling followed by antigen docking to infer BCR conformational epitopes.13,14 These approaches are a logical extension of BCR sequencing in cases where the antigen is known. However, antibody docking is not currently a high-throughput technique, and care must be taken in extracting structural features from BCR models, as the errors are usually highest in the complementarity determining regions (CDRs), which typically mediate interactions with antigens.15 Moreover, the target antigens are not known for the vast majority of BCRs that have been sequenced to date.
Here, we propose an alternative approach for predicting BCR antigen/epitope specificity that is robust to errors in structural modeling, computationally efficient enough for high-throughput sequence analysis and does not require a priori knowledge of the target antigen. Rather than explicitly computing BCR–antigen interactions (e.g. by docking), we attempt to predict whether two or more BCRs are likely to target the same epitope. In order to address this question, we first construct a feature vector describing sequence and structural similarities for a pair of BCRs. Notably, this feature vector does not depend on the antigens themselves, only the BCRs. We then derive a BCR similarity score by training a support vector machine (SVM)-based classifier on feature vectors derived from a set of BCRs with known antigen-bound structures. Finally, we carry out hierarchical clustering based on the pairwise BCR similarity score. Because the feature vector considers sequence and structural features for each CDR region independently, the clustering is robust against modelling errors and can detect functional similarities between BCRs arising from different clonal lineages or even different species. The resulting clusters may be beneficial in comparing the repertoires of different donors or in prioritizing BCR sequences for further experimental analysis.
Methods
BCR notation
We defined segments in each BCR according to the AHo numbering scheme16 as follows:CDR1 (25–40), CDR2 (58–77), CDR3 (109–137), framework (1–24, 41–57, 78–108, 138–149), conserved framework (3–7, 20–24, 41–47, 51–57, 78–82, 89–93, 102–108, 138–144). The ANARCI software17 was used to number BCRs according to the AHo numbering scheme.
Structural alignment
Structural superposition of a pair of BCR receptors was carried out by minimizing the root-mean square deviation (RMSD) of C-alpha atoms in their conserved framework residues. From this superposition, a similarity matrix was computed for each pair of residues as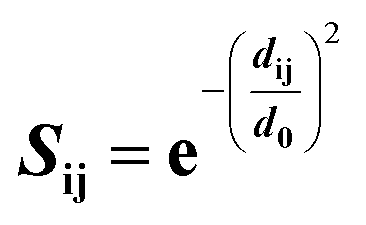 | (1) |
Feature vectors
Feature vectors for the SVM model were defined in terms of pairs of superimposed BCR structures. Given two structural models, 1 and 2, for each segment, we evaluated four quantities: sequence similarity, structural similarity, alignment length and length difference. If we denote an alignment a(k) = [i,j] within the segment, where k is the alignment index (column) and i and j are residue positions in models 1 and 2, the structural similarity was given by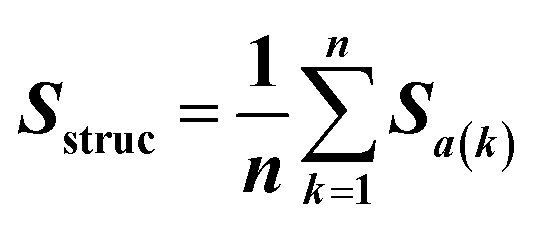 | (2) |
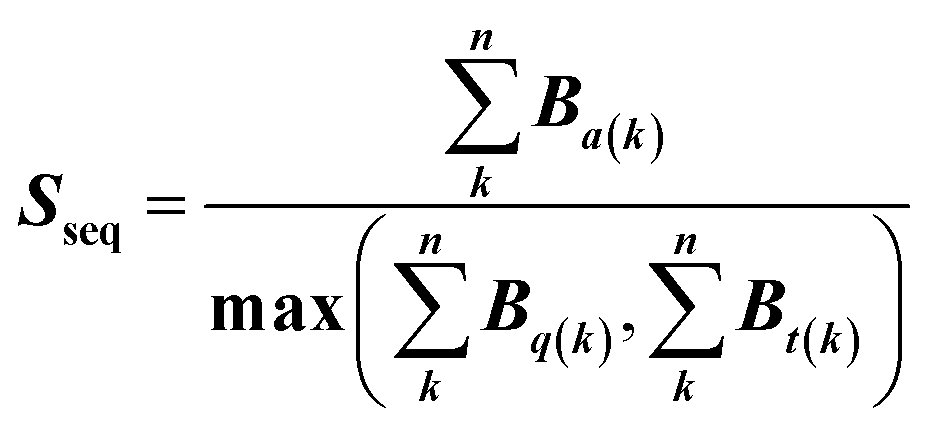 | (3) |
BCR datasets
Epitope-based clustering of PDB BCR entries for SVM training
BCR-antigen chain pairs collected from the PDB were first clustered according to their antigen protein sequences by using Cd-hit28 under a sequence identity cutoff of 40%. For antigen sequences within each cluster, a multiple-sequence alignment (MSA) was built using MAFFT,28 and epitope residues in BCR contact were identified. Epitope residues were defined as those within 3.5 Å of the contacting BCR chain with an accessible surface area (ASA) that was reduced upon BCR binding. Clustered antigen chains were further grouped by their epitope residues. For any two antigens in a cluster, if their epitope residues overlapped, structural alignment was carried out using overlapping epitope residues. If the RMSD between aligned epitope residues was less than 5.0 Å, the two antigens were grouped together. After grouping antigens by their epitope residues, pairwise RMSDs of pairs of bound BCRs were computed by first superimposing the two epitopes, then evaluating the BCR RMSD using pairs of BCR residues identified by sequence alignment. For each epitope-based cluster, average linkage hierarchical clustering was performed using the matrix of BCR RMSDs at a cutoff of 5.0 Å. The resulting clusters of complexes, were composed of similar epitopes with similar BCR binding modes.SVM-based classifier with hierarchical clustering
