
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired carbon dots: from rose petals to tunable emissive nanodots†
Vinay
Sharma
 a,
Sandip K.
Singh
b and
Shaikh M.
Mobin
*abc
a,
Sandip K.
Singh
b and
Shaikh M.
Mobin
*abc
aDiscipline of Biosciences and Bio-Medical Engineering, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, India. E-mail: xray@iiti.ac.in; Tel: +91-731-2438752
bDiscipline of Chemistry, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, India
cDiscipline of Metallurgy Engineering and Materials Science, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, India
First published on 9th January 2019
Abstract
Engineering biomass into functional nanomaterials is captivating. The limitation of versatility in green precursors is pursued by exploring the formation of carbon dots with respect to the contents of the green precursor, i.e. Rosa indica. The intermediates formed at different intervals are analyzed. Moreover, the mechanism of heteroatom-doped Rosa indica derived carbon-dot is proposed.
Deriving inspiration from nature for preparation of functional materials is fascinating. The processing of unique natural composition of biomass can lead to a functional material with interesting properties; one such example is green carbon dots. The field of naturally derived carbon dots has grown exponentially; however, the synthesis mechanism and reproducibility is still a bottleneck due to lack of proper characterization of green precursors, which are versatile and heterogeneous in nature.
The recent trend of using green materials for the synthesis of carbon dots employing edible green materials, such as tomato, banana, milk, orange, cabbage, corn bract and coconut, have gained considerable attention.1 However, the use of non-edible green materials would have apparent benefits, and proper characterization of green precursor can offer better understanding of the synthesis mechanism as well as aid in the reproducibility. Recently, we explored the optical properties of Rosa indica derived carbon dots;2 hence, it is interesting to study its precursors and intermediates.
Importantly, the properties of carbon dots are considerably affected by the initial source of carbon. Chemically synthesized carbon dots suffer from low yield, toxic precursors and the requirement of separate passivation, having implications on capital cost and environment. However, the biomass derived C-dots are beneficial due to their capability of being synthesized on a large-scale, sustainability, easy availability, renewability, green approach and self-passivation. Different source of biomass results in C-dots with largely varying properties because of their inherent combination of organic molecules, assorted functional groups and hetero-atom dopants. A classic example is the use of onion peels resulting in nitrogen, sulfur and phosphorous co-doped C-dots because of the inherent presence of polyphenols and minerals. Polyphenols are known to have anti-oxidation and wound healing properties; hence, onion-derived C-dots were found to be effective for accelerated wound healing.3 Most of the biomass derived C-dots are blue-emitting; however, the use of pulp-free lemon juice results in red-emitting carbon dots evidencing the effect of biomass precursor on the optical properties.4
Herein, we elucidate the conversion mechanism of rose petals to carbon dots in a hydrothermal system by analyzing the composition of precursors, intermediates and the final product. Heteroatom (nitrogen and sulfur) doped carbon-dot (N–S@RCD) using Rosa indica petals provides a renewable and sustainable bio-source of carbon.
Synthesis of N–S@RCD is performed as per our previous report.2 The petals of rose flower were first crushed in a domestic mixer and the rose extract (RE) was filtered. In a typical procedure, 10 mL of RE was mixed with 300 μL of ethylenediamine and 300 mg of L-cysteine. The mixture was stirred for 10 minutes to achieve a homogenous solution (RE-I). The solution was then transferred to a Teflon-lined autoclave for hydrothermal treatment at 180 °C for a period of 5 h. The autoclave was cooled to room temperature and the product suspension was centrifuged to remove large particles. The obtained supernatant (N–S@RCD) was lyophilized to obtain a black sticky product, which was re-suspended in water as per requirement. To elucidate the mechanism of synthesis, four separate reactions in similar conditions with varying reaction times of 1 h, 2 h, 3 h and 4 h were performed. The resulting samples were named NSR-1 h, NSR-2 h, NSR-3 h and NSR-4 h, respectively (Scheme 1).
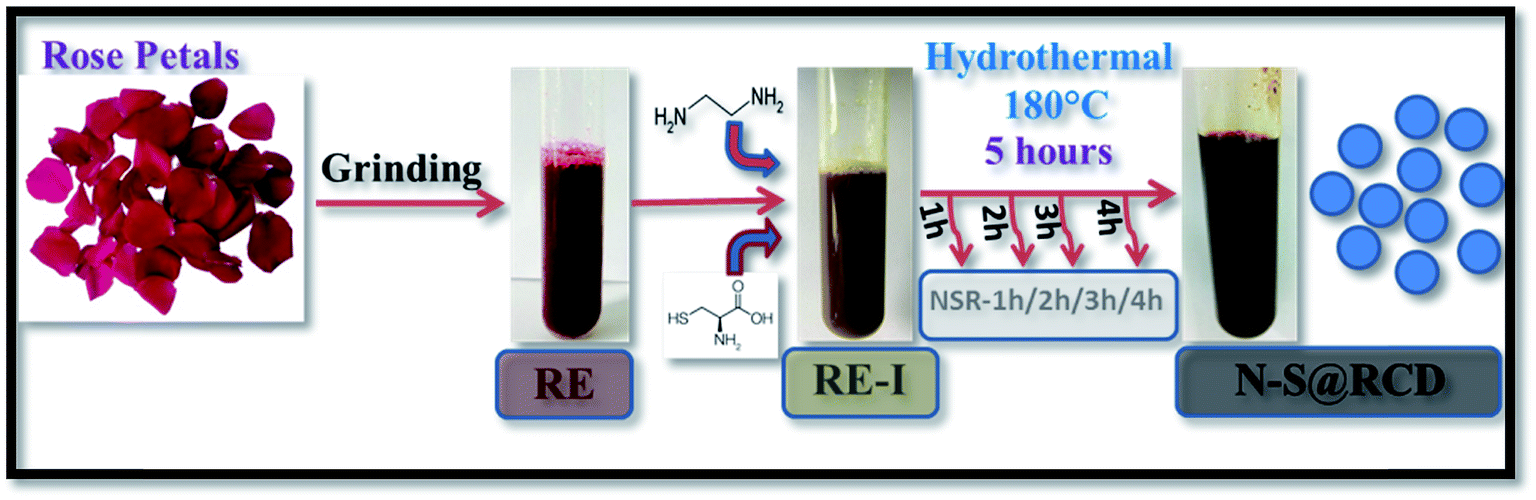 | ||
| Scheme 1 Schematic for synthesis of N–S@RCD and intermediates. | ||
Typical powder X-ray diffraction (PXRD) profile of N–S@RCD shown in Fig. 1(a) exhibits a broad peak centered around 24° (2θ), which represents its amorphous nature. The corresponding d-spacing is 3.7 Å, which is higher than a graphitic [200] plane, indicating the distorted sp2 carbon structure. However, RE and RE-I exhibit broad peak at around 21.4° (2θ) with a similar amorphous nature. The surface functional groups of RE, RE-I and N–S@RCD were identified by attenuated total reflection (ATR) spectroscopy. Fig. 1(b) shows absorption bands at around 3444, 2929, 2870 and 1565 cm−1, corresponding to the stretching vibration of –OH, –NH, –CH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C functional groups, respectively.5 Similar ATR bands with varying intensities between 800 and 1400 cm−1 were observed for all the samples, and were attributed to the stretching vibrations of –C–C–, –C–O–, –C–S– and –C–H groups, respectively.6 However, the most intense band at 1017 cm−1, which is seen only in the RE sample, confirmed the presence of –C–O– groups. Interestingly, the stretching vibration band at 1740 cm−1 was obtained for CO groups in the N–S@RCD sample. Moreover, the stretching bands at 1638 and 1459 cm−1, showing the CN and C–N bands, respectively, are only seen in N–S@RCD.7 These results indicate the N and S doping for the resultant carbon dots.
