
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterfacial complexation driven three-dimensional assembly of cationic phosphorus dendrimers and graphene oxide sheets†
Nadia
Katir
*a,
Anass
Benayad
*b,
Denis
Rouchon
c,
Nathalie
Marcotte
d,
Nabil
El Brahmi
a,
Jean Pierre
Majoral
e,
Mosto
Bousmina
a and
Abdelkrim
El Kadib
*a
aEuromed Research Center, Engineering Division, Euro-Mediterranean University of Fez (UEMF), Route de Meknes, 30000, Fès, Morocco. E-mail: a.elkadib@ueuromed.org
bCEA, LITEN, Department of Nanomaterials, MINATEC, 17 rue des martyrs, F-38054 Grenoble Cedex 09, France
cCEA, LETI, Department of Nanomaterials, Minatec Campus, F-38054 Grenoble Cedex 09, France
dInstitut Charles Gerhardt UMR 5253, CNRS/ENSCM/UM, 8 rue de l'Ecole Normale, Montpellier F-34295 Cedex, France
eLaboratoire de Chimie de Coordination (LCC) CNRS, 205 route de Narbonne, 31077 Toulouse, France
First published on 17th August 2018
Abstract
High content nitrogen, sulfur and phosphorus heteroatoms assembled in tree-like dendrimers (DGn) are confined within the galleries of two-dimensional graphene oxide (GO). The presence of the ternary diethyl-N-ethyl-ammonium groups on the dendrimer peripheries ensures the exfoliation of graphene sheets thereby affording interfacially bridged, three-dimensional heteroatom-enriched graphene-based hybrid nanostructures (DGn-GO). Dendrimer generation (from 1 to 4) that reflects the bulkiness of these conceived nano-trees impacts increasingly the degree of dispersion-exfoliation and sheet desordering. The long-term stability of these aqueous suspensions associated with their handling flexibility allows uniform accommodation of the resulting hybrid materials as flame-retardants in bioplastics.
Introduction
Peeling-off tiny graphene sheets, reported fourteen years ago,1 have initiated an avalanche of investigations primarily focused on their massive production,2 surface-functionalisation,3 nanocomposite manufacturing4 and their subsequent use for energy storage and conversion,5 catalysis,6 gas sensing,7 and in nanomedicine and healthcare applications.8 Owing to their fascinating properties, graphene-based materials stand as prominent contenders for future emerging nanotechnologies, specifically those tightly connected to flexible and wearable electronics.Splitting tactoids or layered graphite stacks into individual, well-exposed thin carbon sheets is a fundamental prerequisite to access high-quality graphene materials.9 The introduction of heteroatoms within these individual sheets by insertion, impregnation or covalent grafting allows for modulating further their physical and chemical properties.3 Physical methods are more relevant to produce high-quality graphene materials but are not, unfortunately, cost-effective because of the need for sophisticated apparatus and energy-associated procedures. Oppositely, the straightforwardness and versatility of liquid-phase synthesis provide a large library of chemically modified graphene materials.
Graphene oxide (GO), obtained by oxidation of graphite, is prone to dispersion in aqueous and liquid solvents and to chemical modification owing to the presence of polar, transformable oxygenated epoxides, alcohols, ketones and carboxylic acids.10 Beyond its molecular-surface functionalisation, more sophisticated nanostructures including graphene oxide coated polymers and biopolymers, liposomes, DNA, polyoxometalate and other assembled graphene-based nanomaterials have been recently reported.11 This hybridisation at the nanoscale brings novel reactivity to graphene sheets and substantially enhances their long-term dispersion and stability. Despite these achievements, further innovative investigations are necessary to assemble these tiny building-blocks into a rationally designed, functional three-dimensional architecture suitable for practical applications.
Having recently revealed many specific behaviours of dendrimers in materials science,12 we envisioned that their supramolecular association with graphene sheets could provide a novo-design concept for new functional hybrid materials.13 Dendrimers differ from their linear polymer congeners by their defined size, shape and topology, molecular diversity from the core to the surface, tunable flexibility or rigidity depending on the spacer installed in their branching units, amphiphilicity, and their well-established “generation effect”.14 More specifically, phosphorus dendrimers display some intrinsic features including their organic–inorganic character, their typically hydrophobic cylcotriphosphazene core, and the high content of nitrogen, sulfur and phosphorus heteroatoms in their framework that impart them with excellent stability.15 The presence of the phosphorus element allows accurate elucidation of the chemical functionalisation undertaken on the outer-sphere of these architectures.16 Their ability to swell in polar solvents17 provides access to hierarchically porous pockets that entrap enzymes and living-cells,18 confine and stabilize nanoparticles19 and prematurely convert amorphous bursts into discrete meso-crystals.12,20 In a conventional medium, this latter phenomenon occurs commonly in the solid state under high temperature conditions.