To train a binary SVM classifier to predict if two BCRs target a common epitope, sequence redundancy within and among epitope-based PDB BCR clusters was first reduced by Cd-hit at a sequence identity cutoff of 90%. Positive samples were collected from a non-redundant set of 138 BCRs in the PDB (Table S1†) for which at least two BCRs targeted the same epitope, as defined above. To this set were added 136 3D models built from the corresponding variable region sequences in order to introduce a realistic level of noise to the training data (two models could not be built by Repertoire Builder because the modeling software uses a stricter criterion for template selection than was used for training/testing data in building the clustering training/testing data.) The purpose for adding models was to train the SVM on data where the most difficult regions (e.g. CDR H3) would exhibit a level of noise that would resemble a real-case use scenario. BCR pairs targeting different epitopes were used as negative samples. Due to the unbalanced number of positive and negative samples, classes were assigned weights inversely proportional to class frequencies in the training data. A radial basis function (RBF) kernel was used for training and optimizing hyperparameters by use of stratified 5-fold cross-validation (CV) as implemented in the Python scikit-learn package.29 Each 5-fold validation was seeded with a different random number in order to generate different training and testing subsets. This process was repeated 5 times in order to assess the stability of the classifier stratified using different data partitions. Here, all testing was carried out using modeled BCRs built such that templates having 90% or more sequence identity to the query were blacklisted in order to simulate a realistic scenario where experimentally determined structures would not be available. A similar performance was obtained when testing was done using Repertoire Builder models built such that templates were blacklisted at 80% sequence identity (Fig. S2D†). Prediction scores returned by CV for all possible BCR pairs in the training data were used to predict clusters by average-linkage hierarchical clustering and a decision threshold, above which two BCRs compared were considered as similar. By varying the decision threshold, the adjusted Rand index30 between reference and predicted clusters was maximized.Structural modeling
BCR structural modeling was carried out using Repertoire Builder31 with default options except where template blacklisting was used, as mentioned in the text (https://sysimm.org/rep_builder/). To analysis and visualize the cluster results, the PyMOL was used (the PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.).ELISA assays
Nunc Maxisorp Immuno plates (Thermo Scientific) were coated with streptavidin (Funakoshi, Japan) in PBS at a concentration of 10 U mL−1. Blocking was carried out using Blocking One solution (Nacalai Tesque, Japan). Plates were sequentially incubated with biotinylated rHA (100 ng/50 μL), sample antibodies (0.5 ng/50 μL) and goat anti-human IgG-HRP (Southern Biotech). Detection was carried out using KPL SureBlue TMB Microwell Peroxidase Substrate (SeraCare) and the reaction was stopped by 1 N HCl. OD450 was read with ARVO X3 (PerkinElmer).Results
SVM training and validation
An SVM was trained to predict whether a given pair of BCRs, share the same antigen and epitope specificity. For SVM training, a set of BCRs with known antigen binding mode was assembled as described in Methods. This resulted in 1090 “True” BCR pairs (i.e. pairs that target the same antigen and epitope) and 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 817 “False” pairs (i.e. pairs that target a different antigen or epitope).
817 “False” pairs (i.e. pairs that target a different antigen or epitope).
The SVM-based classifier was assessed using repeated 5-fold cross-validation (CV). Receiver operating characteristic (ROC) curves for each run (Fig. 1A and B) yielded an average area under the curve (AUC) of 0.981 ± 0.001. The reproducibility of the different CV runs, along with the relatively small size of the feature vector (32 elements) in comparison with the number of unique structures (138), suggests that the SVM model was not overfit. By converting the raw SVM score into a distance, and establishing a threshold for this similarity in hierarchical clustering, we could represent non-singleton clusters either as trees or as networks of BCRs whose similarities fall above the threshold and thus are predicted to target a common antigen and epitope (Fig. S1†). The details are described in Methods.
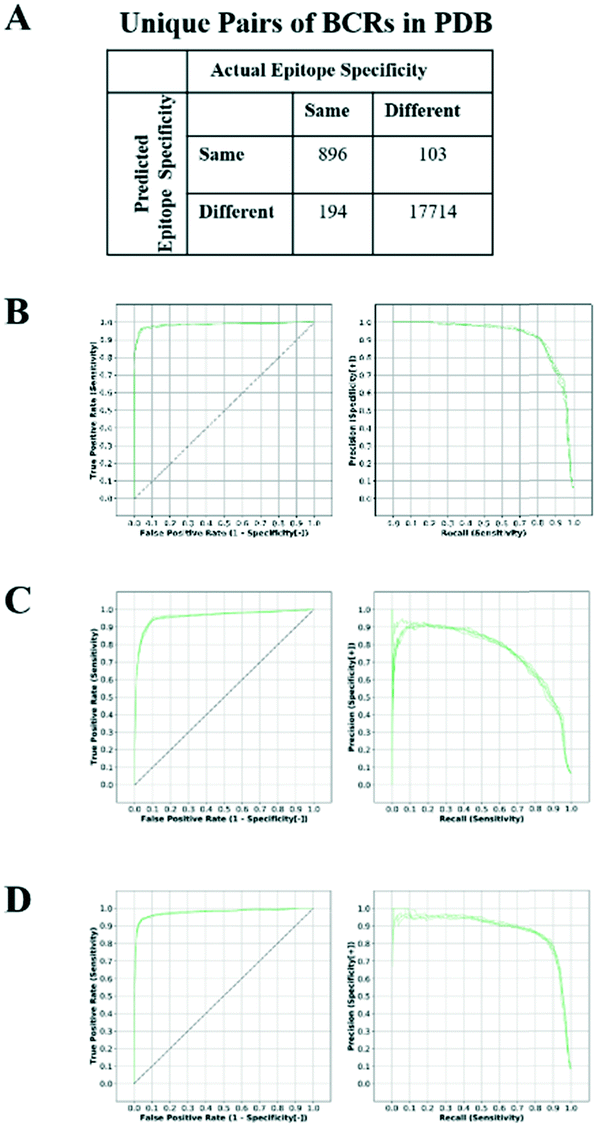 | ||
| Fig. 1 Performance of SVM model on BCRs with known antigen complex structure. A) Confusion matrix showing the numbers of predicted and actual epitope pairs after clustering. The ROC and precision–recall curves were computed using Repertoire Builder models built with a template sequence identity blacklist of 90% for paired (B), heavy chain only (C) and light chain only (D) BCR models. | ||
Contribution of feature vector terms
