
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge injection and transport properties of large area organic junctions based on aryl thin films covalently attached to a multilayer graphene electrode
Clément
Barraud
 *a,
Matthieu
Lemaitre
a,
Roméo
Bonnet
a,
Jacko
Rastikian
a,
Chloé
Salhani
a,
Stéphanie
Lau
b,
Quyen
van Nguyen
bc,
Philippe
Decorse
b,
Jean-Christophe
Lacroix
b,
Maria Luisa
Della Rocca
a,
Philippe
Lafarge
a and
Pascal
Martin
*b
*a,
Matthieu
Lemaitre
a,
Roméo
Bonnet
a,
Jacko
Rastikian
a,
Chloé
Salhani
a,
Stéphanie
Lau
b,
Quyen
van Nguyen
bc,
Philippe
Decorse
b,
Jean-Christophe
Lacroix
b,
Maria Luisa
Della Rocca
a,
Philippe
Lafarge
a and
Pascal
Martin
*b
aMPQ UMR 7162, Université Paris Diderot, Sorbonne Paris Cité, CNRS, F-75013 Paris, France. E-mail: Clement.barraud@univ-paris-diderot.fr
bITODYS UMR 7086, Université Paris Diderot, Sorbonne Paris Cité, CNRS, F-75013 Paris, France. E-mail: pascal.martin@univ-paris-diderot.fr
cDepartment of Advanced Materials Science and Nanotechnology, University of Science and Technology of Hanoi, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, CauGiay, Hanoi, Vietnam
First published on 26th September 2018
Abstract
The quantum interaction between molecules and electrode materials at molecule/electrode interfaces is a major ingredient in the electron transport properties of organic junctions. Driven by the coupling strength between the two materials, it results mainly in the broadening and energy shift of the interacting molecular orbitals. Using new electrode materials, such as the recently developed semi-conducting two-dimensional nanomaterials, has become a significant advancement in the field of molecular/organic electronics that opens new possibilities for controlling the interfacial electronic properties and thus the charge injection properties. In this article, we report the use of atomically thin two-dimensional multilayer graphene films as the base electrode in organic junctions with a vertical architecture. The interfacial electronic structure dominated by the covalent bonding between bis-thienyl benzene diazonium-based molecules and the multilayer graphene electrode has been probed by ultraviolet photoelectron spectroscopy and the results are compared with those obtained on junctions with standard Au electrodes. Room temperature injection properties of such interfaces have also been explored by electron transport measurements. We find that, despite strong variations of the density of states, the Fermi energy and the injection barriers, both organic junctions with Au base electrodes and multilayer graphene base electrodes show similar electronic responses. We explain this observation by the strong orbital coupling occurring at the bottom electrode/bis-thienyl benzene molecule interface and by the pinning of the hybridized molecular orbitals.
1. Introduction
The architecture of the two- and three-terminal building blocks of organic electronic devices, such as electroluminescent diodes and organic transistors, is largely inspired by their inorganic counterparts. Prototypical organic diodes are based on ITO/molecules or conducting polymer/metal (Al, Cu, Au) junctions with the thicknesses of the organic spacer ranging around 100 nm.1 Processing methods are widely developed including spin- or dip coating or evaporation.2 State-of-the art organic diodes now operate at MHz frequencies3,4 and are foreseen to be used in radio frequency identification chips.5,6 Practically, the control and understanding of the properties of the electrode/molecule interface is crucial for reaching high performances and designing functionalities,7 such as electrical rectification. Symmetric current density as a function of bias voltage (J–V) characteristics was widely observed in two-terminal devices and discussed in the case of single molecule junctions,8 monolayers9 or oligomer based organic junctions, as in the case of a large variety of aromatic nanostructures between conducting carbon electrodes.10,11It has been shown that to introduce asymmetry (rectification) in the electronic response of molecular and organic junctions, it is necessary to introduce asymmetry along the transport direction.12,13 This can be achieved either by using different contact electrodes (i.e. with different injection properties) as in organic Schottky diodes and/or by using asymmetric anchoring moieties or molecular structures as originally proposed by Aviram and Ratner.14 Using such artificial systems to create nano-rectifiers has been widely reported and recently reviewed.15 The electrode/molecule coupling also plays a crucial role in defining the amplitude of the rectification ratio (RR).15 Molecules can be either attached to electrodes via strong (chemisorption) or weak interactions (physisorption). Depending on the nature of the interactions on both sides of the junctions, the RR has been demonstrated to vary from few tenths to few thousands.15
Recently, a record RR up to 105 was reported in alkyl-ferrocene-based self-assembled monolayers in contact with a bottom electrode by chemisorption and with a top electrode by physisorption.16 The rectification effect was also shown to be strongly dependent upon the position of the Fc unit (i.e. the molecular structure) between the two electrodes.17 We have shown before that based on the diazonium approach oligo-BisThienyl Benzene (BTB) forms a high quality organic layer and leads to highly stable organic junctions.11,18 We have reported molecular diodes based on diazonium grafting in junctions of the form Au–BTB//Ti/Au where “–” and “//” denote the bottom interface and the top interface respectively.19 Those organic diodes presented a large rectification ratio RR of 1000 and we clearly demonstrated that the rectification behaviour in this configuration is proof of molecular signature even if the two metallic electrodes have different Fermi energies. Several molecules have been tested into junctions and the RR varied from 10−2 to 103 with a very good reproducibility under the same conditions.