Based on the aforementioned achievements and our own endeavours, we became interested in the secondary-structure of phosphorus-dendrimers as a potent efficient exfoliating medium and heteroatom provider for graphene materials. We herein report the liquid-exfoliation of graphene oxide by a set of terminated – ternary diethyl-N-ethyl-ammonium – phosphorus dendrimers. The choice of peripheral ammonium groups was dictated by their favourable interaction with epoxy, alcohol and carboxylic acid, which efficiently stabilize exfoliated sheets.21 The assembled dendrimer–graphene oxide hybrid materials thus obtained were isolated and characterized. Their stability was disclosed and their reactivity with hydrazine as a reducing agent was also undertaken with a special emphasis to elucidate the chemical interactions occurring at the material interface. We also present the wrapping of these exfoliated sheets in a bio-based shell-waste polysaccharide for the preparation of thermally stable, functional phosphorus-based hybrid bioplastics.
Results and discussion
Preparation of DGn-GO
In our experiments, four different generations of cationic phosphorhydrazone dendrimers, denoted as DGn (n = 1–4), were used.22 These dendrimers feature similar core (cyclotriphosphazene) and similar peripheral groups (ternary diethyl-N-ethyl-ammonium) but differ in their size (number of phosphorus layers) and the number of ternary ammonium groups (12 ternary ammoniums for DG1, 24 for DG2, 48 for DG3 and 96 for DG4) (Fig. 1a). | ||
| Fig. 1 (a) Chemical structures of dendrimers DGn. (b) Illustration of the exfoliation of layered graphitic oxide materials by ammonium-terminated phosphorus dendrimers and their subsequent reduction. (c) Structures of GO. (d) Digital photos of DG1-GO and DG1-RGO dispersion in aqueous solution. | ||
Liquid phase exfoliation was performed by subjecting 40 mg of graphene oxide (GO) in 100 mL of water to sonication. A ternary diethyl-N-ethyl-ammonium-terminated dendrimer DGn was then added to the solution and the mixture was stirred for 24 hours at room temperature (see the Experimental section for details). It is noteworthy that immediate flocculation was observed with a significant expansion of the solution volume, typical of the interfacial polyionic complexation between the nanosheets and the cationic dendrimers.23 The solution was then filtered by centrifugation and the precipitate was washed four times with water to remove any soluble, unreacted dendrimers before drying the resulting DGn-GO materials under vacuum at room temperature.
DRIFT analysis of the resulting dark green powder reveals a typical fingerprint of cationic phosphorus dendrimers (Fig. S2, ESI†). Accurate details were drawn from solid-state NMR spectroscopy. While CP MAS 13C NMR of the starting GO displays only three signals nearly at 59, 69 and 132 ppm, attributed respectively to C–O–C, CH2–OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,24 well-resolved signals assignable to the dendritic skeleton are also observed for DGn-GO hybrid materials. The characteristic methylene appears at 8.7 ppm, the N-methylated one at 33 ppm, the adjacent CH2–CH3 at 46 ppm and the aromatics between 122 and 133 ppm. A complete disappearance of the signal attributed to the epoxide groups in GO (at ∼59 to 60 ppm) was observed, pointing out to the ring opening of the latter by nucleophilic species belonging to DGn.24 A slight shift to high values was also observed for the signal belonging to graphenic CH2–OH from 68 to 71 ppm (Fig. S3a, ESI†), owing much probably to the proximal presence of heteroatoms or dynamic sliding of DGn at the carbon layers.24 CP MAS 31P NMR allows discriminating signals of the phosphorus core (∼5 to 8 ppm) and the ones installed within the branches (∼67 to 70 ppm). The Pcoreversus Pbranch intensity in DGn-GO was inverted by increasing the dendrimer generation, consistent with their molecular structures (Fig. S3b, ESI†). X-ray diffraction (XRD) of the starting GO reveals a signal at a 2θ value of 9.68° that reflects the basal distance (d = 0.91 nm) of the periodic layers. A shift to 6.81° was observed once subjected to DG1 (the smallest, first generation phosphorus dendrimer), indicating a significant expansion of the galleries to d = 1.29 nm because of the intercalation of the dendrimer. No signal was observed for more bulky DG2, DG3 and DG4, indicating complete delamination of the GO layers (Fig. S4, ESI†). During sonication, the high shear impact allows for expanding the basal distance where the adsorbed dendrimer on the graphitic surface constitutes the driving force for preventing the restacking of graphene sheets. As the process goes on, the strong chemical interactions occurring at the dendrimer–graphene oxide interface reduce the motion of sheets and provide enough stability to resist the capillary forces acting on their framework during water removal.