C functional groups, respectively.5 Similar ATR bands with varying intensities between 800 and 1400 cm−1 were observed for all the samples, and were attributed to the stretching vibrations of –C–C–, –C–O–, –C–S– and –C–H groups, respectively.6 However, the most intense band at 1017 cm−1, which is seen only in the RE sample, confirmed the presence of –C–O– groups. Interestingly, the stretching vibration band at 1740 cm−1 was obtained for CO groups in the N–S@RCD sample. Moreover, the stretching bands at 1638 and 1459 cm−1, showing the CN and C–N bands, respectively, are only seen in N–S@RCD.7 These results indicate the N and S doping for the resultant carbon dots.
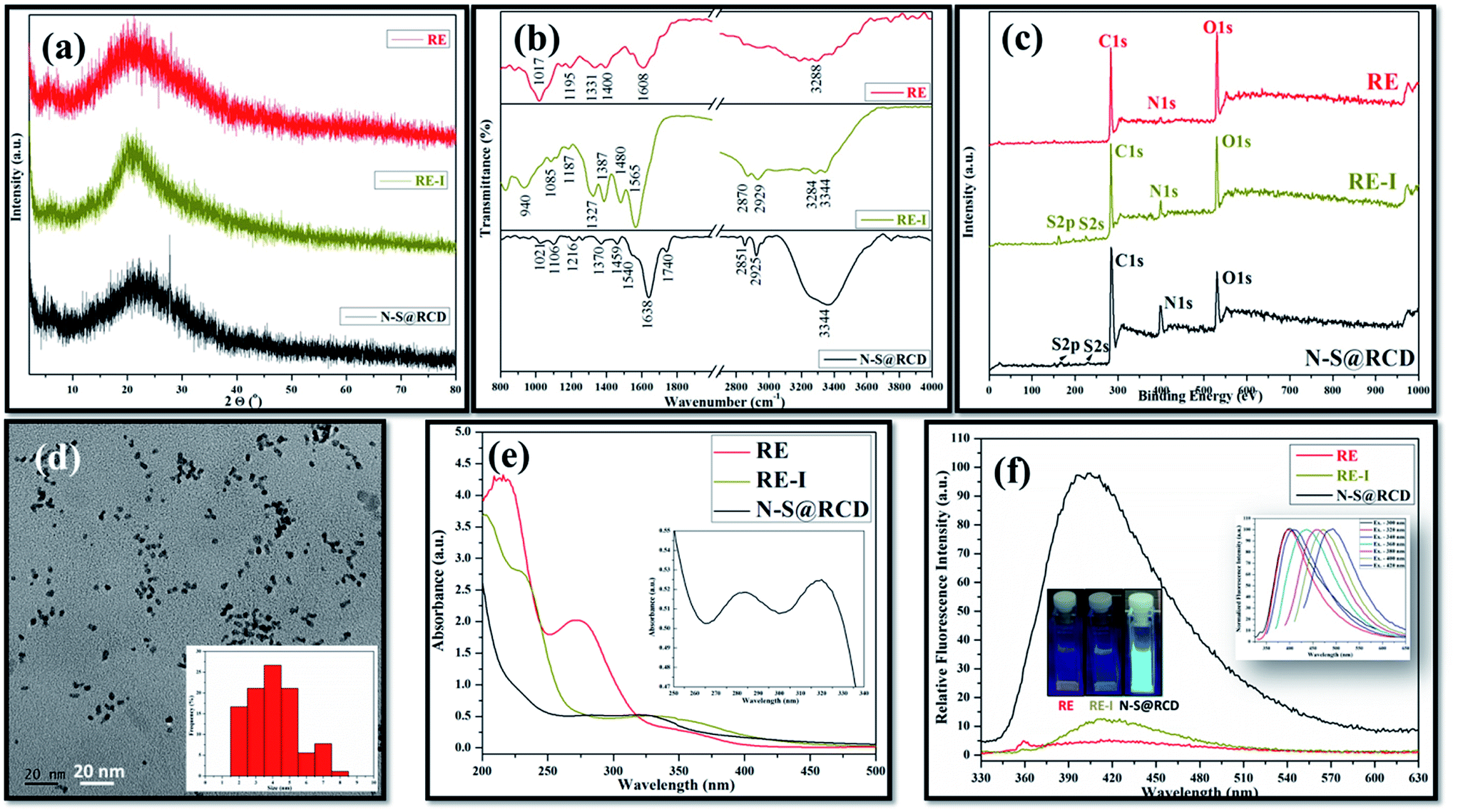 | ||
| Fig. 1 Structural and optical characterization (a) PXRD; (b) ATR spectra; (c) XPS survey spectra; (d) TEM, inset: particle size distribution; (e) UV-vis absorption spectrum, inset: enlarged view of N–S@RCD absorption spectra; and (f) fluorescence spectra of RE, RE-I and N–S@RCD, inset: sample photos under UV light and wavelength tuned emission of N–S@RCD. | ||
In order to provide a more convincing evidence for the elemental composition and the surface states of RE, RE-I and N–S@RCD, X-ray photoelectron spectroscopy (XPS) was carried out. The XPS survey spectrum of N–S@RCD (Fig. 1(c)) shows high intensity peaks at 285.01 eV, 400.02 eV and 530.91 eV, all of which are attributed to C1s, N1s and O1s.8 Additionally, the peaks at 166.98 eV and 230.9 eV were attributed to S2p and S2s, respectively. High resolution C1s spectrum of N–S@RCD (Fig. S1(a)†) was de-convoluted into four peaks and indicated the presence of CC (284.2 eV), C–C (285.3 eV), C–S/C–O/C–N (287.2 eV) and CN/CO (288.0 eV). The N1s spectrum can be fitted in three peaks that are assigned to pyridinic N (398.5 eV), pyrrolic N (399.9 eV) and N–H (402.1 eV), which proves successful incorporation of N atom in the N–S@RCD as shown in Fig. S1(b).† The high-resolution O1s spectrum (Fig. S1(c)†) was de-convoluted into three peaks, which could be attributed to OC (530.5 eV), O–C (531.6 eV) and O–CO (533.8 eV). Furthermore, the S2p spectrum (Fig. S1(d)†) was de-convoluted into two peaks at 163.2 eV (–C–S–) and 167.5 eV (–C–SOx), which represents the successful incorporation of S atoms in the N–S@RCD. The XPS survey spectrum of RE shows prominent peaks corresponding to C1s and O1s, while the intensity of N1s peak is quite low. The obvious presence of N1s and S2s, S2p was evident in RE-I (Fig. 1(c)).
Elemental analysis of RE, RE-I and N–S@RCD samples was carried out and the obtained results were consolidated in Table S1.† Typically, in RE, RE-I and N–S@RCD samples, the C/N ratio is 10.08, 1.47 and 2.91, respectively. Interestingly, after N and S doping within RE, the C/N ratio decreased in RE-I and N–S@RCD samples compared to that in RE. Moreover, both C/N (2.91) and C/S (8.79) ratio increased in N–S@RCD samples compared to those in RE-I.