The F1 score, defined as the harmonic mean of precision (true-positive/predicted-positive) and sensitivity (true-positive/positive), was computed for the models using each feature independently, which gives a rough measure of the information content of the feature of interest (Table 1). From this result, it can be inferred that, on their own, the sequence features contain more information than the structural features.| H1 | H2 | H3 | HFW | L1 | L2 | L3 | LFW | |
|---|---|---|---|---|---|---|---|---|
| Seq | 0.069 | 0.210 | 0.260 | 0.138 | 0.070 | 0.102 | 0.399 | 0.029 |
| Struc | 0.022 | 0.095 | 0.117 | 0.036 | 0.020 | 0.001 | 0.051 | 0.005 |
| LDiff | 0.009 | 0.013 | 0.027 | 0.001 | 0.019 | 0.001 | 0.024 | 0.004 |
| ALen | 0.005 | 0.002 | 0.001 | 0.002 | 0.031 | 0.001 | 0.183 | 0.001 |
One of the surprising observations was that the performance for the third light chain CDR (CDRL3) sequence term (0.399) was greater than that of the third heavy chain CDR (CDRH3; 0.260). Typically, single-chain BCR sequencing studies have focused only on the heavy chain.32,33 We examined SVM models built on only heavy or light chains and found the resulting AUCs were 0.960 (Fig. 1C) and 0.977 (Fig. 1D), respectively. In particular, the precision curve for SVMs trained only on heavy chains dropped sharply beyond a recall value of 0.5. The higher performance on paired BCR models reinforces the importance of paired sequencing technologies. In contrast to the trend observed in the sequence terms, performance for the CDRL3 structure term (0.051) was roughly half of that of CDRH3 (0.117), supporting the importance of accurate CDRH3 structural modeling. In order to further quantify the relative importance of the sequence and structural features, we constructed SVM models using only BCR sequences, BCR sequences with native structural information or BCR sequences with modeled structural information. The resulting AUCs were 0.974 ± 0.002, 0.984 ± 0.001 and 0.983 ± 0.001, respectively (Fig. S2A–C†). These results confirm that, although most of the information in the classifier is contained within the sequence features, structural information does contribute to the overall accuracy of the classifier and this is most evident in the precision–recall curves. When noisier (template blacklist 80%) Repertoire Builder models were used for testing, the benefit of using modeled structures in training was clearer (Fig. S2D†). Therefore, in the end, a classifier trained with a heterogeneous training set containing perfect (PDB entries) and imperfect (modeled) structures was selected in order to include noise from modeled BCRs.
Validation on an independent set of anti-HIV antibody sequences
The diverse sequence and structural space of anti-HIV antibodies and their associated epitopes has been intensively studied.34 The CATNAP database contains several hundred anti-HIV antibodies targeting 16 classes of epitopes.35 We reduced this set down to 104 non-redundant paired heavy–light chain variable domain sequences with less than 95% sequence identity to known structures or to other sequences in the set. 3D modeling by Repertoire Builder, followed by clustering revealed a high degree of agreement with CATNAP-annotated epitopes (Fig. 2). After removing singleton clusters, we observed 373 BCR pairs with the same epitope annotations (“Positives”), and 1643 BCR pairs with different epitope annotations (“Negatives”). Out of the 1643 Negatives, 1638 pairs were correctly predicted with an overall specificity (true-negative/negative) of 99.70%. Of the 373 positives 231 pairs were correctly predicted with an overall sensitivity (true-positive/positive) of 61.93%. This imbalance was due primarily to the fact that antibodies targeting the V3 loop region in the HIV gp120 envelope protein did not form a single cluster. In fact, 85% of false-negative predictions involved anti-V3 antibodies. | ||
| Fig. 2 Comparison between predicted and CATNAP anti-HIV epitope assignments. A) Confusion matrix showing the numbers of predicted and actual epitope pairs. B) Tree representation of BCR clusters. With the exception of the diagonal, the colors of intersecting squares (red/blue) indicate matched/ mismatched CATNAP epitope annotations. Black arrows in the tree leaves indicate “gp120 V3 loop-binding” annotated antibodies. | ||
We next prepared two sets of experimentally-determined BCRs: PDB BCRs, the “HIV Group” and the “Control Group”, as described in Methods. We then computed clusters for the 104 CATNAP models with the HIV and control groups, and counted the number of PDB entries from each group that clustered with CATNAP models. For the HIV Group, we observed 48 PDB entries that clustered with CATNAP models (“True-Positives”) and 56 entries that did not (“False-Negatives”). In contrast, for the control group, we observed 13 PDB entries that clustered with CATNAP models (“False-Positives”) and 580 entries that did not (“True-Negatives”). From these values we could obtain a specificity 97.80% of and a sensitivity of 46.17%, which qualitatively agrees with the analysis of the CATNAP epitope annotations above.