C,24 well-resolved signals assignable to the dendritic skeleton are also observed for DGn-GO hybrid materials. The characteristic methylene appears at 8.7 ppm, the N-methylated one at 33 ppm, the adjacent CH2–CH3 at 46 ppm and the aromatics between 122 and 133 ppm. A complete disappearance of the signal attributed to the epoxide groups in GO (at ∼59 to 60 ppm) was observed, pointing out to the ring opening of the latter by nucleophilic species belonging to DGn.24 A slight shift to high values was also observed for the signal belonging to graphenic CH2–OH from 68 to 71 ppm (Fig. S3a, ESI†), owing much probably to the proximal presence of heteroatoms or dynamic sliding of DGn at the carbon layers.24 CP MAS 31P NMR allows discriminating signals of the phosphorus core (∼5 to 8 ppm) and the ones installed within the branches (∼67 to 70 ppm). The Pcoreversus Pbranch intensity in DGn-GO was inverted by increasing the dendrimer generation, consistent with their molecular structures (Fig. S3b, ESI†). X-ray diffraction (XRD) of the starting GO reveals a signal at a 2θ value of 9.68° that reflects the basal distance (d = 0.91 nm) of the periodic layers. A shift to 6.81° was observed once subjected to DG1 (the smallest, first generation phosphorus dendrimer), indicating a significant expansion of the galleries to d = 1.29 nm because of the intercalation of the dendrimer. No signal was observed for more bulky DG2, DG3 and DG4, indicating complete delamination of the GO layers (Fig. S4, ESI†). During sonication, the high shear impact allows for expanding the basal distance where the adsorbed dendrimer on the graphitic surface constitutes the driving force for preventing the restacking of graphene sheets. As the process goes on, the strong chemical interactions occurring at the dendrimer–graphene oxide interface reduce the motion of sheets and provide enough stability to resist the capillary forces acting on their framework during water removal.
SEM and TEM analyses were undertaken to visualise these objects in the micro- and nano-scale domain. SEM reveals crumpled layers that are connected to each other to form continuous stacks of several tens of nanometers with smooth compact layers inside (Fig. 2a, b and S5a, ESI†). This aligned densification at the microscale can be rooted in the occurrence of both physical and chemical cross-linking between cationic dendrimers and negative GO sheets. Individual, separated or uncovered graphene layers are not observed indicating that in principle, all sheets are entangled by phosphorus dendrimers, resulting in three-dimensional hybrid materials. EDX mapping confirms the presence of phosphorus, nitrogen, and sulphur supplied from the starting dendrimers and their homogenous distribution through the whole material network (Fig. 2c). TEM analyses show that DGn-GO materials are built from transparent thin layers where the intimate self-assembly of the two building-blocks affords wrinkled and crumpled nanostructures (Fig. 2d). The presence of aligned tubular layers (300 nm to 2 μm in length and 8 to 12 nm in width) as revealed by HRTEM analysis indicates an extension of the self-assembly mechanism beyond the nanoscale level (Fig. 2e, f and S5b, ESI†). This result finds its root in the dual role of the cationic dendrimer that acts both as an exfoliating medium and as a cross-linking agent owing to the presence of abundant reticular anchoring points on its periphery. A quite similar pattern was previously reported by assembling polyethylenimine and graphene oxide nano-architectures.25
 | ||
| Fig. 2 Microscopic analyses of DG4-GO. (a and b) SEM of DG4-GO. (c) EDX mapping of phosphorus (up) and sulfur (bottom). (d) TEM and (e and f) HRTEM of DG4-GO. | ||
The resulting solid materials can be re-dispersed in water and the precipitation–dispersion process can be repeated several times without graphene sedimentation or dendrimer leaching. UV-visible analyses of the suspended DGn-GO hybrids display a holistic bond with maxima at 286–290 nm attributed to the corresponding dendrimer, with a tail extending to the visible region due to the presence of conjugated aromatics belonging to graphene oxide sheets (Fig. S6, ESI†).24 Compared to native DGn that displayed a wavelength maximum at 285 nm, DGn-GO maxima of these materials were shifted to higher values with a significant broadening of the band that may indicate the presence of both covalent and physical interactions as well as free terminal groups within the dendrimer skeleton.