The particle shape, size and nature of N–S@RCD were investigated via transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM). As shown in Fig. 1(d), N–S@RCD consists of small spherical particles with an average particle diameter of 4.51 ± 1.46 nm. The particle size distribution is shown in the inset of Fig. 1(d), which is calculated using 90 particles. HR-TEM micrograph (Fig. S2†) confirms the spherical shape of N–S@RCD; moreover, well-resolved lattice fringes were obtained for some particles.
To investigate the optical behavior of RE, RE-I, and N–S@RCD, UV-visible spectroscopy and fluorescence spectroscopy were carried out. The UV-visible spectrum of RE shows peaks at 215 nm, 272 nm and an edge at 350 nm; however, the spectrum of RE-I shows broad edges at 230 nm and 330 nm (Fig. 1(e)). The UV-visible spectrum of N–S@RCD shows two absorption peaks at around 281 nm and 320 nm, corresponding to π–π* and n–π* transitions, respectively (Fig. 1(e) inset). The N–S@RCD exhibits excellent water dispersibility and light yellow color under ambient light and gives a fair blue fluorescence under UV irradiation using a 375 nm trans-illuminator, as shown in the inset of Fig. 1(f). The fluorescence spectrum of N–S@RCD shows emission maxima at 397 nm for the excitation at 320 nm, while RE and RE-I are non-fluorescent. The N–S@RCD also exhibited a tuned excitation wavelength, emission maxima and emission intensity (Fig. 1(f) inset), which is an apparent feature of many reported C-dots. Quantum yield of N–S@RCD using quinine sulfate as a reference was found to be 9.6%, which is respectable in comparison to other reported green synthesized carbon dots (Table S2†).
To gain more insights into the mechanism of synthesis of carbon dots from rose petals, electron spin ionization-mass spectroscopy (ESI-MS) and nuclear magnetic resonance (NMR) were performed. The LC-MS chromatogram of RE, RE-I and N–S@RCD samples were recorded by dissolving 1.0 mg samples in CH3OH (5.0 mL) at room temperature (25 ± 2 °C). The LC-MS chromatograms of RE and RE-I samples, which are shown in Fig. S3,† indicates low molecular weights of 413 and 481 g mol−1, respectively. Moreover, the N–S@RCD sample derived from hydrothermal treatment of RE-I has a relatively high molecular weight (665 g mol−1); it may be plausible that polycondensation of low molecular weight moieties (for e.g. furfural, 96 g mol−1; 5-hydroxymethyl furfural (5-HMF), 132 g mol−1) during hydrothermal treatment leads to a high molecular weight of N–S@RCD. The effect of hydrothermal treatment on polycondensation of molecules is a known phenomenon and is stated in several reports.9 Moreover, the obtained molecular weights of RE, RE-I and N–S@RCD samples were comparable with those reported in literature.