In this article, we focus on the electrode's properties. New atomically thin electrodes made of two-dimensional materials, such as graphene, have been recently proposed and developed to replace more traditional metallic electrodes.20 Graphene, multilayer graphene (MG) and graphite were recently used as electrodes to enable contact with rubrene single crystals,21 self-assembled monolayers22–24 and even single molecules.25–30 The advantages of using graphene and MG as electrodes in molecular electronics are numerous. In terms of device operation graphene and MG are less sensitive to oxidation, diffusion and/or to electro-migration. They are also transparent in the visible light range, dopable and are envisioned to serve as a good platform for molecule adsorption.31
The strength of the chemical bonding between a graphene/graphite electrode and molecules was demonstrated to be a key ingredient over the global electronic response of single-molecule junctions.29
Here we demonstrate electrical rectification effects in the case of MG-(oligo(1-(2-bisthienyl) benzene, named BTB) diazonium-based thin film//Ti/Au junctions. We show that rectification is directly linked to the strong orbital coupling at the MG–BTB interface like in the well-studied case of Au–BTB interfaces18 and to pinning of the hybridized molecular orbitals at the top and bottom electrode/molecule interfaces. Our observation is strongly supported by a comparative analysis of those two device interfaces by electron transport measurements and ultraviolet photoelectron spectroscopy.
2. Experimental section
2.1 Multilayer graphene transfer
MG was purchased on Ni films. The Ni/MG bilayer was first covered with a thin layer of PMMA 950K deposited by spin-coating (4000 rpm) and baked at 110 °C for 10 minutes. The Ni/MG/PMMA trilayer was then gently deposited at the surface of a FeCl3 bath in order to dissolve the Ni substrate. After dissolution, the floating MG/PMMA bilayer was transferred to a deionized H2O bath for 5 minutes. MG was cleaned in successive baths following the recipe described in ref. 32 (i.e., RCA clean). It was then transferred to a deionized H2O bath and then gently “fished” using the substrate. After transfer, the sample was slowly baked from room temperature to 110 °C in order to evaporate the remaining water. The PMMA film was finally removed by immersing the sample in a warm (50 °C) acetone bath for 5 minutes and in an isopropanol bath for 2 minutes.2.2 Electro-grafting
1-(2-Bisthienyl)-4-aminobenzene (BTB) was synthesized following a published procedure.33 For electrochemical experiments, a conventional one-compartment, three-electrode cell was employed. A CHi760C potentiostat (CH Instruments, Austin, TX) was used. The auxiliary electrode was a platinum grid. A SCE (3 M KCl) in an ACN/LiClO4 bridge was used as a reference electrode.2.3 Experimental setup
DC electrical measurements were performed by applying a DC voltage signal to the junction top electrode by a low-noise voltage source (Yokogawa GS200). The output current was measured by a low-noise current–voltage amplifier connected to the bottom electrode. The amplified output was measured using a digital voltmeter (Agilent 34405A). The sweep rate was 100 mV s−1.2.4 Spectroscopy characterization
XPS and UPS analyses were performed in an ultrahigh vacuum system (VG Scientific ESCALAB 250) with a base pressure of 2 × 10−10 mbar. XPS was performed using an Al KR X-ray source (1486.6 eV) and a microfocused monochromatic and magnetic lens. The spectra were acquired in the constant analyzer energy mode with pass energies of 150 and 40 eV for the survey and the narrow regions, respectively. UPS was carried out using monochromatic He I (21.21 eV) emission together with a toroidal mirror monochromator.2.5 Yield and reproducibility
A high percentage (80%) of working and reproducible devices is found in the case of Au–BTB//Ti/Au junctions in agreement with previous reports.19 The yield of working devices is reduced when MG is inserted (around 20%). It may be explained by the relatively high peak-to-peak roughness (<10 nm) due to folds of the MG electrode compared to a more standard evaporated Au electrode (<1 nm) as revealed by the AFM image presented in Fig. 1(c). All the J–V characteristics presented are stable over periods of several hours and do not show a significant hysteresis under the used experimental conditions.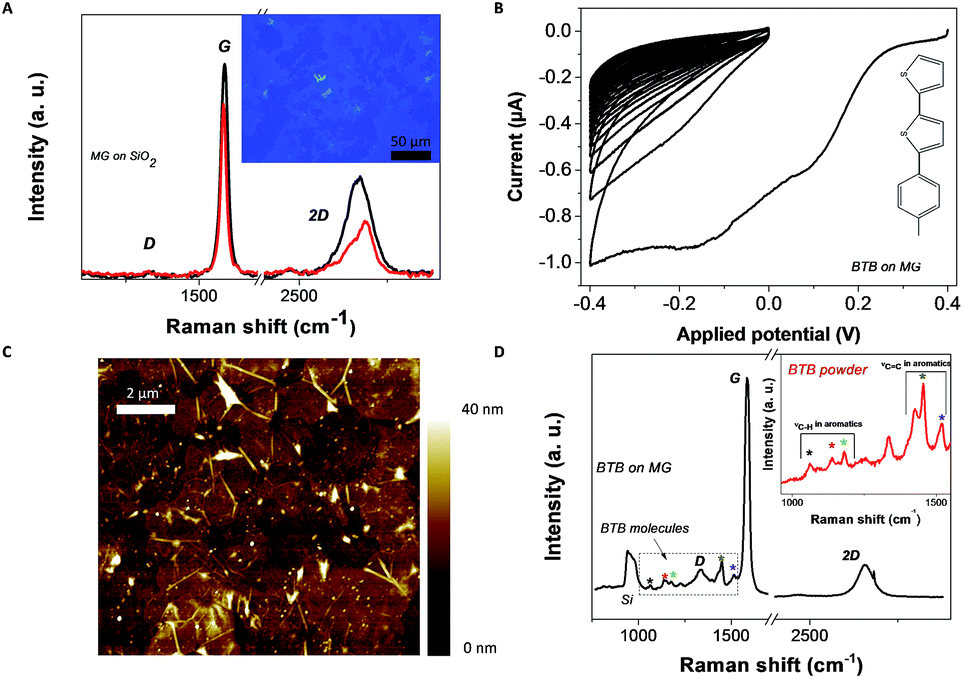 | ||
| Fig. 1 (a) Two Raman spectra acquired at different locations on MG after transfer on a 280 nm SiO2 substrate (laser wavelength: 633 nm). Inset: optical image of a MG film deposited over a 500 nm SiO2 substrate. (b) Cyclic voltammograms of (1-(2-bisthienyl)-4-aminobenzene) (BTAB) electro-reduction in acetonitrile (10 cycles at 0.1 V s−1) over the MG electrode. Inset: molecular structure of the BTB molecule. (c) Atomic force microscopy topography image of a MG grafted with BTB molecules acquired under ambient conditions in tapping mode (10 × 10 μm2). (d) Raman spectrum acquired on MG/BTB surfaces highlighting the region between 1000 cm−1 and 1550 cm−1 where the resonances associated with the oligo(BTB) molecules are observed. Inset: Raman spectrum acquired on a high-purity 1-(2-bisthienyl) benzene amine powder revealing similar resonances. | ||