A significant improvement of the thermal stability was noticed for DGn-GO compared to the pristine GO as illustrated by their TGA profiles. For instance, the T40 corresponding to the loss of 40 wt% was shifted from 254 °C for GO to around 480 °C for DG1-GO, DG2-GO and DG3-GO. An impressive T40 of 502 °C was reached for DG4-GO (Fig. S11, ESI†).
Preparation of DGn-RGO
Having ascertained the exfoliation of graphene oxide sheets by phosphorus dendrimers and the tight presence of the latter within the tectonic nanostructure, one important question was how the presence of phosphorus dendrimers affects the accessibility to the graphene surface during reduction. Although complete restoration of graphene from graphene oxide is quite impossible (even under forcing conditions),10,26 reduced graphene oxide (RGO) displayed improved electrical conductivity and thermal and chemical stability, making the corresponding hybrid materials of strong interest.27 With this in mind, we subjected DGn-GO aqueous suspensions to hydrazine treatment followed by filtration and extensive washing before drying the resulting solid materials under vacuum. At this stage, two experimental observations must be noted: (i) while the initial exfoliation step did not affect the dark green colour of the starting graphene oxide, a dramatic change to a more pronounced black material was noticed during hydrazine treatment; (ii) simultaneously, while DGn-GO flocculated in solution, significant volume shrinkage was observed during hydrazine treatment, strongly suggesting an alteration of the ionic interfacial complexation of the two building blocks. Similar to their parent materials, the resulting DGn-RGO solids can be isolated and re-dispersed in water with good temporal stability. The precipitation–dispersion cycle can be repeated at least five times without leaching of the dendrimer skeleton from the graphenic surface material. It should be noted that DG1-RGO provides the less-stable dispersion among the DGn-RGO series, which can be correlated with the smallest size of the starting dendrimer.SEM and TEM analyses of DGn-RGO reveal a similar morphology to DGn-GO parent materials, although significant densification of layers and slight shrinkage of the network seem to occur during hydrazine treatment (Fig. 3a, b and S7a, ESI†). HRTEM analyses show elongated core–shell crumbled nanostructures of several hundreds of nanometers in length with an interior width of 24 to 54 nm covered outside by a dense layer of 6 to 8 nm (Fig. 3d–f and S7b, ESI†). Comparing the topology of DGn-RGO and DGn-GO, the variation concerns only the width and not the length of the elementary nanostructures. This reorganisation emanates probably from an interfacial folding of several elementary structures during hydrazine treatment; the reduction step is suspected to consume polar oxygenated groups and consequently minimise interfacial electrolytic complexation.
 | ||
| Fig. 3 Microscopic analysis of DG4-RGO. (a and b) SEM of DG4-RGO. (c and d) TEM and (e and f) HRTEM of DG4-RGO. | ||
XRD reveals a slight decrease of the basal plane in DG1-RGO (from 1.29 nm to 1.04 nm) that can be explained by a decrease of the layer thickness during oxygen removal from the surface. The basal plane was still however high in DG1-RGO compared to native GO (d = 0.91). The absence of any appreciable peak for DGn-RGO (n = 2 to 4) ruled out any restacking of graphene sheets and confirmed the total disordering and exfoliation of graphene sheets in these materials (Fig. S4, ESI†).
Chemical interplay at the DGn-GO and DGn-RGO interface
The interfacial chemical interplay was accurately examined by coupling a set of analytical tools including RAMAN, X-ray photon spectroscopy (XPS) and solid-state NMR spectroscopy.MAS CP 13C NMR of DGn-RGO revealed the first evidence for oxygen removal from the GO surface. A typical signal belonging to the graphene oxide at ∼70 ppm completely disappeared as illustrated in the ESI (Fig. S3a, ESI†). MAS CP 31P NMR of the four materials displays the two typical signals of the core and the branching units (respectively at 4.8 to 7.5 ppm and at 67 to 69 ppm) thereby indicating the intactness of the dendrimer skeleton during chemical reduction and precipitation–dispersion cycles (Fig. S3b, ESI†).