Analysis of anti-hemagglutinin mouse BCR sequences
Influenza hemagglutinin (HA) is a trimeric molecule consisting of a membrane-proximal stem region, a solvent-exposed head region and a joint region lying in between the head and the stem regions. The head region is more polymorphic and immunogenic, while the more conserved stem region is the target of broadly neutralizing anti-flu antibodies.6 Based on known anti-HA crystal structures, nearly every surface of the antigen is a potential epitope (Fig. 3).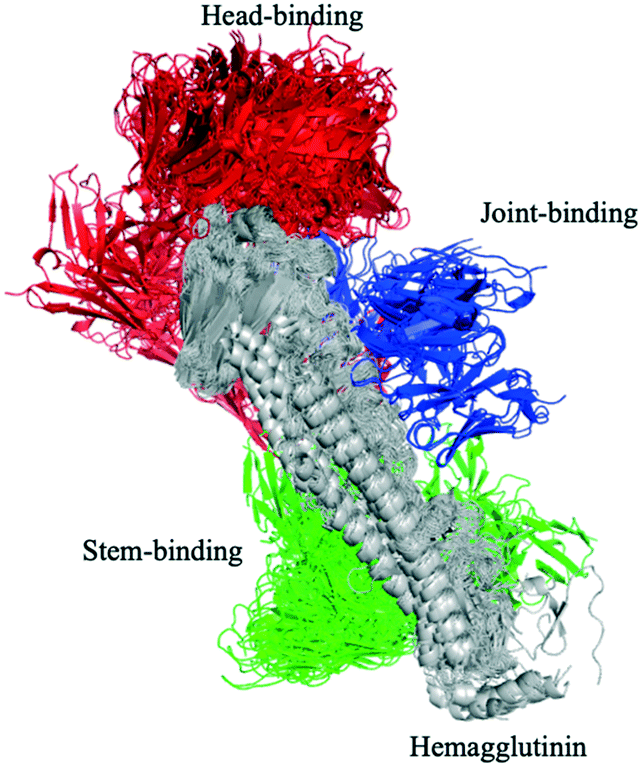 | ||
| Fig. 3 Known antibody structure cover surface of hemagglutinin (HA). HA is a symmetric trimer and the structures have been rotated into a common frame allowing the HA head, joint and stem regions to be visible. The anti-HA antibodies that bind with different epitope of HA are annotated as head-binding (red), joint-binding (blue) and stem-binding (green) respectively. | ||
As an independent test set we utilized 31 mouse anti-HA BCR lineage-representative sequences, none of which displayed obvious (>90%) sequence identity to known PDB entries as described in Methods. By combining the results of ELISA assays using stem-only36 and full-length HA, we assigned each mouse antibody to a “stem-binding” (i.e. binding to both the stem-only and full-length HA probes) and “full-length-binding” (i.e., those binding to only the full-length HA probe) epitope class. 3D models built from these sequences yielded a total of 3 non-singleton clusters: one cluster containing 11 full-length-binding sequences, one containing 3 full-length-binding sequences and one containing 2 stem-binding sequences. These results are consistent with the high specificity (low false-positive rate) and low sensitivity (high false-negative rate) of the proposed clustering method (Fig. 4A). Five clones (170105-025, 170105-032, 170105-034, 170105-056, 170105-082) which are depicted as light green circles without a black outline, were predicted as full-length-binding. The clones that are depicted as dark green circles with a black outline were full-length-binding, as confirmed by the ELISA assay (Fig. 4B). Subsequently, ELISA assays were performed and the five clones predicted to target to full-length HA (Fig. 4C) rather than stem HA (Fig. 4D), were validated.
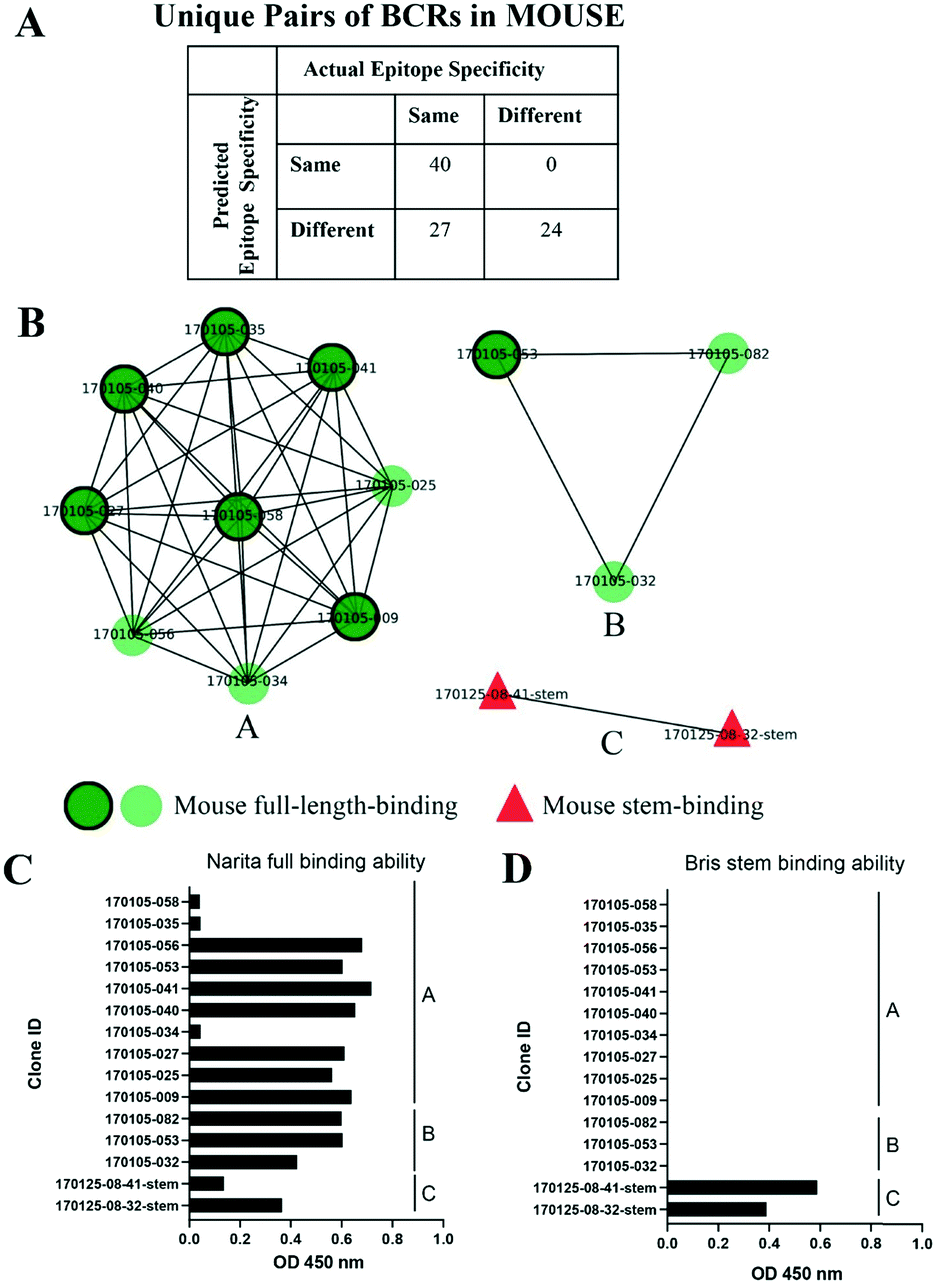 | ||
| Fig. 4 Clusters of mouse post-vaccination BCRs. A) Confusion matrix showing the numbers of predicted and actual epitope pairs of mouse post-vaccination BCRs. B) Clusters of mouse post-vaccination BCRs. Clusters were represented as networks that Repertoire Builder models were characterized by their ELISA-based annotations as stem-binding (red triangles) or full-length-binding (dark green circles with a black outline or light green circles without a black outline). Clusters are differentially labeled by alphabet. C/D) ELISA to detect the binding ability of full-length (C) or stem (D) HA protein of antibodies. | ||
We next prepared two PDB sets as with the HIV analysis above. These consisted of 40 non-redundant anti-HA antibody sequences (“HA Group”) with known structure and 662 antibody sequences with antigens unrelated to HA (“Control Group”). We computed clusters for mouse 3D models with the two groups of structures and counted the number of PDB entries in each group that clustered with anti-HA mouse models. For the HA Group, we observed 2 PDB entries that clustered with mouse models (“True-Positives”) and 38 entries that did not (“False-Negatives”). In contrast, for the Control Group, we observed 14 PDB entries that clustered with mouse