3. Results and discussion
MG–BTB//Ti/Au junctions were fabricated by first transferring a MG layer (purchased from Graphene Supermarket®) over an insulating 280 nm SiO2 substrate. The transfer technique is adapted from ref. 32 and is described in the Experimental section. Two Raman spectra acquired on MG after transfer (laser wavelength: 633 nm) in two different locations are shown in Fig. 1(a), together with an optical microscope image (inset of Fig. 1(a)) of a 500 nm SiO2 substrate covered by the transferred MG. The observed changes in the shape and relative heights and widths of the G and 2D peaks reveal that the number of graphene layers is not uniform over the entire MG film.34 This is expected as the MG thickness is specified from Graphene Supermarket® to be equal to 4 monolayers in average with local variations between 1 and 7 monolayers. The absence (black line) or the weak (red line) D peak arising around 1350 cm−1 confirms the excellent structural quality of the MG after transfer. The MG is then patterned by optical lithography and O2 plasma (70 W during 120 s) in long stripes of 10 mm long and 40 μm wide.After fabrication of the patterned MG electrodes, electrochemical grafting of BTB was carried out in solution by cyclic voltammetry. Fig. 1(b) shows a cyclic voltammetry characteristic of the reduction of the 1-(2-bisthienyl) benzene diazonium cation at the surface of a MG electrode. It shows an irreversible wave during the first scan [+0.4 V;−0.4 V], which corresponds to the formation of aryl radicals35 at the proximity of the MG electrode. In the following scans, the current strongly drops to zero as the conducting surface of the electrode becomes passivated by the growth of an oligo-BTB thin layer.33 BTB thin films are electro-active and can be easily p-doped at a potential close to 0.5 V/SCE (saturated calomel electrode).33 As a consequence, the conductance of a BTB thin film can be switched and diode-like behavior with a high rectification ratio is observed in the electrochemical response of outer-sphere redox probes.36 At the end of the process, the MG surface is fully covered by the organic film as previously detailed for graphene and graphite surfaces.37 No trace of diazonium salt was detected by X-ray photoelectron spectrometry analysis within the resolution of the detector (not shown). The final thickness and roughness of the BTB film were determined by atomic force microscopy as shown in Fig. 1(c) and were found to be around 10 ± 1 nm. After electro-grafting, the MG electrode was again characterized by Raman spectroscopy. Fig. 1(d) shows the Raman spectrum (laser wavelength: 633 nm) acquired on the MG/BTB surface. While the main features from the graphitic structure, the D, G and 2D resonances, can still be identified, additional resonances (individually marked with colored *) are observed in the frequency range of 1000 cm−1 to 1550 cm−1. They are attributed to vibrational modes (stretching vibration for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatics (1520 cm−1 and 1450 cm−1) and C–H aromatics (1060 cm−1, 1140 cm−1 and 1180 cm−1)) within the oligo-BTB film.38,39 This is further probed by measuring the Raman spectrum of high-purity 1-(2-bisthienyl) benzene amine powder (inset of Fig. 1(d)), where clearly resonances at identical frequencies are observed. In the Raman spectra of Fig. 1(d), the sp2 graphitic structure is still evident as only the topmost layer of MG is chemically functionalized37 leaving the remaining layer of the MG structure intact. The topmost layer is sp3 hybridized giving rise to the increase of the visibility of the D peak around 1350 cm−1. This result is confirmed by measuring the resistance (not shown) of a 2.5 mm long – 40 μm wide functionalized MG stripe. A value of ∼25 kΩ is extracted in good agreement with standard electrical characterizations of MG layers.40 The top electrode is finally patterned in cross-bar geometry by optical lithography. A thin film of Ti (1 nm)/Au (50 nm) is then evaporated at pressure <10−7 mbar and with a low rate <0.1 nm s−1 leading to the MG–BTB//Ti/Au organic/inorganic heterostructures without titanium oxide formation. As we have shown previously,10,18,19,41–43 such an organic layer was sufficiently robust to allow the direct evaporation of metallic atoms. The titanium layer acts as an adhesion layer for gold evaporation but also as a blocking layer to avoid gold atom diffusion through the organic layer.