We further investigated the variation observed in Raman spectroscopy, where the D (associated with the order/disorder of the system) and G (an indicator of the stacking structure) bands are the dominant vibrational modes observed respectively at 1354 and 1601 cm−1.26,28 The ratio of the intensities of the two bands (D/G) is often used to determine the overall stacking of the sheets, and high D/G ratios indicate a high degree of exfoliation/disorder (Fig. S8, ESI†). The intensity ratio of the starting GO increased from 1.15 to 1.25 when intercalated with DG1 and to 1.36 for the full exfoliation reached in the case of the bulky DG4-GO dendrimer. The highest exfoliation and disorder caused by DG4 are further supported by the appearance of a new D′ band as a shoulder at 1619 cm−1 (Fig. 4). Upon reduction, a further increase of the D/G ratio was observed for these materials compared to the starting GO (1.48 for DG1-RGO, 1.54 for DG2-RGO, 1.58 for DG3-RGO and 1.37 for DG4-RGO) consistent with previous literature.26 Moreover, all these materials displayed an additional D′ band characteristic of increased disorder. Such perturbation, not observed in the case of graphene oxide exfoliated ammonium-terminated chitosan,21 indicates that more disordering was experienced in the case of ternary ammonium-terminated dendrimers owing to their increased globular and radial (instead of linear) size bulkiness. The unique structure of phosphorus dendrimers and the highest ramification on the external surface maximise its contact with graphene sheets. The highest entropy reached under these circumstances allows for performing both parallel and perpendicular exfoliation depending on the thermodynamically favourable conformation.29 The non-variation of the D/G ratio in the special case of DG4 before (1.36 for DG4-GO) and after hydrazine treatment (1.37 for DG4-RGO) while keeping the D′ band indicates that the highest disorder was already reached during the dispersion step. The increased number of functional groups allows for the total consumption/binding of the dendrimer to the sheets and the significant wrapping of the graphene surface left few oxygenated groups accessible/available during hydrazine treatment. A negligible alteration (reduction) of the material surface was consequently observed in this case.
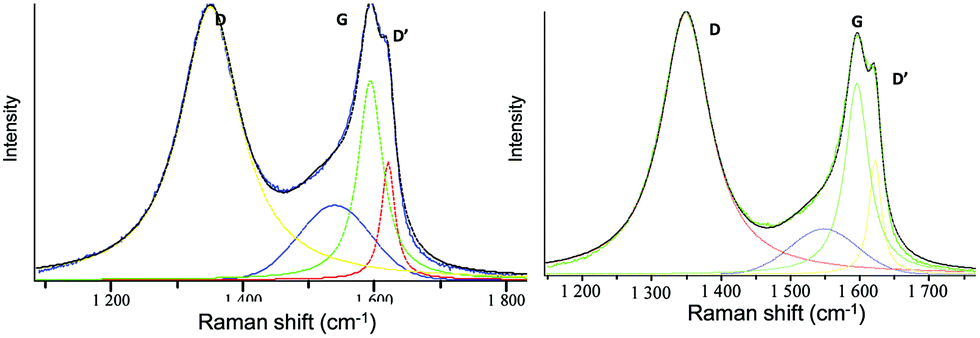 | ||
| Fig. 4 RAMAN analysis of DG4-GO (left) and DG4-RGO (right). | ||
Insight into the DGn-GO and DGn-RGO interfacial interplay was gained by XPS analyses. Pristine graphene oxide displays signals of carbon deconvoluted at 284.4 (CC/C–C), 286.5 (C–O) and 288.5 eV (–CO/COO) and a minor π–π* plasmonic related band at 289.6 eV. The oxygen binding signal appears at 532.2 eV. We note the presence of an S (2p) doublet at 168.3–169.5 eV, attributed to sulfonic or sulfate groups generated during Hummer oxidation. In DGn-GO (n = 1, 4), binding energies of carbon, nitrogen, oxygen, sulfur and phosphorus were observed. The C(sp2–sp3)/C–O ratio was increased with a decrease of the COO ratio and the π–π* indicative of a chemical reaction of peripheral ternary ammoniums with oxygenated groups as well as more disordering of carbon sheets because of their exfoliation (Table S1, ESI†). Oxygen binding shows two oxygen types, C–O and C–O–P bonding. Nitrogen also reveals two peaks at 398.9 eV of N–P and C–N and at 401.2 eV typical of quaternized nitrogen atoms. Sulfur (2p) spectra also confirm the presence of PS fragments belonging to the dendrimers at 161.3–162.5 eV alone with residual sulfonate at higher binding energies (168.3–169.5 eV). Phosphorus binding reveals a broad signal corresponding to both N2PO and N4PS at 132.4 and 133.8 eV (Fig. 5 and S9, ESI†). Lastly, chlorides (the counter ions of the ternary ammonium) remain on the material surface (with a binding energy of 197.0 eV) despite extensive washing of the material framework. However, its concentration is below the spectrometer sensitivity (0.1 at%).