The 1H NMR spectra of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD are shown in Fig. 2(a, c, e, g, i & k). The peaks seen in the spectra of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples in the range δ = 0.5–1.75 ppm were employed to distinguish the presence of sp3/sp2/sp alkyl groups. Additionally, the characteristic peaks belonging to alkyl chains attached to CO groups such as aldehyde/ketone/ester (aryl/alkyl–CHO; aryl/alkyl–CO–aryl/alkyl; aryl/alkyl–CH2–COO–aryl/alkyl) or allylic protons (Ar–CH3) in the range δ = 2.0–3.0 ppm were also observed in all samples. This study confirms that in the RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples, there is a probability that aryl rings are linked with alkyl/aryl moieties across ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–O–C, C–C/–C(O)–O–C– (ether, ester, etc.) and C–C linkages. The NMR peaks seen in the range δ = 3.5–4.5 ppm in the spectra of RE, NSR-1 h/2 h/3 h/4 h linkages and N–S@RCD samples with varying concentration confirms the presence of alkoxy group such as –OCH3; –OCH2–aryl; alkyl and aryl/alkyl–COOCH2–aryl/alkyl groups. Thus, this study emphasized that RE and NSR-1 h/2 h/3 h/4 h samples contain C–O–C and –C(O)–O–C– linkages through which aryl/alkyl units are probably linked together. A significant peak observed in the range δ = 3.5–4.5 ppm for the RE sample derived from rose petal suggests the presence of higher concentration of alkyl (sp3) type moieties. However, as the reaction time of intermediates (1 h to 4 h) increased, minimal peaks with decreased intensities in the range of δ = 3.5–4.5 ppm (Fig. 3(c, e, g & i)) were observed. Moreover, inferior intensity of these peaks in the spectrum of the N–S@RCD sample shows that the structure of N–S@RCD is not similar as that of RE. It may be plausible that during the hydrothermal treatment, the alkyl groups condensed into the aryl moieties. The peaks characteristic to alkene-type moiety linked to aryl or alkyl groups (Ar/R′–CHCH–) were observed in the range of δ = 4.5–6.0 ppm in the spectra of RE, NSR-1 h, NSR-2 h, NSR-4 h intermediates and N–S@RCD sample. However, these peaks appeared with varying intensities, which indicates the competition for the transformation of alkyl hydroxyl into alkene groups via dehydration pathways under hydrothermal conditions. The conversion of alkyl alcohols into alkene via dehydration pathways is known in literature.10 However, proton peaks for alkene groups disappeared in the spectrum of NSR-3 h intermediate. Similar broad and hump peaks were perceived in the spectra of NSR-1 h, NSR-2 h, NSR-4 h intermediates and N–S@RCD sample. These peaks appeared because of the hydroxyl groups associated with the polysaccharide-rich initial bio-precursor; also, changes occurred in the peak intensities and positions because of different chemical environments around the hydroxyl groups. Moreover, the presence of aryl moieties was confirmed in the spectra of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples by perceiving peaks between δ = 6.0 and 8.5 ppm. Significantly, the aryl peaks in N–S@RCD spectrum exhibited higher intensity compared to those in the RE and NSR-1 h/2 h/3 h/4 h spectra. This also confirms that during the hydrothermal treatment, the alkyl groups were converted into aryl moieties. Observance of these aryl units in the spectra of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples was corroborated by ATR (∼1600 ± 10 and ∼1520 ± 10 cm−1) and UV-Vis spectra (273 ± 2, 280 ± 2 nm and 315 ± 2 nm). Furthermore, as the reaction time increased from 1 h to 5 h, the UV-Vis spectra showed a consistent enhancement in the peak intensities (Fig. S4†).
C–O–C, C–C/–C(O)–O–C– (ether, ester, etc.) and C–C linkages. The NMR peaks seen in the range δ = 3.5–4.5 ppm in the spectra of RE, NSR-1 h/2 h/3 h/4 h linkages and N–S@RCD samples with varying concentration confirms the presence of alkoxy group such as –OCH3; –OCH2–aryl; alkyl and aryl/alkyl–COOCH2–aryl/alkyl groups. Thus, this study emphasized that RE and NSR-1 h/2 h/3 h/4 h samples contain C–O–C and –C(O)–O–C– linkages through which aryl/alkyl units are probably linked together. A significant peak observed in the range δ = 3.5–4.5 ppm for the RE sample derived from rose petal suggests the presence of higher concentration of alkyl (sp3) type moieties. However, as the reaction time of intermediates (1 h to 4 h) increased, minimal peaks with decreased intensities in the range of δ = 3.5–4.5 ppm (Fig. 3(c, e, g & i)) were observed. Moreover, inferior intensity of these peaks in the spectrum of the N–S@RCD sample shows that the structure of N–S@RCD is not similar as that of RE. It may be plausible that during the hydrothermal treatment, the alkyl groups condensed into the aryl moieties. The peaks characteristic to alkene-type moiety linked to aryl or alkyl groups (Ar/R′–CHCH–) were observed in the range of δ = 4.5–6.0 ppm in the spectra of RE, NSR-1 h, NSR-2 h, NSR-4 h intermediates and N–S@RCD sample. However, these peaks appeared with varying intensities, which indicates the competition for the transformation of alkyl hydroxyl into alkene groups via dehydration pathways under hydrothermal conditions. The conversion of alkyl alcohols into alkene via dehydration pathways is known in literature.10 However, proton peaks for alkene groups disappeared in the spectrum of NSR-3 h intermediate. Similar broad and hump peaks were perceived in the spectra of NSR-1 h, NSR-2 h, NSR-4 h intermediates and N–S@RCD sample. These peaks appeared because of the hydroxyl groups associated with the polysaccharide-rich initial bio-precursor; also, changes occurred in the peak intensities and positions because of different chemical environments around the hydroxyl groups. Moreover, the presence of aryl moieties was confirmed in the spectra of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples by perceiving peaks between δ = 6.0 and 8.5 ppm. Significantly, the aryl peaks in N–S@RCD spectrum exhibited higher intensity compared to those in the RE and NSR-1 h/2 h/3 h/4 h spectra. This also confirms that during the hydrothermal treatment, the alkyl groups were converted into aryl moieties. Observance of these aryl units in the spectra of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples was corroborated by ATR (∼1600 ± 10 and ∼1520 ± 10 cm−1) and UV-Vis spectra (273 ± 2, 280 ± 2 nm and 315 ± 2 nm). Furthermore, as the reaction time increased from 1 h to 5 h, the UV-Vis spectra showed a consistent enhancement in the peak intensities (Fig. S4†).