C aromatics (1520 cm−1 and 1450 cm−1) and C–H aromatics (1060 cm−1, 1140 cm−1 and 1180 cm−1)) within the oligo-BTB film.38,39 This is further probed by measuring the Raman spectrum of high-purity 1-(2-bisthienyl) benzene amine powder (inset of Fig. 1(d)), where clearly resonances at identical frequencies are observed. In the Raman spectra of Fig. 1(d), the sp2 graphitic structure is still evident as only the topmost layer of MG is chemically functionalized37 leaving the remaining layer of the MG structure intact. The topmost layer is sp3 hybridized giving rise to the increase of the visibility of the D peak around 1350 cm−1. This result is confirmed by measuring the resistance (not shown) of a 2.5 mm long – 40 μm wide functionalized MG stripe. A value of ∼25 kΩ is extracted in good agreement with standard electrical characterizations of MG layers.40 The top electrode is finally patterned in cross-bar geometry by optical lithography. A thin film of Ti (1 nm)/Au (50 nm) is then evaporated at pressure <10−7 mbar and with a low rate <0.1 nm s−1 leading to the MG–BTB//Ti/Au organic/inorganic heterostructures without titanium oxide formation. As we have shown previously,10,18,19,41–43 such an organic layer was sufficiently robust to allow the direct evaporation of metallic atoms. The titanium layer acts as an adhesion layer for gold evaporation but also as a blocking layer to avoid gold atom diffusion through the organic layer.
An optical image of the devices accompanied by a schematic of a polarized junction is depicted in Fig. 2(a). All the junctions were electrically characterized using standard DC measurement techniques. Details about the experimental setup can be found in the Experimental section. Room temperature J–V characteristics for five different MG–BTB//Ti/Au junctions with area ranging from 5 × 40 μm2 to 20 × 40 μm2 are plotted in Fig. 2(b). A rectifying effect is revealed with a rectification ratio RR = |J(−2 V)/J(+2 V)| up to 100. The current density in the conducting states at negative voltages (i.e. current flowing from the bottom electrode to the top electrode) reaches values up to 0.15 A cm2. The measured resistance of the devices is of the order of 1 to 100 MΩ depending on the bias voltage. It remains well above the series resistance of graphene such that it should not impact the measurements in the DC regime.
 | ||
| Fig. 2 (a) Optical image of MG–BTB//Ti/Au junctions with various dimensions of junctions. A schematic view of a polarized junction is presented in the black box. (b) Average single sweep J–V characteristic measured at room temperature for MG–BTB//Ti/Au junctions with standard deviation (over five characterized devices). Dimensions of the measured junctions were ranging from 5 × 40 μm2 to 20 × 40 μm2. | ||
To gain insights on the electronic properties of the different hybrid interfaces, we have performed ultraviolet photoelectron spectroscopy (UPS) of a bare MG surface and of a MG–BTB interface. As mentioned already, BTB molecules are known to be p-type molecules and transport occurs preferentially through the occupied molecular orbitals.10,18 For UPS spectroscopy the MG–BTB bilayer was fabricated following the same procedure with a smaller BTB thickness (3–4 nm) in order to get relevant information for the injection barrier. The secondary electron cut-off and valence band spectra for MG (black line) and for MG–BTB (red line) are represented in Fig. 3(a) and (b) respectively. The very small density of states at the Fermi energy of MG gives the observed rather weak increase of the signal towards higher binding energies compared to standard metals.44 From these measurements, we can extract the vacuum level shift (ΔE = 0.2 eV), generally interpreted as the presence of dipoles at the electrode/molecule interfaces45 and the injection barrier 3–4 nm away from the interface (φMG = 1.1 eV). Both values of vacuum level shift and injection barrier are comparable with reported values of BTB and other diazonium compounds grafted on pyrolized photoresist film by Sayed et al.10 The extracted work function WF of the MG surface was 4.4 eV consistent with the literature46–48 and was found to be very close to the WF of a Ti surface (4.3 eV),49 the top electrode of our devices. These values are reported on the energy diagram of the MG–BTB interface presented in Fig. 3(c). First the value of WF indicates that the two electrodes are at the same energy level at 0 V. In such a case, without taking into account the coupling at the interfaces, we should expect symmetric J–V characteristics