models (“False-Positives”) and 648 entries that did not (“True-Negatives”). From these values we could obtain a specificity 97.88% and a sensitivity of only 5%. One explanation for the very low sensitivity observed here is that the known anti-HA PDB entries do not cover the actual mouse anti-HA repertoire, at least to a degree required by the SVM similarity score. A second issue is that the distinction of “stem-binding” and “full-length-binding” based only on ELISA assays is not very precise or quantitative. On the other hand, an encouraging observation is that the two clusters that did form—a stem-binding anti-HA PDB entry (4nm8) that clustered with a stem-binding mouse model and a head-binding anti-HA PDB entry (4hg4) that matched with a full-length binding mouse model were consistent with experimental results.
Analysis of human BCR sequences acquired post flu-vaccination
We next analyzed a set of 8986 models built from human BCR lineage representative sequences acquired post flu vaccination using various seasonal flu vaccines, as described in Methods. 3D modeling followed by clustering resulted in a total of 1276 non-singleton clusters, 125 (9.8%) of which contained cells derived from multiple donors. The number of clusters as a function of cluster size, s, was well-approximated by an exponential function 35589e−1.748s (Fig. S3†). If this distribution is general for other large-scale sequence datasets it may help to identify clusters that are over-represented. This set of BCRs was derived from both B cells and plasmablasts, the latter of which are expected to be enriched in cells responding to the vaccine.37 Our initial hypothesis was that, since the plasmablasts are mostly expected to be vaccine-responsive, the distribution of the SVM similarity scores would be skewed to higher values than that of the B cells. However, although there were a small number of high-scoring pairs of plasmablast-derived BCRs, we did not observe a significant difference in the cumulative distribution of similarity scores for pairs of plasmablasts compared to pairs of BCRs (Fig. S4†).We next examined the clustering of the human BCR sequences with anti-HA antibody structures from known PDB entries. We observed a total of 13 clusters comprised of a total of 15 human BCRs and at least one PDB anti-HA antibody: 5 clusters consisting of 6 human sequences with 5 anti-head PDB entries, 2 clusters of human sequences with 2 anti-joint PDB entries and 6 clusters consisting of 7 human sequences with 9 anti-stem PDB entries (Fig. 5). Then, we checked each cluster by structural visualization using PyMOL. In each cluster, human BCRs overlapped well with the PDB entries (Fig. S5†). As with the mouse data, we observed that anti-HA antibodies in the PDB did not cover the sequence or structural space of these naturally occurring human BCRs.
 | ||
| Fig. 5 Clusters containing human and known anti-HA PDB entries. Clusters (labeled A to M) were represented as networks and PDB entries were characterized by their HA-binding modes as head (red circles), joint (blue squares) or stem (green triangles). Human BCRs are shown as yellow diamonds. | ||
We next examined the clustering of the human BCR sequences with the mouse anti-HA models. We observed 13 clusters between human and mouse BCRs (Fig. 6), comprised of a total of 22 human BCRs. Here, 8 of the clusters contained full-length mouse models and 5 contained stem models. Similarly, clusters were also checked by structural visualization (Fig. S6†). Human BCRs were overlapped with mouse BCRs well in accordance with the prediction results.