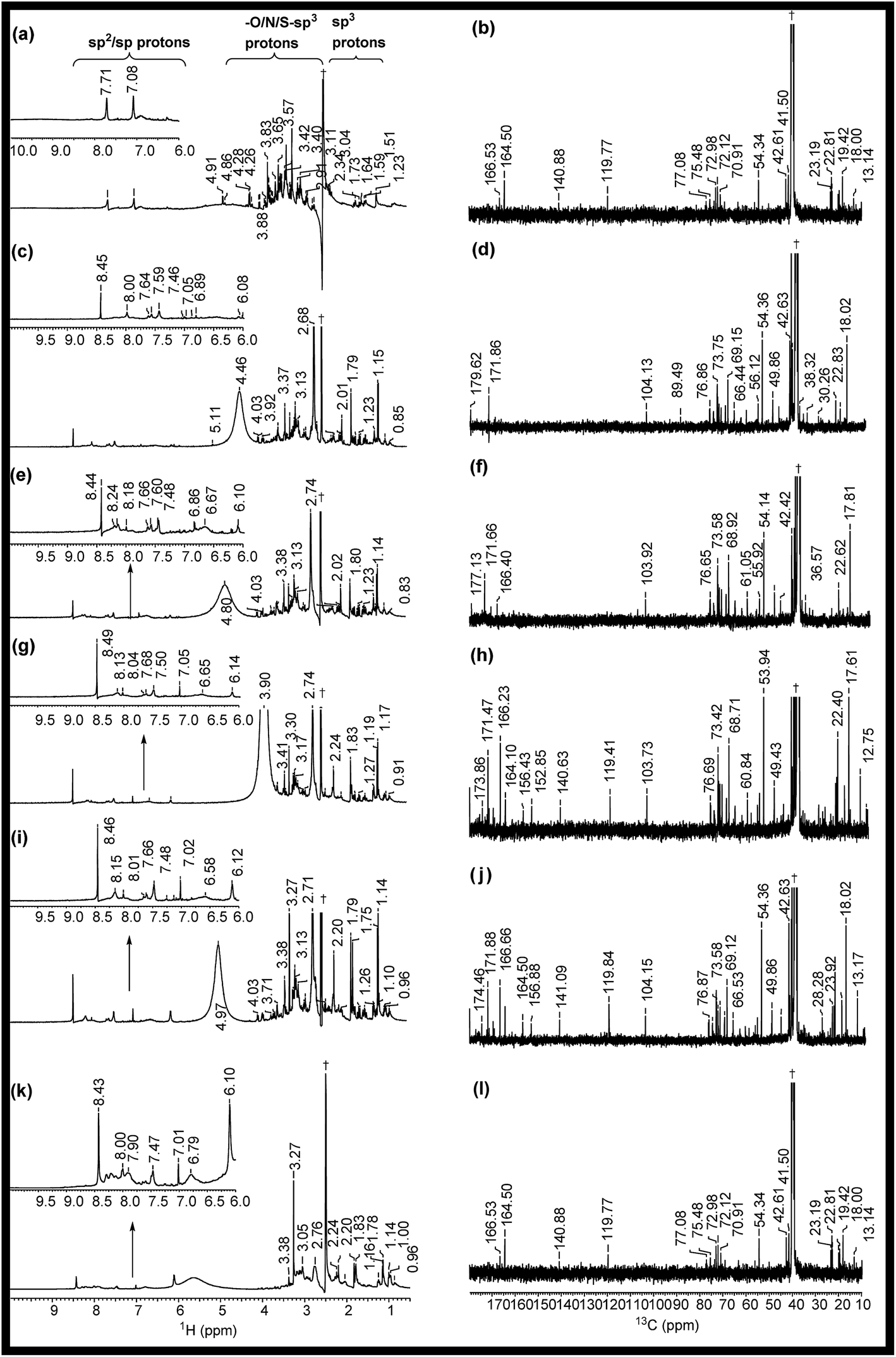 | ||
| Fig. 2 1H, 13C (1D) NMR spectra of (a, b) RE, (c, d) NSR-1 h, (e, f) NSR-2 h, (g, h) NSR-3 h, (i, j) NSR-4 h intermediates and (k, l) N–S@RCD. The spectra were recorded in dimethyl sulfoxide-d6 solvent and δ = 2.50 and 39.50 ppm were considered as reference peaks. †Peak for the dimethyl sulfoxide-d6 solvent. | ||
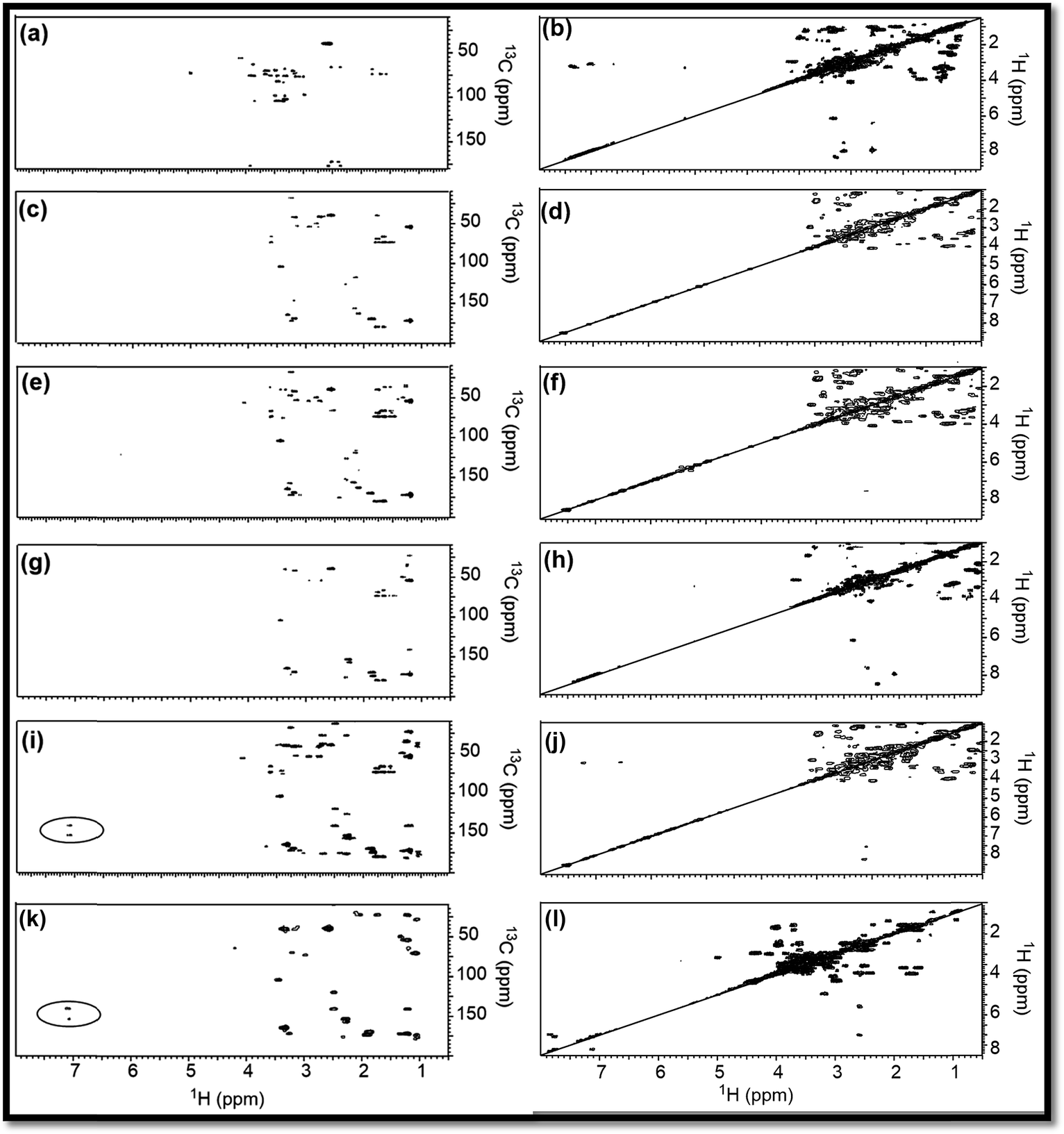 | ||
| Fig. 3 HMBC, COSY NMR (2D) spectra of (a, b) RE, (c, d) NSR-1 h, (e, f) NSR-2 h, (g, h) NSR-3 h, (i, j) NSR-4 h intermediates and (k, l) N–S@RCD. The spectra were recorded in dimethyl sulfoxide-d6 solvent and the δ (13C/1H) = 39.50/2.50 ppm is considered as a reference signal. | ||
To validate the observations from 1H NMR, 13C NMR spectra for RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples (Fig. 2(b, d, f, h, j & l)) were also recorded. For all samples, peaks of the alkyl chains (–CH3; –CH2–; CH–; C; CH3CO–) were observed in the range δ = 10.0–50.0 ppm. However, variation in the peak intensities confirmed that their concentrations in RE, NSR-1 h/2 h/3 h/4 h, and N–S@RCD samples were dissimilar. For instance, RE had higher intensity peaks for –CH3; –CH2–; CH–; C; CH3CO– (δ = 10–50 ppm) compared to NSR-1 h/2 h/3 h/4 h and N–S@RCD samples.11 As the reaction time extended, intensities and number of peaks decreased, which is attributed to polycondensation (Fig. 2(d, f, h & j)). It is plausible because of the aromatization of alkyl groups and formation of aryl groups during the hydrothermal treatment at a higher temperature (180 °C). The observance of linkages between two carbons (Ar/C–X–C/Ar) in RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples via heteroatoms (X = O, N & S) was confirmed by observing peaks in the range of δ = 50.0–90.0 ppm. The dissimilarity in intensities of the peaks in all samples confirms that each sample has a different alkyl and aryl structure. Presence of sp2 alkene/or aryl moieties was ensured by observing peaks between δ = 100 and 140 ppm. The observation of variable aryl moieties is corroborated via1H NMR (δ = 6.0–8.5 ppm), ATR (∼1600 and ∼1510 cm−1) and UV-Vis spectra (280 ± 5 nm and 320 ± 5 nm). Peaks in the range of 170–180 ppm, which correspond to carboxyl/amide groups, were seen in the spectrum of the N–S@RCD samples.2,3
To support the explanations derived from 1D NMR, 2D NMR homo (1H/1H) and hetero (13C/1H) correlation were studied with the help of HMBC and COSY NMR, respectively, for RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples (Fig. 3). By employing this study, the structure of RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD was elucidated between the homo (H–H) and hetero (C–H) signals because of the sp3, sp2 and sp carbon–hydrogen or hydrogen–hydrogen atomic correlations. Moreover, varying chemical compositions of the major inter-structural linkages (C–X–C and C–C) and moieties (alkyl and aryl) were present in all samples.
As observed in the 2D (HMBC) NMR spectra shown in Fig. 3(a, c, e, g, i & k), major concentration of saturated alkyl chain and heteroatoms next to carbon signals were observed in the range δC/δH = 10.0–50.0/0.5–2.50 ppm and δC/δH = 50.0–90.0/2.5–6.0 ppm, respectively, for all samples.12 Moreover, few signals for aryl moieties between δC/δH = 100.0–140.0/6.0–8.5 ppm were seen in NSR-1 h/2 h/3 h/4 h and N–S@RCD samples (Fig. 3(i & k)), which further confirms that the alkyl groups were converted into aryl moieties during the hydrothermal treatment.
Although 1D (1H and 13C) and 2D (HMBC) NMR studies confirmed the variable intensities of alkyl and aryl signals in RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD, the mode of proton–proton homo interaction was studied by employing 2D (COSY) NMR for RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples. As shown in Fig. 3(b, d, f, h, j, & l), saturated alkyl and heteroatoms next to carbon atom intra-molecular H–H interaction was detected in RE, NSR-1 h/2 h/3 h/4 h and N–S@RCD samples. Moreover, in N–S@RCD, few signals for aryl proton interactions were detected. It is understood that N–S@RCD sample contains both alkyl and aryl moieties (Fig. 3l).