and no rectification appears. However the rectification observed indicates that the coupling at both interfaces is different and we attempt to explain this behaviour in the following. The first molecular monolayer is expected to be strongly coupled to MG and molecular orbitals can be broadened by several eV50 and on resonance with the Fermi energy of the MG electrode.51 In the specific case of diazonium molecules, a strong C–C chemical bond is expected between the molecule and the carbon-based surface.10 In case of chemisorption of molecules on metals, orbitals' broadening up to few eV has been calculated for instance, for Co/Alq3 surfaces.51 It then tends to decay within the molecular layer as also shown for a Co/Alq3interface and experimentally measured for N,N′-bis-(1-naphthyl)-N,N′-diphenyl1-1,1-biphenyl1-4,4′-diamine (α-NPD) deposited on Au.52
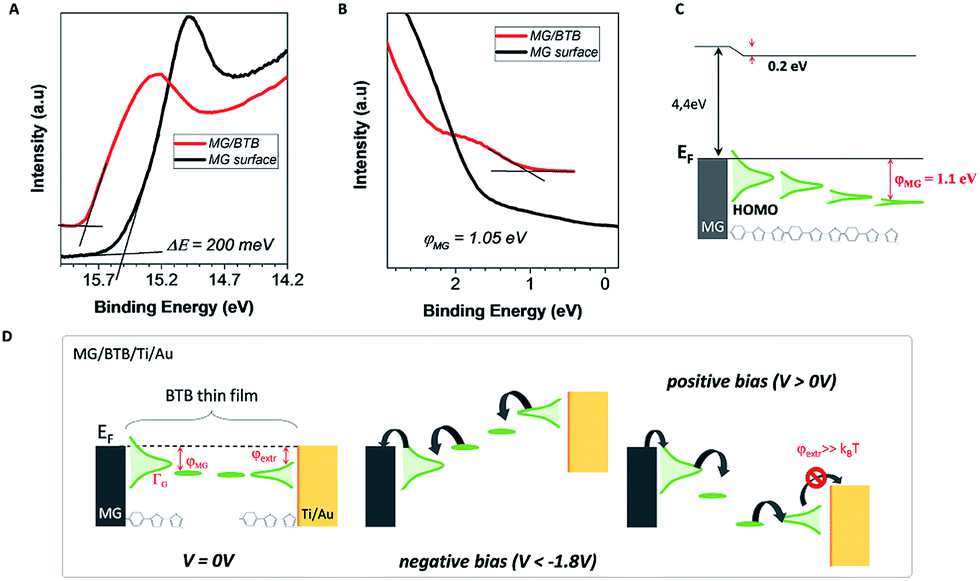 | ||
| Fig. 3 (a) and (b) UPS spectra acquired on a bare MG surface (black line) and on a MG/BTB surface (red line). The spectra were taken with the sample biased at −5 V to clear the detector work function. (c) Schematic of the interfacial electronic structure with the reported values of dipoles and barrier. The HOMO of BTB is strongly broadened and shifted in contact with the MG electrode. The orbital broadening decays with respect to the distance from the electrode. (d) Proposed energy level diagrams for a MG–BTB//Ti/Au junction based on our results. The black arrows indicate the charge transport direction. (Left) Electronic structure of the junction at zero bias voltage. (Middle) At negative bias voltages in the passing state. (Right) At positive bias voltages. In the last case, the extraction energy barrier (φextr) is higher than the thermal energy preventing the current flow. | ||
The top BTB//Ti/Au interface has been recently characterized by XPS highlighting the presence of carbide moieties19 and a HOMO broadening is thus expected. Based on electron transport measurements and UPS analysis, we propose in Fig. 3(d) a transport mechanism for MG-based organic junctions at zero, negative, and positive bias voltages that explains the rectification observed in MG-based organic junctions. Pinning of the HOMO at both interfaces is considered with a strong broadening at the MG interface due to the strong C–C coupling, it results in a current flow at a negative bias voltage below −1.8 V. On the opposite, at the BTB/Ti/Au top interface, the extraction energy barrier is higher than the thermal energy preventing the current flow at a positive bias voltage.19 Hopping transport via polaronic states is considered in between the represented interfacial molecular orbitals.18 Note that the LUMO level is not represented due to the large HOMO–LUMO gap (3.1 eV) of the BTB molecule.19
To highlight the effect of the orbital coupling, we compare now the J–V characteristics presented in Fig. 2(c) to the J–V characteristics measured under the same conditions on Au–BTB//Ti/Au, with BTB film of identical thicknesses. We show in Fig. 4(a) and (b) a schematic of an Au–BTB//Ti/Au junction and four different room temperature J–V characteristics. They clearly show a rectification effect similar to that of MG based electrode junctions in Fig. 2(c), with RR up to 170 at ± 2 V and current densities up to 0.3 A cm2. The similar electronic responses of the two systems suggest that the coupling intensity at the MG–BTB interface is similar to the one observed at Au–BTB interfaces, opening the avenue for diazonium-based organic films over MG electrodes as a possible platform for molecular junctions. We have thus performed UPS analysis of Au surfaces and Au–BTB interfaces to compare with the data extracted from MG-based surfaces. The secondary electron cut-off and valence band spectra are given in Fig. 4(c) and (d). The Au surface spectrum (black line) is in agreement with the literature.44,53 UPS spectra show very sharp steps at the Fermi energy due to the strong density of states present in the metal in opposition to MG (Fig. 3(b) – black line). This sharp step disappears as soon as the BTB film is inserted due to the much lower density of states of the organic layer. The measured injection barrier of the Au–BTB interface is ∼1.2 eV, approximately 150 meV higher than the one measured for the MG/BTB case. The value of interfacial dipoles is ∼1.6 eV so largely different compared to the MG–BTB case but comparable with pentacene, Alq3 or α-NPD52 or thiols on Au electrodes.54 These values are reported in the schematics in Fig. 4(e).