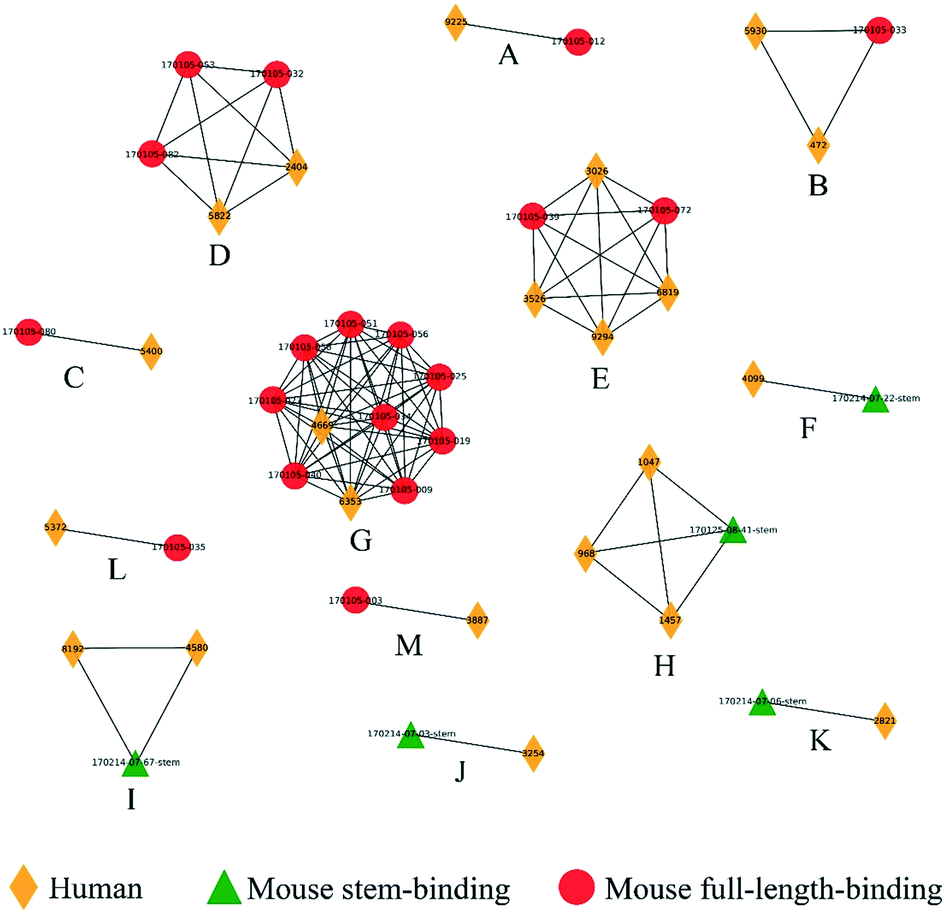 | ||
| Fig. 6 Clusters composed of human and mouse BCRs obtained post flu vaccination. Clusters (labeled A to M) were represented as networks and human, mouse stem-binding and mouse full-length-binding BCRs were represented by yellow diamonds, red circles and green triangles, respectively. | ||
In summary, although the number of clusters containing human and known anti-HA BCRs was rather low, none of the observed clusters was inconsistent with the available experimental information, in spite of the fact that neither the mouse nor the human data were used in training the classifier. These results demonstrate that SVM score is sensitive enough to detect similarity between BCRs from difference species.
Conclusions
In order to sustain their biochemical functions, proteins experience strong evolutionary pressure to maintain their three-dimensional structures, leading to the well-known paradigm that structure is more conserved than sequence. As proteins, whose main function is molecular recognition, BCRs are also subjected to strong sequence and structural constraints. However, because the process by which BCRs evolve is so unique among proteins (i.e. new protein sequences are continuously generated during the lifetime of the host through combinatorial assembly of genes followed by somatic hypermutation), it is not clear how the relative importance of sequence and structural features contribute to the overall fitness of a particular BCR. Moreover, since sequence and structural variation is generally concentrated in a very local part of the protein (i.e., the CDRs), it is not clear how sequence and structural similarity should be quantified in a way that reflects functional similarity.Here, we have chosen to use the conserved framework residues as a common reference frame, and to define BCR similarity in terms of a set of pairwise features, which describe sequence, structure and length. We first show that, given a mixture of experimentally-determined antibody crystal structures and models exhibiting a realistic level of noise, we can cluster models of BCRs with known antigens and epitopes with an AUC of 0.981. Moreover, we find that, although sequence features are the most information-rich, removal of either the crystal structures, the models, or both, results in a degradation of the clustering performance.
The high accuracy of the clustering is an important proof of concept, but is not a realistic test of the performance of the methodology in a real-world setting using high-throughput sequencing of BCR repertoires. However, the large-scale datasets that have been published to date do not contain functional annotations on the targeted antigens and epitopes. Therefore, we next examined BCR sequence datasets that contain partial annotations. In the CATNAP dataset, epitopes were taken from standard definitions of neutralizing antibodies rather than precise residue positions. In the set of mouse-derived BCRs, the binding to HA stem or full-length regions was determined using a rather non-quantitative binding assay (ELISA). In the much larger set of human-derived BCRs obtained post vaccination, the sequences were simply expected to be enriched in anti-HA binders, but HA-binding was not confirmed experimentally. By clustering different combinations of data sets we were able to assess whether BCRs annotated as HA head-, stem- or joint-binders formed clusters that contradicted any known epitope annotations. In spite of the fact that only PDB-derived data was used in the training of the SVM, no spurious clusters were observed. On the other hand, broad classes of epitopes, such as the gp120 V3 loop or HA head/stem designations do not necessarily cluster together. This situation leads to an apparent imbalance in specificity (high) and sensitivity (low). The most straightforward remedy for this situation is to increase the amount of training data. However, since the throughput of experimentally-determined structures is unlikely to change qualitatively in the near future, we must consider other types of training data that can scale with high-throughput BCR sequencing methods. One approach is to utilize multiplex immunoassays, which yield complimentary information to residue-level epitope information. In spite of these limitations, the current results provide evidence that the SVM model is not overfit and is robust. In the future we aim to experimentally validate the clusters in order to measure the actual true/false positive rates. Potential extensions of the proposed clustering method include BCR conformational epitope prediction, antibody–antigen docking and clustering of T cell receptors (TCRs). It is our hope that, together, these methods will contribute to the discovery of diagnostic and therapeutic lymphocyte receptors.
Conflicts of interest
S. L and D. M. S own equity in KOTAI Biotechnologies Inc. K. Y is an employee of and owns equity in KOTAI Biotechnologies Inc.Acknowledgements
We would like to thank all members of the Systems Immunology Lab for helpful discussions. We would also like to thank Ken J. Ishii, Masatoshi Momota, Takuya Yamamoto, Yoshimasa Takahashi, Yu Adachi, Hidehiro Fukuyama, and Chie Kawai for their assistance with mouse BCR analysis. This research was supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number 17am0101108j0001.References
- H. H. Wandall, O. Blixt, M. A. Tarp, J. W. Pedersen, E. P. Bennett, U. Mandel, G. Ragupathi, P. O. Livingston, M. A. Hollingsworth, J. Taylor-Papadimitriou, J. Burchell and H. Clausen, Cancer Res., 2010, 70, 1306–1313 CrossRef CAS PubMed.
- R. Lepore, P. P. Olimpieri, M. A. Messih and A. Tramontano, Nucleic Acids Res., 2017, 45(W1), W17–W23 CrossRef CAS PubMed.
- S. Friedensohn, T. A. Khan and S. T. Reddy, Trends Biotechnol., 2017, 35, 203–214 CrossRef CAS PubMed.
- G. Yaari and S. H. Kleinstein, Genome Med., 2015, 7, 121 CrossRef PubMed.
- S. Sakakibara, T. Arimori, K. Yamashita, H. Jinzai, D. Motooka, S. Nakamura, S. Li, K. Takeda, J. Katayama, M. A. El Hussien, M. Narazaki, T. Tanaka, D. M. Standley, J. Takagi and H. Kikutani, Sci. Rep., 2017, 7, 16428 CrossRef PubMed.
- M. G. Joyce, A. K. Wheatley, P. V. Thomas, G. Y. Chuang, C. Soto, R. T. Bailer, A. Druz, I. S. Georgiev, R. A. Gillespie, M. Kanekiyo, W. P. Kong, K. Leung, S. N. Narpala, M. S. Prabhakaran, E. S. Yang, B. Zhang, Y. Zhang, M. Asokan, J. C. Boyington, T. Bylund, S. Darko, C. R. Lees, A. Ransier, C. H. Shen, L. Wang, J. R. Whittle, X. Wu, H. M. Yassine, C. Santos, Y. Matsuoka, Y. Tsybovsky, U. Baxa, N. C. S. Program, J. C. Mullikin, K. Subbarao, D. C. Douek, B. S. Graham, R. A. Koup, J. E. Ledgerwood, M. Roederer, L. Shapiro, P. D. Kwong, J. R. Mascola and A. B. McDermott, Cell, 2016, 166, 609–623 CrossRef CAS PubMed.
- J. F. Scheid, H. Mouquet, B. Ueberheide, R. Diskin, F. Klein, T. Y. Oliveira, J. Pietzsch, D. Fenyo, A. Abadir, K. Velinzon, A. Hurley, S. Myung, F. Boulad, P. Poignard, D. R. Burton, F. Pereyra, D. D. Ho, B. D. Walker, M. S. Seaman, P. J. Bjorkman, B. T. Chait and M. C. Nussenzweig, Science, 2011, 333, 1633–1637 CrossRef CAS PubMed.
- E. Polychronidou, I. Kalamaras, A. Agathangelidis, L. A. Sutton, X. J. Yan, V. Bikos, A. Vardi, K. Mochament, N. Chiorazzi, C. Belessi, R. Rosenquist, P. Ghia, K. Stamatopoulos, P. Vlamos, A. Chailyan, N. Overby, P. Marcatili, A. Hatzidimitriou and D. Tzovaras, BMC Bioinf., 2018, 19, 414 CrossRef CAS PubMed.
- B. J. DeKosky, O. I. Lungu, D. Park, E. L. Johnson, W. Charab, C. Chrysostomou, D. Kuroda, A. D. Ellington, G. C. Ippolito, J. J. Gray and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2636–2645 CrossRef CAS PubMed.
- A. Kovaltsuk, J. Leem, S. Kelm, J. Snowden, C. M. Deane and K. Krawczyk, J. Immunol., 2018, 201, 2502–2509 CrossRef CAS PubMed.
- M. I. J. Raybould, C. Marks, K. Krawczyk, B. Taddese, J. Nowak, A. P. Lewis, A. Bujotzek, J. Shi and C. M. Deane, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4025–4030 CrossRef CAS PubMed.
- J. M. Chandonia and S. E. Brenner, Science, 2006, 311, 347–351 CrossRef CAS PubMed.
- J. Dunbar, K. Krawczyk, J. Leem, C. Marks, J. Nowak, C. Regep, G. Georges, S. Kelm, B. Popovic and C. M. Deane, Nucleic Acids Res., 2016, 44, W474–478 CrossRef CAS PubMed.
- B. D. Weitzner, J. R. Jeliazkov, S. Lyskov, N. Marze, D. Kuroda, R. Frick, J. Adolf-Bryfogle, N. Biswas, R. L. Dunbrack, Jr. and J. J. Gray, Nat. Protoc., 2017, 12, 401–416 CrossRef CAS PubMed.
- J. C. Almagro, A. Teplyakov, J. Luo, R. W. Sweet, S. Kodangattil, F. Hernandez-Guzman and G. L. Gilliland, Proteins, 2014, 82, 1553–1562 CrossRef CAS PubMed.
- A. Honegger and A. Pluckthun, J. Mol. Biol., 2001, 309, 657–670 CrossRef CAS PubMed.
- J. Dunbar and C. M. Deane, Bioinformatics, 2016, 32, 298–300 CAS.
- O. Gotoh, J. Mol. Biol., 1982, 162, 705–708 CrossRef CAS PubMed.
- S. B. Needleman and C. D. Wunsch, J. Mol. Biol., 1970, 48, 443–453 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- J. Dunbar, K. Krawczyk, J. Leem, T. Baker, A. Fuchs, G. Georges, J. Shi and C. M. Deane, Nucleic Acids Res., 2014, 42, D1140–1146 CrossRef CAS PubMed.
- Y. Matsuzaki, K. Sugawara, M. Nakauchi, Y. Takahashi, T. Onodera, Y. Tsunetsugu-Yokota, T. Matsumura, M. Ato, K. Kobayashi, Y. Shimotai, K. Mizuta, S. Hongo, M. Tashiro and E. Nobusawa, J. Virol., 2014, 88, 12364–12373 CrossRef PubMed.
- T. Tiller, C. E. Busse and H. Wardemann, J. Immunol. Methods, 2009, 350, 183–193 CrossRef CAS PubMed.
- L. von Boehmer, C. Liu, S. Ackerman, A. D. Gitlin, Q. Wang, A. Gazumyan and M. C. Nussenzweig, Nat. Protoc., 2016, 11, 1908–1923 CrossRef CAS PubMed.
- K. Katoh and D. M. Standley, Mol. Biol. Evol., 2013, 30, 772–780 CrossRef CAS PubMed.
- Y. C. Tan, L. K. Blum, S. Kongpachith, C. H. Ju, X. Cai, T. M. Lindstrom, J. Sokolove and W. H. Robinson, Clin. Immunol., 2014, 151, 55–65 CrossRef CAS PubMed.
- J. DeFalco, M. Harbell, A. Manning-Bog, G. Baia, A. Scholz, B. Millare, M. Sumi, D. Zhang, F. Chu, C. Dowd, P. Zuno-Mitchell, D. Kim, Y. Leung, S. Jiang, X. Tang, K. S. Williamson, X. Chen, S. M. Carroll, G. Espiritu Santo, N. Haaser, N. Nguyen, E. Giladi, D. Minor, Y. C. Tan, J. B. Sokolove, L. Steinman, T. A. Serafini, G. Cavet, N. M. Greenberg, J. Glanville, W. Volkmuth, D. E. Emerling and W. H. Robinson, Clin. Immunol., 2018, 187, 37–45 CrossRef CAS PubMed.
- W. Li and A. Godzik, Bioinformatics, 2006, 22, 1658–1659 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- L. Hubert and P. Arabie, J. Classif., 1985, 2, 193–218 CrossRef.
- D. Schritt, S. Li, J. Rozewicki, J. Billaud, J. Nieri, K. Katoh, K. Yamashita, W. Volkmuth, G. Cavet and D. M. Standley, Mol. Syst. Des. Eng., 2019 10.1039/c9me00020h.
- B. Briney, A. Inderbitzin, C. Joyce and D. R. Burton, Nature, 2019, 566, 393–397 CrossRef PubMed.
- F. Horns, C. Vollmers, C. L. Dekker and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1261–1266 CrossRef CAS PubMed.
- A. P. West, Jr., L. Scharf, J. F. Scheid, F. Klein, P. J. Bjorkman and M. C. Nussenzweig, Cell, 2014, 156, 633–648 CrossRef PubMed.
- H. Yoon, J. Macke, A. P. West, Jr., B. Foley, P. J. Bjorkman, B. Korber and K. Yusim, Nucleic Acids Res., 2015, 43, W213–219 CrossRef CAS PubMed.
- A. Impagliazzo, F. Milder, H. Kuipers, M. V. Wagner, X. Zhu, R. M. Hoffman, R. van Meersbergen, J. Huizingh, P. Wanningen, J. Verspuij, M. de Man, Z. Ding, A. Apetri, B. Kukrer, E. Sneekes-Vriese, D. Tomkiewicz, N. S. Laursen, P. S. Lee, A. Zakrzewska, L. Dekking, J. Tolboom, L. Tettero, S. van Meerten, W. Yu, W. Koudstaal, J. Goudsmit, A. B. Ward, W. Meijberg, I. A. Wilson and K. Radosevic, Science, 2015, 349, 1301–1306 CrossRef CAS PubMed.
- J. Wrammert, K. Smith, J. Miller, W. A. Langley, K. Kokko, C. Larsen, N. Y. Zheng, I. Mays, L. Garman, C. Helms, J. James, G. M. Air, J. D. Capra, R. Ahmed and P. C. Wilson, Nature, 2008, 453, 667–671 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00021f |
| ‡ Equal contribution. |
| § Current address: KOTAI Biotechnologies Inc, 3-1 Yamadaoka, Suita, Osaka 565-0871. |
| This journal is © The Royal Society of Chemistry 2019 |