In this text, assorted bulk (PXRD, elemental analysis, etc.) and molecular (UV-Vis, fluorescence, ATR, 1D and 2D NMR, etc.) level physico-chemical characterization techniques were employed to anticipate the polycondensation during the hydrothermal treatment. The plausible mechanism for the formation of carbon dots from rose petals involves carbonisation of polysaccharide and other constituents present in rose petals, which was corroborated by the PXRD ([200] plane, sp2 graphene carbon), ATR (2929, 1638 and 1459 cm−1, –NH, CN and C–N respectively), XPS (400.02 eV, N1s), elemental analysis, (C/N ratio of RE, RE-I and N–S@RCD 10.08, 1.47 & 2.91 respectively) and other analytical techniques. Moreover, the decomposition of petal constituents by hydrothermal treatment involved hydrolysis and dehydration, which led to the synthesis of primarily C5 and C6 sugars and furan (furfural, 5-hydroxymethyl furfural, etc.), respectively. Dehydration of RE was established via ATR (1740, and 1638 cm−1, CO and CN respectively), UV-Vis (281 nm and 320 nm corresponding to π–π* and n–π*) and the appearance of peaks and signals in 1D (1H, 6.0–8.5 ppm and 13C, 100–140 ppm) and 2D (HMBC & COSY) NMR. This absorbance enhancement in UV-Vis spectra of NSR-1 h to NSR-4 h intermediates agrees with the hydrolysis and dehydration of RE via hydrothermal treatments and is renovated into sugars and aromatic moieties, respectively. The presence of cysteine helps in the acid hydrolysis, whereas ethylenediamine helps in the dehydration of polysaccharide extracted from rose petal as detected via the aforementioned assorted measurement techniques. Subsequently, polymerisation and polycondensation occurred, which were demonstrated by various bulk and molecular level analytical techniques. Furthermore, nitrogen- and sulphur-containing aromatic clusters were formed by aromatization and carbonisation, which was confirmed by NMR (1D and 2D), ATR, UV-Vis and XPS analyses. Once the aromatic clusters reached a critical concentration, a nuclear burst took place and N–S co-doped carbon dots were formed (Fig. 4).13–15
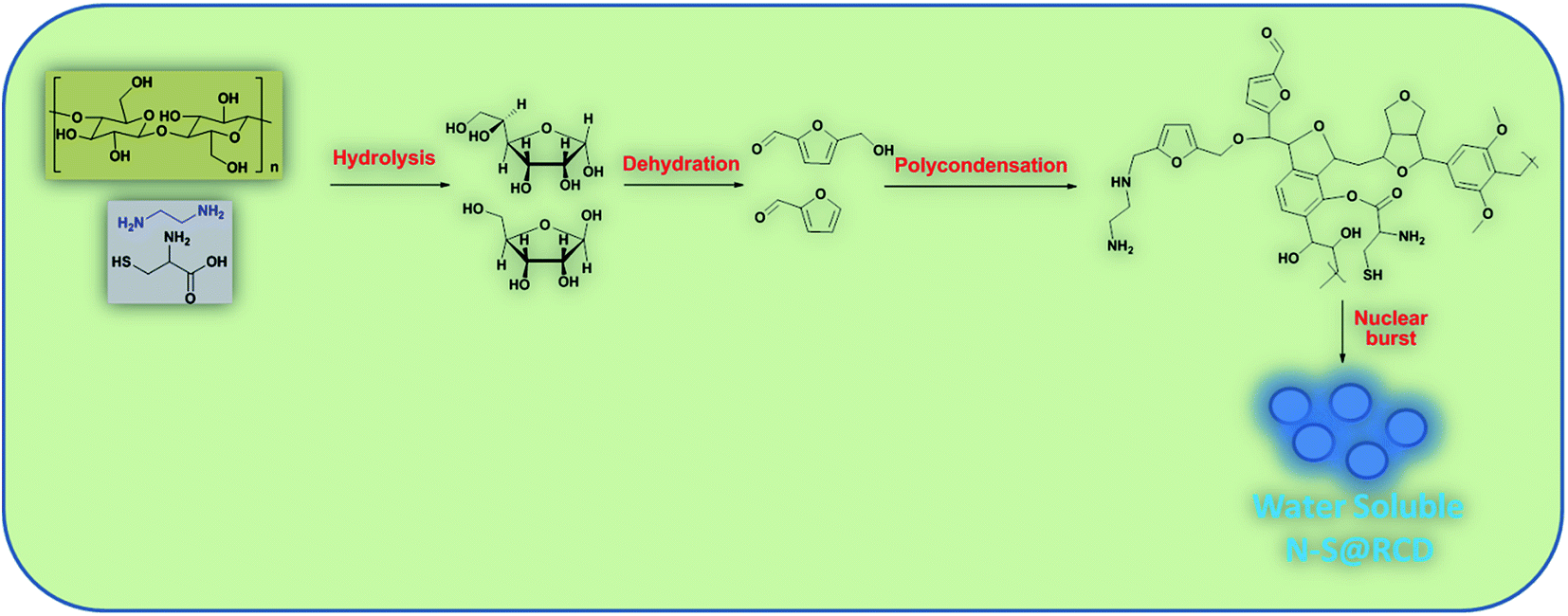 | ||
| Fig. 4 Formation mechanism of N–S@RCD from rose petals. | ||
To correlate the characteristics of carbon dots produced by different green biomass precursors, different precursor derived C-dots are summarised in Table 1 and are compared in terms of QY, particle size, elemental composition, and optical properties.
| Source | QY (%) | Size | Elemental content (%) | Elemental ratio | Excitation wavelength (nm) | Emission wavelength (nm) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| C | N | O | N/C | O/C | ||||||
| Lemon juice | 28 | 4.6 | 60.9 | 15.4 | 23.7 | 0.25 | 0.38 | 533 | 631 | 4 |
| Orange juice | 26 | 2.5 | Not provided | 0.16 | 360 | 441 | 13 | |||
| Human pee | 5.3 | 2–4 | Not provided | 325 | 392 | 16 | ||||
| Bee pollens | 6.1–12.8 | 1.1–2.1 | 42.97 | 3.89 | 39.35 | 0.09 | 0.91 | 360 | 435 | 17 |
| Coffee grounds | 3.8 | 5 | Not provided | 365 | 440 | 18 | ||||
| Cocoon silk | 38 | 70 | 56.34 | 12.82 | 25.21 | 0.22 | 0.44 | 330 | 414 | 19 |
| Papaya | 18.39 | 2–6 | 74.1 | 1.7 | 23.5 | 0.02 | 0.31 | 370 | 450 | 20 |
| Peach gum | 28.46 | 2–5 | 47.6 | 14.1 | 28.6 | 0.29 | 0.6 | 370 | 445 | 21 |
| Crab shell | 12.86–19.84 | 4–4.5 | Not provided | 350 | 425 | 22 | ||||
| Lactobacillus plantarum | 16 | 3.1 | 73.4 | 9.8 | 16.3 | 0.13 | 0.22 | 480 | 514 | 23 |
| Water chestnut and onion | 12 | 2–4 | Not provided | 370 | 475 | 24 | ||||
| Eutrophic algal blooms | 13 | 8.5 ± 5.6 | 59 | — | 41 | — | 0.69 | 360 | 438 | 25 |
| Cow manure | 65 | 2–7 | Not provided | 360 | 450 | 26 | ||||
| Sugarcane juice | 10.86 | 10.7 | 67 | 7.8 | 22.8 | 0.11 | 0.34 | 330 | 445 | 27 |
| Cabbage | 16.5 | 2–6 | 66.5 | 4.61 | 28.73 | 0.069 | 0.43 | 345 | 432 | 28 |
In conclusion, the present study demonstrates the conversion of rose petals to nitrogen and sulfur co-doped carbon dots exhibiting interesting optical properties viz. appreciable quantum yield, stable fluorescence and wavelength tuned emission. Hydrothermal formation of N–S@RCD was through aromatization and carbonization of the contents of rose extract, which was elucidated using various physico-chemical analyses. The use of green precursor or biomass offers advantages of sustainability, low cost and large-scale synthesis. Excellent photo-physical properties of N–S@RCD make it an efficient and potential candidate for various applications in sensing and biomedical imaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Sophisticated Instrumentation Centre, IIT Indore for all the characterization facilities. V. S. gratefully thanks UGC, New Delhi for research fellowship. S. K. S. thanks IIT Indore for Institute Postdoctoral fellowship. SMM thanks SERB-DST (EMR/2016/001113), Govt. of India and CSIR-New Delhi for research grants (01(2935)/18/EMR-II). We sincerely acknowledge MNCF, IISc Bangalore and ACMS, IIT Kanpur for providing XPS facility. We acknowledge CMET, Pune for TEM facility.References
- V. Sharma, P. Tiwari and S. M. Mobin, J. Mater. Chem. B, 2017, 5, 8904–8924 RSC.
- V. Sharma, N. Kaur, P. Tiwari, A. K. Saini and S. M. Mobin, Carbon, 2018, 139, 393–403 CrossRef CAS.
- K. Bankoti, A. P. Rameshbabu, S. Datta, B. Das, A. Mitra and S. Dhara, J. Mater. Chem. B, 2017, 5, 6579–6592 RSC.
- H. Ding, Y. Ji, J.-S. Wei, Q.-Y. Gao, Z.-Y. Zhou and H.-M. Xiong, J. Mater. Chem. B, 2017, 5, 5272–5277 RSC.
- S. K. Singh and P. L. Dhepe, Green Chem., 2016, 18, 4098–4108 RSC.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- J. Zhou, Y. Yang and C. Zhang, Chem. Commun., 2013, 49, 8605–8607 RSC.
- D. Sun, R. Ban, P.-H. Zhang, G.-H. Wu, J.-R. Zhang and J.-J. Zhu, Carbon, 2013, 64, 424–434 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- C. Bockisch, E. D. Lorance, H. E. Hartnett, E. L. Shock and I. R. Gould, ACS Earth Space Chem., 2018, 2, 821–832 CrossRef CAS.
- S. K. Singh and P. L. Dhepe, Bioresour. Technol., 2016, 221, 310–317 CrossRef CAS PubMed.
- S. K. Singh, S. Banerjee, K. Vanka and P. L. Dhepe, Catal. Today, 2018, 309, 98–108 CrossRef CAS.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- N. R. Pires, C. M. W. Santos, R. R. Sousa, R. C. M. de Paula, P. L. R. Cunha, J. P. A. Feitosa, N. R. Pires, C. M. W. Santos, R. R. Sousa, R. C. M. de Paula, P. L. R. Cunha and J. P. A. Feitosa, J. Braz. Chem. Soc., 2015, 26, 1274–1282 CAS.
- J. B. Essner, C. H. Laber, S. Ravula, L. Polo-Parada and G. A. Baker, Green Chem., 2015, 18, 243–250 RSC.
- J. Zhang, Y. Yuan, G. Liang and S.-H. Yu, Adv. Sci., 2015, 2, 1500002 CrossRef PubMed.
- P.-C. Hsu, Z.-Y. Shih, C.-H. Lee and H.-T. Chang, Green Chem., 2012, 14, 917–920 RSC.
- W. Li, Z. Zhang, B. Kong, S. Feng, J. Wang, L. Wang, J. Yang, F. Zhang, P. Wu and D. Zhao, Angew. Chem., Int. Ed. Engl., 2013, 52, 8151–8155 CrossRef CAS PubMed.
- N. Wang, Y. Wang, T. Guo, T. Yang, M. Chen and J. Wang, Biosens. Bioelectron., 2016, 85, 68–75 CrossRef CAS PubMed.
- J. Liao, Z. Cheng and L. Zhou, ACS Sustainable Chem. Eng., 2016, 4, 3053–3061 CrossRef CAS.
- Y.-Y. Yao, G. Gedda, W. M. Girma, C.-L. Yen, Y.-C. Ling and J.-Y. Chang, ACS Appl. Mater. Interfaces, 2017, 9, 13887–13899 CrossRef CAS PubMed.
- F. Lin, C. Li, L. Dong, D. Fu and Z. Chen, Nanoscale, 2017, 9, 9056–9064 RSC.
- Y. Hu, L. Zhang, X. Li, R. Liu, L. Lin and S. Zhao, ACS Sustainable Chem. Eng., 2017, 5, 4992–5000 CrossRef CAS.
- V. Ramanan, S. K. Thiyagarajan, K. Raji, R. Suresh, R. Sekar and P. Ramamurthy, ACS Sustainable Chem. Eng., 2016, 4, 4724–4731 CrossRef CAS.
- C. D'Angelis do E. S. Barbosa, J. R. Corrêa, G. A. Medeiros, G. Barreto, K. G. Magalhães, A. L. de Oliveira, J. Spencer, M. O. Rodrigues and B. A. D. Neto, Chem.–Eur. J., 2015, 21, 5055–5060 CrossRef PubMed.
- V. Sharma, N. Kaur, P. Tiwari and S. M. Mobin, J. Photochem. Photobiol., B, 2018, 182, 137–145 CrossRef CAS PubMed.
- A.-M. Alam, B.-Y. Park, Z. K. Ghouri, M. Park and H.-Y. Kim, Green Chem., 2015, 17, 3791–3797 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00105g |
| This journal is © The Royal Society of Chemistry 2019 |