 | ||
| Fig. 4 (a) Schematic of an Au–BTB//Ti/Au organic junction. (b) Average single sweep J–V characteristic measured at room temperature for Au–BTB//Ti/Au junctions with standard deviation (over ten characterized devices). Dimensions of the junctions are 20 × 20 μm2. (c) and (d) UPS spectra acquired on a bare Au surface (black line) and on an Au–BTB surface (red line). The spectra were taken with the sample biased at −5 V to clear the detector work function. (e) Schematic of the interfacial electronic structure with the reported values of dipoles and barrier. | ||
This comparative study between the MG–BTB//Ti/Au and the Au–BTB//Ti/Au devices reveals surprisingly that the electronic responses (J–V) are weakly dependent on the bottom electrode nature, on their density of states at the Fermi energy, on their interfacial dipole with the molecules and on the injection barriers. We can note that the current density for both devices is in the same order (slightly smaller value for the graphene junction) and the rectification ratio is up to 100 for both. These values are comparable with some of the recently reviewed organic rectifiers1 mainly working as Schottky diodes or p–n junctions with thicker organic films (∼100 nm)55,56 and higher operating voltages (>±5 V).3,57
Our results partially rule out the recent mechanism proposed to explain rectification reported in a slightly different system: graphite/amino-benzene-based molecule/Au tip junction using a scanning tunneling microscope.29 This mechanism relies on the rapidly varying and highly dispersive nature of the density of states of graphite around the Fermi energy. Focusing on the electrode/molecule interface, our results agree more with the recent work of Sayed et al. in similar systems.10 They have shown that the injection barrier of PPF/diazonium-based molecule interfaces was independent of the molecular nature of the grafted layer.10 The effect was attributed to a dubbed “compression” of the molecular orbitals. Our results go a step further in this interpretation by demonstrating that the strong electronic coupling given by the BTB-diazonium moieties dominates the injection properties of the base electrode/BTB interface. They unravel the important role of the “diazonium” approach in organic electronics.
4. Conclusions
We demonstrated the use of atomically thin multilayer graphene as a soft, transparent and transferrable electrode for molecular junctions. A clear rectification effect is reported in MG–BTB//Ti/Au junctions at room temperature comparable to what was observed in Au–BTB//Ti/Au junctions whereas the WF of the MG electrodes is closer to the WF of the top electrode. This is explained by the strong orbital coupling present at the MG–BTB interface and by the pinning of hybridized molecular orbitals at the MG–BTB and BTB//Ti/Au interfaces. A more detailed BTB-thickness dependent UPS study is needed in order to get the evolution of the position and broadening of the different unoccupied molecular orbitals. Moreover, a theoretical support would be interesting to precisely determine the interfacial band structure. This work is a further step towards all carbon molecular junctions which are already proved to more stable than those using classical Au electrodes.10 Such devices are already used in clipping elements in overdrive circuits.58Author contribution
All authors contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge C. Manquest, P. Filloux and S. Suffit for technical supports within the clean-room of the Laboratoire Matériaux et Phénomènes Quantiques (UMR 7162) at the Université Paris Diderot and M. Marsi at the Université ParisSaclay for helpful discussions about UPS data. This work is supported by the 2DSPIN project from the “Ville de Paris” Emergence program. ANR (Agence Nationale de la Recherche) and CGI (Commissariat à l'Investissement d’Avenir) are gratefully acknowledged for their financial support of this work through Labex SEAM (Science and Engineering for Advanced Materials and devices) ANR 11 LABX086, ANR 11IDEX 05 02. This work has also been supported by the Region Ile-de-France in the framework of DIM Nano-K (SMS project).Notes and references
- T. M. Kraft, P. R. Berger and D. Lupo, Flexible Printed Electron., 2017, 2, 033001 CrossRef.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- C. M. Kang, H. Shin and C. Lee, MRSCommun., 2017, 7, 755–769 CAS.
- E. Cantatore, T. C. T. Geuns, G. H. Gelinck, E. Van Veenendaal, A. F. A. Gruijthuijsen, L. Schrijnemakers, S. Drews and D. M. De Leeuw, IEEE J. Solid-State Circuits, 2007, 42, 84–92 Search PubMed.
- J. Semple, D. G. Georgiadou, G. Wyatt-Moon, G. Gelinck and T. D. Anthopoulos, Semicond. Sci. Technol., 2017, 32, 123002 CrossRef.
- A. Dodabalapur, Mater. Today, 2006, 9, 24–30 CrossRef CAS.
- D. Cahen, A. Kahn and E. Umbach, Mater. Today, 2005, 8, 32–41 CrossRef CAS.