 | ||
| Fig. 5 XPS analyses (from left to right: binding energy of carbon, nitrogen oxygen and phosphorus elements) of (a) DG4-GO and (b) DG4-RGO. | ||
Hydrazine treatment induces a significant alteration of the material-surface composition as substantiated by XPS and in full agreement with NMR and RAMAN analyses. Indeed, the C 1s peak at 284.4 eV becomes predominant, attributed mainly to carbon sp2, while C–O peaks appear only as a shoulder with C–N and COO consistent with the removal of oxygenated species from the surface. A significant change in the spectral shape of O 1s is noticed with C–O–P being more abundant compared to C–O related bonds. In the meantime, a significant NH+ related peak intensity decrease is observed alongside with the removal of chloride and sulphate species from the material surface during hydrazine treatment. The remanence of the phosphorus signal indicates the non-alteration of the phosphorus environment upon reduction. The atomic ratio of the chemical species is provided in the ESI section (Table S1, ESI†) and the suggested mechanism is further discussed in detail (S10, ESI†).
In UV-visible spectroscopy, significant to lower values were observed for DGn-RGO with respect to the parent DGn-GO materials (for instance from 290 nm to 285 nm and from 290 nm to 279 nm for DG2-RGO and DG3-RGO, respectively). This shift indicates a significant alteration of the interfacial properties of DGn-RGO compared to the starting DGn-GO materials (Scheme 1).24
 | ||
| Scheme 1 Chemical interplay occurring at the dendrimer–graphene oxide interface. (a) Typical chemical structure of the first generation phosphorus dendrimer DG1 and oxygenated graphene oxide sheets. (b) Plausible mechanism occurring during interfacial complexation of ternary diethyl-N-amino-ammonium chloride arms and (c and d) the subsequent chemical reduction. See S10 in the ESI† for further discussion. | ||
CS@DGn-(R)GO nanocomposite bioplastics
The stable exfoliation reached in these modified graphene sheets associated with their improved properties prompted their use as flame-retardant nano-fillers for soft-bioplastics. Among the various polymers suitable as film-forming matrices, we considered chitosan CS (a biodegradable glucosamine polymer derived from bio-waste generated in fishery plants, which is available in enormous quantity on earth) especially useful as a packaging material e.g. for healthcare, food and agriculture.30Our bioplastic was prepared in two steps starting first by solubilising medium molecular weight chitosan in aqueous acidic solution followed by the addition of 3 wt% of the filler (GO, DGn-GO, and DGn-RGO) dispersed in aqueous solution. Casting of the mixture under air in a Petridish enables the formation of good-quality, crack-free films (Fig. 6). Interestingly, despite the bulkiness of the as-prepared nano-fillers, SEM analysis reveals excellent homogenisation of the film, particularly for CS@DGn-RGO materials that exhibit a smooth surface. No phase separation or filler aggregation was observed, which is an excellent preliminary result toward the rational design of functional, mechanically robust bioplastics.
 | ||
| Fig. 6 (a) Intercalation of DGn-RGO within the fibrillary network of chitosan. (b) Aqueous dispersion of DG4-GO and the resulting transparent CS@DG4-GO film (top) and (d) the corresponding SEM analysis. (c) Aqueous dispersion of DG4-RGO and the resulting transparent CS@DG4-RGO film (top) and (e) the corresponding SEM analysis. | ||
Thermogravimetric analysis (TGA) allows monitoring the bioplastic degradation. Previously, we noticed the detrimental use of graphene oxide as a filler for bioplastics and evidenced that oxygenated groups catalytically accelerate the C–C bond scission of the biopolymer.21 To our delight, we herein discovered that the degradation of chitosan was significantly delayed in the case of CS@DGn-GO and even, substantial improvement was gained for CS@DGn-RGO. In practical terms, the half weight degradation of chitosan denoted as T50 was measured to be 325 °C. Improvement of CS thermal properties was noticed by the use of DG1-GO and DG4-GO where T50 was found to be 340 °C and 345 °C, respectively. The performance was even pushed up for CS@DG4-RGO where its T50 reached nearly 400 °C, corresponding to an impressive ΔT of 75 °C with respect to non-modified bioplastics (Fig. S11, ESI†).
Experimental
Synthesis of DGn-GO and DGn-RGO materials
Conclusions
In this contribution, high content heteroatoms (nitrogen, phosphorus, and sulfur) assembled in a tree-like dendritic network are confined within the layered galleries of graphene oxide. The globular structure of cation-terminated dendrimers enables them to act both as an exfoliating medium and a cross-linking agent to afford novel-extended nanostructures with highly disordered carbon sheets. Hydrazine treatment allowed substantial surface alteration of the ionic interfacial complexation without restacking of the graphene layers. Stable dispersion was obtained and as a proof of concept, the resulting functional ink was used as a nano-filler for aqueous-phase processing of bioplastics where significant improvement of the thermal stability was noticed. Considering the molecular diversity of DGn-GO and DGn-RGO materials, their synergistic functionality and the promising use of phosphorus dendrimers themselves in nanotechnology, these results appear promising for the development of multifunctional and biologically active flame-retardant hybrid mats and bioplastics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
UEMF is warmly acknowledged for the financial support. We thank Vincent Collière for technical assistance (SEM and TEM analyses).Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- (a) M. Yi and Z. Shen, J. Mater. Chem. A, 2015, 3, 11700–11715 RSC; (b) Y. L. Zhong, Z. Tian, G. P. Simon and D. Li, Mater. Today, 2015, 18, 73–78 CrossRef CAS.
- (a) V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed; (b) J. E. Johns and M. C. Hersam, Acc. Chem. Res., 2013, 46, 77–86 CrossRef CAS PubMed; (c) M. Quintana, E. Vazquez and M. Prato, Acc. Chem. Res., 2013, 46, 138–148 CrossRef CAS PubMed.
- K. Hu, D. D. Kulkarni, I. Choi and V. V. Tsukruk, Prog. Polym. Sci., 2014, 39, 1934–1972 CrossRef CAS.
- J.-Y. Liu, X.-X. Li, J.-R. Huang, J.-J. Li, P. Zhou, J.-H. Liu and X.-J. Huang, J. Mater. Chem. A, 2017, 5, 5977–5994 RSC.
- (a) S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed; (b) S. Navalon, A. Dhakshinamoorthy, M. Alvaro, M. Antonietti and H. Garcia, Chem. Soc. Rev., 2017, 46, 4501–4529 RSC.
- (a) R. Zhang and W. Chen, Biosens. Bioelectron., 2017, 89(Part 1), 249–268 CrossRef CAS PubMed; (b) A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS PubMed.
- (a) K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC; (b) C. Cheng, S. Li, A. Thomas, N. A. Kotov and R. Haag, Chem. Rev., 2017, 117, 1826–1914 CrossRef CAS PubMed.
- (a) S. Ravula, S. N. Baker, G. Kamath and G. A. Baker, Nanoscale, 2015, 7, 4338–4353 RSC; (b) A. Ciesielski and P. Samorì, Adv. Mater., 2016, 28, 6030–6051 CrossRef CAS PubMed; (c) J. I. Paredes and S. Villar-Rodil, Nanoscale, 2016, 8, 15389–15413 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- (a) P. T. Yin, S. Shah, M. Chhowalla and K.-B. Lee, Chem. Rev., 2015, 115, 2483–2531 CrossRef CAS PubMed; (b) T. Premkumar and K. E. Geckeler, Prog. Polym. Sci., 2012, 37, 515–529 CrossRef CAS; (c) X. Wang, E. N. Kalali, J.-T. Wan and D.-Y. Wang, Prog. Polym. Sci., 2017, 69, 22–46 CrossRef CAS; (d) R. Zappacosta, M. Di Giulio, V. Ettorre, D. Bosco, C. Hadad, G. Siani, S. Di Bartolomeo, A. Cataldi, L. Cellini and A. Fontana, J. Mater. Chem. B, 2015, 3, 6520–6527 RSC; (e) J. Suarez-Guevara, V. Ruiz and P. Gomez-Romero, Phys. Chem. Chem. Phys., 2014, 16, 20411–20414 RSC.
- (a) Y. Brahmi, N. Katir, J. A. M. Agullo, A. Primo, M. Bousmina, J. P. Majoral, H. Garcia and A. El Kadib, Dalton Trans., 2015, 44, 15544–15556 RSC; (b) K. Milowska, A. Rybczyńska, J. Mosiolek, J. Durdyn, E. M. Szewczyk, N. Katir, Y. Brahmi, J.-P. Majoral, M. Bousmina, M. Bryszewska and A. El Kadib, ACS Appl. Mater. Interfaces, 2015, 7, 19994–20003 CrossRef CAS PubMed; (c) Y. Brahmi, N. Katir, M. Ianchuk, V. Colliere, E. M. Essassi, A. Ouali, A.-M. Caminade, M. Bousmina, J. P. Majoral and A. El Kadib, Nanoscale, 2013, 5, 2850–2856 RSC.
- (a) A. El Kadib, N. Katir, M. Bousmina and J. P. Majoral, New J. Chem., 2012, 36, 241–255 RSC; (b) A.-M. Caminade and J.-P. Majoral, Chem. Soc. Rev., 2010, 39, 2034–2047 RSC.
- (a) D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Macromolecules, 1986, 19, 2466–2468 CrossRef CAS; (b) R. Roy and T. C. Shiao, Chem. Soc. Rev., 2015, 44, 3924–3941 RSC; (c) A. Sheikhi, N. Li, T. G. M. van de Ven and A. Kakkar, Environ. Sci.: Water Res. Technol., 2016, 2, 71–84 RSC; (d) N. Katir, N. El Brahmi, A. El Kadib, S. Mignani, A.-M. Caminade, M. Bousmina and J. P. Majoral, Chem.–Eur. J., 2015, 21, 6400–6408 CrossRef CAS PubMed; (e) A.-M. Caminade, A. Ouali, R. Laurent, C.-O. Turrin and J.-P. Majoral, Chem. Soc. Rev., 2015, 44, 3890–3899 RSC.
- (a) A.-M. Caminade, A. Hameau and J.-P. Majoral, Dalton Trans., 2016, 45, 1810–1822 RSC; (b) C.-O. Turrin, V. Maraval, J. Leclaire, E. Dantras, C. Lacabanne, A.-M. Caminade and J.-P. Majoral, Tetrahedron, 2003, 59, 3965–3973 CrossRef CAS.
- (a) A.-M. Caminade, R. Laurent, C.-O. Turrin, C. Rebout, B. Delavaux-Nicot, A. Ouali, M. Zablocka and J.-P. Majoral, C. R. Chim., 2010, 13, 1006–1027 CrossRef CAS; (b) N. Katir, N. El Brahmi, N. Marcotte, J. P. Majoral, M. Bousmina and A. El Kadib, Macromolecules, 2016, 49, 5796–5805 CrossRef CAS.
- J. Leclaire, Y. Coppel, A.-M. Caminade and J.-P. Majoral, J. Am. Chem. Soc., 2004, 126, 2304–2305 CrossRef CAS PubMed.
- (a) K. Ciepluch, N. Katir, A. El Kadib, M. Weber, A.-M. Caminade, M. Bousmina, J. Pierre Majoral and M. Bryszewska, J. Lumin., 2012, 132, 1553–1563 CrossRef CAS; (b) K. Ciepluch, M. Weber, N. Katir, A.-M. Caminade, A. El Kadib, B. Klajnert, J. P. Majoral and M. Bryszewska, Int. J. Biol. Macromol., 2013, 54, 119–124 CrossRef CAS PubMed.
- N. Katir, A. El Kadib, V. Colliere, J. P. Majoral and M. Bousmina, Chem. Commun., 2014, 50, 6981–6983 RSC.
- (a) N. Katir, Y. Brahmi, J. P. Majoral, M. Bousmina and A. El Kadib, Chem. Commun., 2015, 51, 17716–17719 RSC; (b) Y. Brahmi, N. Katir, A. Hameau, A. Essoumhi, E. M. Essassi, A.-M. Caminade, M. Bousmina, J.-P. Majoral and A. El Kadib, Chem. Commun., 2011, 47, 8626–8628 RSC.
- S. Frindy, A. Primo, H. Ennajih, A. el kacem Qaiss, R. Bouhfid, M. Lahcini, E. M. Essassi, H. Garcia and A. El Kadib, Carbohydr. Polym., 2017, 167, 297–305 CrossRef CAS PubMed.
- C. Loup, M. A. Zanta, A. M. Caminade, J. P. Majoral and B. Meunier, Chem.–Eur. J., 1999, 5, 3644–3650 CrossRef CAS.
- J. Zou and F. Kim, ACS Nano, 2012, 6, 10606–10613 CrossRef CAS PubMed.
- F. T. Johra, J.-W. Lee and W.-G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- J. Zou and F. Kim, Nat. Commun., 2014, 5, 5254 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, Chem. Soc. Rev., 2012, 41, 6944–6976 RSC.
- M. S. Dresselhaus, A. Jorio, A. G. Souza Filho and R. Saito, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 5355–5377 CrossRef CAS PubMed.
- J. Yang, X. Yang and Y. Li, Curr. Opin. Colloid Interface Sci., 2015, 20, 339–345 CrossRef CAS.
- (a) M. Aider, Food Sci. Technol., 2010, 43, 837–842 CAS; (b) M. Z. Elsabee and E. S. Abdou, Mater. Sci. Eng., C, 2013, 33, 1819–1841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of dendrimers, NMR spectra, DRIFT, XRD, UV-visible, SEM, EDX, TEM, XPS, TGA, and RAMAN analysis of materials. See DOI: 10.1039/c8na00047f |
| This journal is © The Royal Society of Chemistry 2019 |