- M. Taniguchi, M. Tsutsui, R. Mogi, T. Sugawara, Y. Tsuji, K. Yoshizawa and T. Kawai, J. Am. Chem. Soc., 2011, 133, 11426–11429 CrossRef CAS PubMed.
- X. Shi, Z. Dai and Z. Zeng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 235412 CrossRef.
- S. Y. Sayed, J. A. Fereiro, H. Yan, R. L. McCreery, A. Johan, A. J. Bergren and A. Johan, Proc. Natl. Acad. Sci., 2012, 109, 11498–11503 CrossRef CAS PubMed.
- H. Yan, A. J. Bergren, R. McCreery, M. L. Della Rocca, P. Martin, P. Lafarge and J. C. Lacroix, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5326–5330 CrossRef CAS PubMed.
- V. Mujica, M. A. Ratner and A. Nitzan, Chem. Phys., 2002, 281, 147–150 CrossRef CAS.
- C. Van Dyck and M. A. Ratner, Nano Lett., 2015, 15, 1577–1584 CrossRef CAS PubMed.
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- R. M. Metzger, Chem. Rev., 2015, 115, 5056–5115 CrossRef CAS PubMed.
- X. Chen, M. Roemer, L. Yuan, W. Du, D. Thompson, E. del Barco and C. A. Nijhuis, Nat. Nanotechnol., 2017, 12, 797–803 CrossRef CAS PubMed.
- L. Yuan, N. Nerngchamnong, L. Cao, H. Hamoudi, E. del Barco, M. Roemer, R. K. Sriramula, D. Thompson and C. A. Nijhuis, Nat. Commun., 2015, 6, 6324 CrossRef CAS PubMed.
- P. Martin, M. L. Della Rocca, A. Anthore, P. Lafarge and J.-C. Lacroix, J. Am. Chem. Soc., 2012, 134, 154–157 CrossRef CAS PubMed.
- Q. van Nguyen, P. Martin, D. Frath, M. L. Della Rocca, F. Lafolet, C. Barraud, P. Lafarge, V. Mukundan, D. James, R. L. McCreery and J. C. Lacroix, J. Am. Chem. Soc., 2017, 139, 11913–11922 CrossRef PubMed.
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2008, 8, 2458–2462 CrossRef CAS PubMed.
- C.-H. Lee, T. Schiros, E. J. G. Santos, B. Kim, K. G. Yager, S. J. Kang, S. Lee, J. Yu, K. Watanabe, T. Taniguchi, J. Hone, E. Kaxiras, C. Nuckolls and P. Kim, Adv. Mater., 2014, 26, 2812–2817 CrossRef CAS PubMed.
- S. Leitherer, P. B. Coto, K. Ullmann, H. B. Weber and M. Thoss, Nanoscale, 2017, 9, 7217–7226 RSC.
- Y. Jang, H. Jeong, D. Kim, W.-T. Hwang, J.-W. Kim, I. Jeong, H. Song, J. Yoon, G.-C. Yi, H. Jeong and T. Lee, Nanotechnology, 2016, 27, 145301 CrossRef PubMed.
- Y. Jang, S. J. Kwon, J. Shin, H. Jeong, W. T. Hwang, J. Kim, J. Koo, T. Y. Ko, S. Ryu, G. Wang, T. W. Lee and T. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 42043–42049 CrossRef CAS PubMed.
- J. O. Island, A. Holovchenko, M. Koole, P. F. A. Alkemade, M. Menelaou, N. Aliaga-Alcalde, E. Burzurí and H. S. J. van der Zant, J. Phys.: Condens. Matter, 2014, 26, 474205 CrossRef CAS PubMed.
- F. Prins, A. Barreiro, J. W. Ruitenberg, J. S. Seldenthuis, N. Aliaga-Alcalde, L. M. K. Vandersypen and H. S. J. van der Zant, Nano Lett., 2011, 11, 4607–4611 CrossRef CAS PubMed.
- E. Burzurí, J. O. Island, R. Díaz-Torres, A. Fursina, A. González-Campo, O. Roubeau, S. J. Teat, N. Aliaga-Alcalde, E. Ruiz and H. S. J. Van Der Zant, ACS Nano, 2016, 10, 2521–2527 CrossRef PubMed.
- Q. Zhang, L. Liu, S. Tao, C. Wang, C. Zhao, C. González, Y. J. Dappe, R. J. Nichols and L. Yang, Nano Lett., 2016, 16, 6534–6540 CrossRef CAS PubMed.
- A. V. Rudnev, V. Kaliginedi, A. Droghetti, H. Ozawa, A. Kuzume, M. Haga, P. Broekmann and I. Rungger, Sci. Adv., 2017, 3, e1602297 CrossRef PubMed.
- T. Kim, Z.-F. Liu, C. Lee, J. B. Neaton and L. Venkataraman, Proc. Natl. Acad. Sci., 2014, 111, 10928–10932 CrossRef CAS PubMed.
- C. Jia, B. Ma, N. Xin and X. Guo, Acc. Chem. Res., 2015, 48, 2565–2575 CrossRef CAS PubMed.
- X. Liang, B. A. Sperling, I. Calizo, G. Cheng, C. A. Hacker, Q. Zhang, Y. Obeng, K. Yan, H. Peng, Q. Li, X. Zhu, H. Yuan, A. R. Hight Walker, Z. Liu, L. M. Peng and C. A. Richter, ACS Nano, 2011, 5, 9144–9153 CrossRef PubMed.
- C. Fave, Y. Leroux, G. Trippé, H. Randriamahazaka, V. Noel, J.-C. Lacroix, G. Trippé, H. Randriamahazaka, V. Noel and J.-C. Lacroix, J. Am. Chem. Soc., 2007, 129, 1890–1891 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. Mesnage, X. Lefèvre, P. Jégou, G. Deniau and S. Palacin, Langmuir, 2012, 28, 11767–11778 CrossRef CAS PubMed.
- C. Fave, V. Noel, J. Ghilane, G. Trippé-Allard, H. Randriamahazaka and J. C. Lacroix, J. Phys. Chem. C, 2008, 112, 18638–18643 CrossRef CAS.
- J. Greenwood, T. H. Phan, Y. Fujita, Z. Li, O. Ivasenko, W. Vanderlinden, H. Van Gorp, W. Frederickx, G. Lu, K. Tahara, Y. Tobe, H. Uji-i, S. F. L. Mertens and S. De Feyter, ACS Nano, 2015, 9, 5520–5535 CrossRef CAS PubMed.
- S. Huang, X. Ling, L. Liang, Y. Song, W. Fang, J. Zhang, J. Kong, V. Meunier and M. S. Dresselhaus, Nano Lett., 2015, 15, 2892–2901 CrossRef CAS PubMed.
- T. Kupka, R. Wrzalik, G. Pasterna and K. Pasterny, J. Mol. Struct., 2002, 616, 17–32 CrossRef CAS.
- A. Venugopal, L. Colombo and E. M. Vogel, Appl. Phys. Lett., 2010, 96, 013512 CrossRef.
- T. Fluteau, C. Bessis, C. Barraud, M. L. Della Rocca, P. Martin, J.-C. Lacroix and P. Lafarge, J. Appl. Phys., 2014, 116, 114509–114514 CrossRef.
- V. Rabache, J. Chaste, P. Petit, M. L. Della Rocca, P. Martin, J.-C. Lacroix, R. L. McCreery and P. Lafarge, J. Am. Chem. Soc., 2013, 135, 10218–10221 CrossRef CAS PubMed.
- C. Bessis, M. L. Della Rocca, C. Barraud, P. Martin, J. C. Lacroix, T. Markussen and P. Lafarge, Sci. Rep., 2016, 6, 20899–20906 CrossRef CAS PubMed.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- M. Baldo and S. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 085201 CrossRef.
- H.-K. Seo, T.-S. Kim, C. Park, W. Xu, K. Baek, S.-H. Bae, J.-H. Ahn, K. Kim, H. C. Choi and T.-W. Lee, Sci. Rep., 2015, 5, 16710 CrossRef CAS PubMed.
- S. Seo, E. Hwang, Y. Cho, J. Lee and H. Lee, Angew. Chem., Int. Ed., 2017, 56, 12122–12126 CrossRef CAS PubMed.
- P. Song, S. Guerin, S. J. R. Tan, H. V. Annadata, X. Yu, M. Scully, Y. M. Han, M. Roemer, K. P. Loh, D. Thompson and C. A. Nijhuis, Adv. Mater., 2018, 30, 1706322 CrossRef PubMed.
- D. E. Eastman, Phys. Rev. B: Condens. Matter Mater. Phys., 1970, 2, 1–2 CrossRef.
- H. Vasquez, R. Oszwaldowski, P. Pou, J. Ortega, F. Flores and A. Kahn, Europhys. Lett., 2004, 65, 802 CrossRef.
- A. Droghetti, P. Thielen, I. Rungger, N. Haag, N. Großmann, J. Stöckl, B. Stadtmüller, M. Aeschlimann, S. Sanvito and M. Cinchetti, Nat. Commun., 2016, 7, 12668 CrossRef CAS PubMed.
- A. Kahn, N. Koch and W. Gao, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2529–2548 CrossRef CAS.
- S. Braun, W. R. Salaneck and M. Fahlman, Adv. Mater., 2009, 21, 1450–1472 CrossRef CAS.
- D. Alloway, M. Hofmann, D. L. Smith, N. E. Gruhn, A. Graham, R. Colorado, V. H. Wysocki, T. R. Lee, P. Lee and N. R. Armstrong, J. Phys. Chem. B, 2003, 107, 11690–11699 CrossRef CAS.
- A. Haldi, A. Sharma, W. J. Potscavage and B. Kippelen, J. Appl. Phys., 2008, 104, 064503 CrossRef.
- Y. Ai, S. Gowrisanker, H. Jia, M. Quevedo-Lopez, H. N. Alshareef, R. M. Wallace and B. E. Gnade, Thin Solid Films, 2013, 531, 509–512 CrossRef CAS.
- K. E. Lilja, T. G. Bäcklund, D. Lupo, T. Hassinen and T. Joutsenoja, Org. Electron., 2009, 10, 1011–1014 CrossRef CAS.
- A. J. Bergren, L. Zeer-Wanklyn, M. Semple, N. Pekas, B. Szeto and R. L. McCreery, J. Phys.: Condens. Matter, 2016, 28, 094011 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |