
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic self-wiring in nanopores and its application in direct electron transfer biofuel cells†
Alexander
Trifonov
,
Andreas
Stemmer
and
Ran
Tel-Vered
 *
*
Nanotechnology Group, ETH Zürich, Säumerstrasse 4, CH-8803 Rüschlikon, Switzerland. E-mail: teran@ethz.ch
First published on 6th September 2018
Abstract
A synthetic enzymatic activity in nanopores leading to the direct fabrication of modified electrodes applicable as biosensors and/or biofuel cell elements is reported. We demonstrate the heterogeneous enzymatic implanting of platinum nanoclusters, PtNCs, in glucose oxidase, GOx, immobilized on mesoporous carbon nanoparticles, MPCNP-modified surface. As the pores confine the growth of the clusters, the PtNC@GOx/MPCNP assembly becomes electrically wired to the matrix, demonstrating direct electron transfer, DET, bioelectrocatalytic properties that correlate with the applied duration of synthesis and cluster size. This inside-out nanocluster growth from the cofactor to the matrix is investigated and further compared to a reversed outside-in strategy which follows the electrochemical deposition of the Pt clusters inside the pores and their electrically induced expansion towards the FAD center of the enzyme. While the inside-out and outside-in methodologies provide, for the first time, synthetic bidirectional direct wiring routes of an enzyme to a surface, we highlight an asymmetry in the wiring efficiency associated with the different assemblies. The results indicate the existence of a shorter gap between the FAD cofactor and the PtNCs in the enzymatically implanted assembly, resulting in elevated bioelectrocatalytic currents, lower overpotential, and a higher turnover rate, 2580 e− s−1. The implanted assembly is then coupled to a bilirubin oxidase-adsorbed MPCNP cathode to yield an all-DET biofuel cell. Due to the superior electrical contact of the inside-out-synthesized anode, this cell demonstrates enhanced discharge potential and power outputs as compared to similar systems employing electrochemically synthesized outside-in-grown PtNC-GOx/MPCNPs or even GOx-modified MPCNPs diffusionally mediated by ferrocenemethanol.
Introduction
Due to its attractive characteristics to bioelectronics, direct electron transfer (DET)-based enzymatic electrocatalysis has been a focus of intensive research in recent years.1,2 The current realization of challenges and opportunities lying ahead in the field of bioelectronics,3–6 as well as rapid progress in the fields of material science and nanotechnology,7–9 fuels the pursuing of novel direct charge transfer pathways to be implemented in third generation biosensors and enzymatic biofuel cells. DET is indeed beneficial for such applications from various practical standpoints. One apparent advantage of DET stems from avoiding the usage of electron relay units in mediating the charge transport between the electrode surface and the redox cofactors buried in the insulated interior of protein structures.10 The mediator units, occasionally employed as diffusional relays added to the electrolyte solution,11–13 interfere with the frequent practical requirement for an integrative design of the assembly as they freely travel in the electrolyte or may leak out from a hosting surface. These problems consequently limit the applicability of diffusive mediators in implanted devices due to potential health hazards. Alternative methodologies to mediate redox enzymes with molecular relays include their tailoring to the protein backbone of the enzymes,14–16 which is often found to degrade the natural activity of biocatalysts through inflicting severe changes to their tertiary structure. Other studies employed contact adsorption and/or electrochemical deposition to co-immobilize redox enzymes, molecular relays, and metallic nanoparticles, yielding effective bioelectrocatalysis.17–20 Frequently used are also redox active mediators which are grafted onto and/or constitute repetitive monomeric units in polymer hydrogels.21–23 While the surface concentration of the relaying species in such assemblies is relatively high, these systems are prone to diffusional limitations of the enzymatic substrates and their products, hindering the charge transport and affecting the performance of the electrocatalysis. In contrast, DET bioelectrocatalysis features mediatorless integrated assemblies that facilitate a typically seamless transfer of biocatalyzed electrons between enzymatic cofactors and current collectors. While the implementation of these DET wiring paradigms is still limited to specific redox enzymes,1,2,10 efforts are made to extend the existing synthetic routes in order to explore a broader spectrum of analytes and fuels for biosensors and biofuel cell applications, respectively.To date, the majority of the studies demonstrating DET bioelctrocatalytic transformations have used conductive nanomaterials to modify current collector surfaces, on which respective redox enzymes were then immobilized. Using this methodology, DET was reported on metallic nanoparticles,24–26 metal oxides,27,28 conducting polymers and related substances,29–31 or composite materials.32,33 Frequently used are also carbonaceous nanostructures including nanotubes,34–36 nanoparticles,37,38 graphene,39,40 and others,41–44 on which several specific enzymes show long range electron transfer upon direct adsorption.45,46 Somewhat harder to execute is the reconstitution method47–49 which involves an initial separation of the redox cofactor from the native enzymatic protein shell, followed by its attachment to an electrically conducting nano-unit (or a molecular relay in the case of non-DET supporting assemblies). Under these conditions, the apo-enzyme, that is the protein shell lacking the cofactor, becomes an optimally linkable enzymatic unit which can be further functionalized to electrode surfaces providing the shortest possible path for electron transfer.
Recently, several new synthetic approaches to induce DET have been proposed. In one,50 a mesoporous carbon nanoparticle, MPCNP, electrode was used as a host matrix to platinum ions which were entrapped and enzymatically capped in the pores of the nanoparticles. Following the application of a reductive potential on the surface PtNCs were formed, facilitating, in the presence of glucose oxidase, GOx, as the enzymatic cap, a DET glucose oxidation bioelectrocatalytic pathway. In different studies, Au,51 or Pt,52 nanoclusters, NCs, were enzymatically grown near the FAD cofactor of glutathione reductase or GOx, respectively. The hybrids were then tailored to conductive surfaces to reveal direct bioelectrocatalytic currents. In contrast to them, we present a heterogeneous inside-out enzymatically catalyzed synthetic step which generates electronically conductive nanoclusters extending from the vicinity of the redox site of an enzyme towards its pore-confined host matrix. Through the entrapment of PtCl62− ions and glucose inside the pores of the MPCNP-modified glassy carbon, GC, surface and their capping with GOx, we demonstrate the surface-localized time-dependent anaerobic enzymatic growth of a PtNC@GOx/MPCNP-modified GC anodic composite assembly. The intimate contact formed between the clusters and the interior of the confining pores leads to an electrically conductive path between the FAD cofactor and the underlying carbonaceous matrix, and thus to effective DET bioelectrocatalysis towards glucose oxidation. This phenomenon is mechanistically investigated and compared to the outside-in electrochemical synthetic approach, highlighting the shorter tunneling gap generated between the surface-localized, enzymatically grown PtNCs and the FAD center. The kinetically and thermodynamically enhanced electrocatalytic features evidenced by the implanted PtNC@GOx/MPCNP assembly are then reflected through the construction and discharge of an all-DET biofuel cell that uses bilirubin oxidase-adsorbed MPCNPs as a cathode.
Results and discussion
Platinum nanoclusters, PtNCs, were enzymatically grown inside glucose oxidase immobilized to mesoporous carbon nanoparticles, MPCNP-modified glassy carbon electrodes. This methodology promoted a time-controlled enzymatic reaction in which pore-entrapped glucose was oxidized by the pore-capping GOx, concurrently with the reduction of pore-containing platinum ions to metallic clusters and regeneration of the reduced redox active center of the enzyme, FADH2, back to its oxidized FAD state. The growth mechanism of the platinum nanoclusters is described in eqn (1) and (2). The first reaction is GOx-catalyzed and yields the reduced flavin cofactor, FADH2, acting as a strong reducing agent (EFADH2/FAD ∼ −0.43 V vs. Ag/AgCl at pH = 7.0). At this negative thermodynamic value, the FADH2 drives the chemical reduction of the platinum ions (E° < +0.48 V vs. Ag/AgCl, through the PtCl42− intermediate), eqn (2), to form a nanometric metal cluster which extends from the vicinity of the cofactor to the nanopore.| Glucose + FAD + H2O → FADH2 + gluconic acid | (1) |
| 2FADH2 + PtCl62− → 2FAD + Pt0 + 4H+ + 6Cl− | (2) |
As the growth stage is performed under nearly anaerobic conditions in which O2, the natural acceptor of GOx, is eliminated from the system through extensive N2 purging, eqn (2) becomes favorable over eqn (3), and the PtCl62− ions act as the sole acceptor of the FADH2 electrons.
| FADH2 + O2 → FAD + H2O2 | (3) |
Following the synthetic stage, the total protein content of GOx on the modified MPCNP matrix was evaluated by a series of colorimetric assays, see Fig. S1† and details in the Experimental section, yielding a surface coverage of 3.5 ± 0.2 × 10−10 mol mg−1 MPCNPs. The assays further facilitated the determination of the enzymatic activity of the surface-bound GOx to be 88 ± 3% of the native enzyme in solution.
The enzymatic generation of the PtNC-implanted GOx assembly on the MPCNPs, PtNC@GOx/MPCNPs, was initially probed by STEM imaging. Following the enzymatic growth, the samples were gently peeled off of the GC surface, dispersed in solution and loaded onto TEM grids. Fig. 1 exemplifies the resulting images obtained for PtNC@GOx/MPCNP samples grown for 15 and 60 minutes. As can be seen, in both of the cases an abundance of Pt nanoclusters is evident on the surface of the carbon nanoparticles. Through a systematic analysis of the nanocluster sizes collected from several imaged samples, we determined the size distribution histograms shown in the figure. Evidently, the 4-fold increase in the duration of the synthesis yielded an increased mean diameter of the PtNCs and led to a broader distribution of particle sizes.
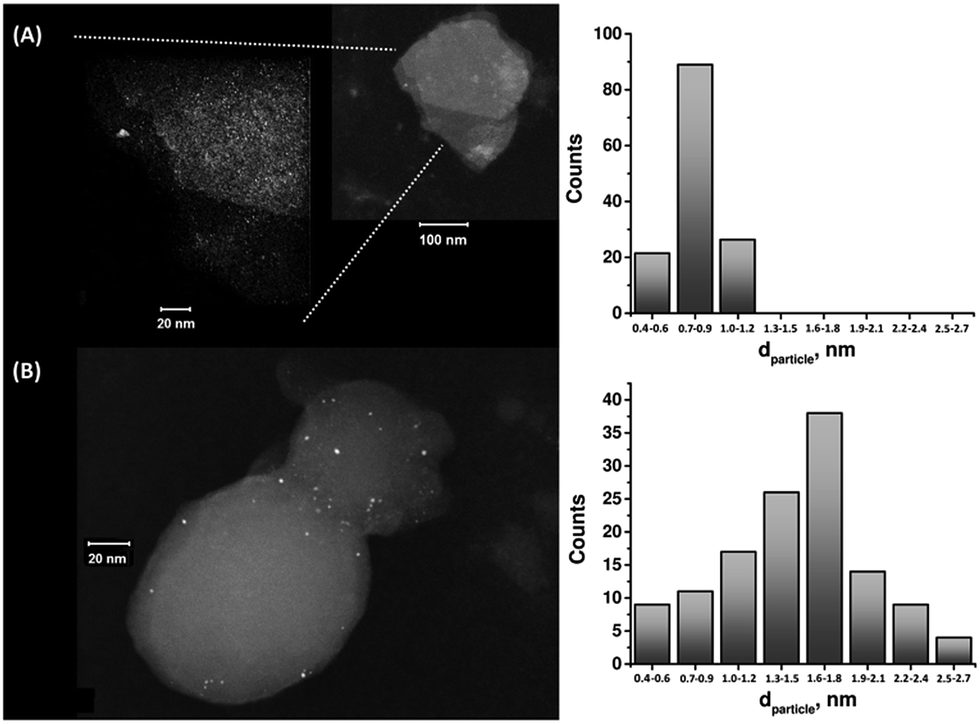 | ||
| Fig. 1 (A) Left: a STEM image corresponding to the 15 minutes enzymatically inside-out-grown PtNC@GOx/MPCNP assembly. Right: a histogram based on the analysis of multiple STEM images, showing the size distribution of the PtNCs in the 15 minutes enzymatically grown assembly. (B) The same as (A) but for a 60 minutes grown assembly. In all cases the particle size was rounded to the closest mentioned value in the histogram. | ||
It should be noted that in parallel to these experiments, attempts to synthesize the PtNCs for 60 minutes in the absence of the glucose substrate, or by replacing GOx with the redox-inactive albumin enzymatic cap, yielded no metallic nanoclusters on the carbonaceous supports, Fig. S2(A) and (B), ESI.† Fig. S3† depicts a STEM image corresponding to a cluster of PtNC@GOx hybrids synthesized in solution for 60 minutes and loaded onto the TEM grid. Evidently, within this growth time-interval and in the absence of the spatial confinement imposed by the pores, the PtNCs may be further enlarged, to diameters exceeding 20 nm.
As the composition of the PtNCs was locally verified during the imaging stage using energy dispersive X-ray spectroscopy, we have also scanned the voltammetric responses of the resulting PtNC@GOx/MPCNP electrodes in 0.2 M H2SO4 to trace down the presence of metallic platinum, Fig. S4.† Evidently, when glucose was deliberately excluded from the pores during the preparation stage, no significant faradaic response was observed, curve (a). Upon incorporation of the substrate into the pores and increasing the duration of the synthesis from 15 to 60 minutes, a buildup of amperometric responses is evident at potentials E < 0.0 V vs. Ag/AgCl. This region, typically governed by the adsorption and desorption of hydrogen atoms prior and following the Pt0-catalyzed proton reduction reaction, PRR, eqn (4),53 seemed to be broadened and lacking/masking some of the distinctive adsorption peaks.
| 2H+ + 2e− → H2 (Pt catalyzed) | (4) |
As the strong voltammetric responses for H2 evolution, at E < −0.23 V vs. Ag/AgCl, and consequential desorption band, at ca. E = −0.2 V, are clearly evident, they provide a spectral fingerprint for the presence of the platinum clusters. We relate the broadening effect to the high concentration of Cl− ions locally released during the chemical stage of the growth, eqn (2). The ions strongly adsorb on the surface of the Pt0, thus interfering with the single crystal contribution(s) of the H adsorption/desorption state(s), an effect that has been previously described.54,55 The increased content of Pt0 with the growth time is predominantly indicated by the increase in the amperometric H desorption response, Fig. S4,† curves (b) and (c). The integration of the charge associated with this process allowed the estimation of the loading of the metal on the surface, which corresponds to 7.2 nmol Pt per mg MPCNPs following 1 hour of synthesis. Interestingly, this loading is ca. 5 times higher than the one observed for the 15 minutes grown PtNC@GOx/MPCNP assembly, in a good correlation with their relative surface areas estimated from the STEM histogram in Fig. 1, assuming perfect spherical shapes. Also, using the weighted average particle size from the histograms in Fig. 1, and the surface loading of both GOx and the PtNCs, a molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 GOx:PtNC was determined. The excess of GOx is explained on the grounds of the distribution of the enzyme units over the MPCNP surface. Orientations in which the FAD centers are not aligned towards or close enough to the nanopores, where the nanocluster formation takes place, are expected to lead to unreacted GOx. Two important conclusions can also be derived from the observed voltammograms: (i) in all cases the enzymatically implanted PtNCs were large enough to exhibit bulk electrical conductivity,56 in agreement with their STEM-evaluated sizes, and (ii) under the conditions employed, the enzymatically grown PtNCs support the formation of a direct electrical contact with the carbonaceous matrix, apparently due to spatial confinements over the direction of their growth inside the pores of the MPCNP matrix. Furthermore, the coulometric responses allowed us to follow the time-dependent growth of the PtNCs, Fig. S5.† The clearly evident monotonic increase in the growth rate is attributed to the gradually elevated surface area available for the reduction of the remaining platinum ions in the pores. It should also be noted at this point that the duration of the syntheses was technically limited by the flow rates of N2 employed for drying up the samples. As the minimal distance between one of the two FAD centers of the GOx and the outer periphery of the enzyme shell is commonly believed to be 14.1 Å,57,58 a question may be raised regarding the feasibility of the smaller clusters, associated with the 15 minutes synthesis, to become attached to the carbon terminal of the nanopore. The indiscernible onset potential observed for the PRR with both the 60 and 15 minutes grown PtNCs suggests however the existence of a direct contact between the Pt and C terminals, supporting minimal or no overpotential for shuttling the electrons over possible insulating gaps. We thus speculate that the observed current responses in the presence of the smaller clusters rely on the existence of a tunneling gap at the other terminal, namely between the FAD redox site and the PtNCs. As the diameters of PtCl62− and glucose are comparable, no steric effect hindering the accessibility of the ions to the cofactor is expected. Nevertheless, the presence of the negatively charged glutamic acid, Glu412, as one of the three amino acids (together with the neutrally charged His516 and His559) intimately involved in the direct catalytic process of the glucose oxidation on GOx,59,60 and the distribution of other negatively charged amino acids along the diffusion path of PtCl62− to the FAD site (GOx isoelectric point 4.2) are expected to contribute to the electrostatic separation of the cluster from the cofactor. The gap, which indirectly facilitates the protruding of the clusters from the enzyme's exterior to come into contact with the MPCNPs, will next be shown to play an important role in explaining the bioelectrocatalytic responses of the PtNC@GOx/MPCNP assembly during the direct oxidation of glucose.
:1 GOx:PtNC was determined. The excess of GOx is explained on the grounds of the distribution of the enzyme units over the MPCNP surface. Orientations in which the FAD centers are not aligned towards or close enough to the nanopores, where the nanocluster formation takes place, are expected to lead to unreacted GOx. Two important conclusions can also be derived from the observed voltammograms: (i) in all cases the enzymatically implanted PtNCs were large enough to exhibit bulk electrical conductivity,56 in agreement with their STEM-evaluated sizes, and (ii) under the conditions employed, the enzymatically grown PtNCs support the formation of a direct electrical contact with the carbonaceous matrix, apparently due to spatial confinements over the direction of their growth inside the pores of the MPCNP matrix. Furthermore, the coulometric responses allowed us to follow the time-dependent growth of the PtNCs, Fig. S5.† The clearly evident monotonic increase in the growth rate is attributed to the gradually elevated surface area available for the reduction of the remaining platinum ions in the pores. It should also be noted at this point that the duration of the syntheses was technically limited by the flow rates of N2 employed for drying up the samples. As the minimal distance between one of the two FAD centers of the GOx and the outer periphery of the enzyme shell is commonly believed to be 14.1 Å,57,58 a question may be raised regarding the feasibility of the smaller clusters, associated with the 15 minutes synthesis, to become attached to the carbon terminal of the nanopore. The indiscernible onset potential observed for the PRR with both the 60 and 15 minutes grown PtNCs suggests however the existence of a direct contact between the Pt and C terminals, supporting minimal or no overpotential for shuttling the electrons over possible insulating gaps. We thus speculate that the observed current responses in the presence of the smaller clusters rely on the existence of a tunneling gap at the other terminal, namely between the FAD redox site and the PtNCs. As the diameters of PtCl62− and glucose are comparable, no steric effect hindering the accessibility of the ions to the cofactor is expected. Nevertheless, the presence of the negatively charged glutamic acid, Glu412, as one of the three amino acids (together with the neutrally charged His516 and His559) intimately involved in the direct catalytic process of the glucose oxidation on GOx,59,60 and the distribution of other negatively charged amino acids along the diffusion path of PtCl62− to the FAD site (GOx isoelectric point 4.2) are expected to contribute to the electrostatic separation of the cluster from the cofactor. The gap, which indirectly facilitates the protruding of the clusters from the enzyme's exterior to come into contact with the MPCNPs, will next be shown to play an important role in explaining the bioelectrocatalytic responses of the PtNC@GOx/MPCNP assembly during the direct oxidation of glucose.
As the PtNCs are grown by an enzymatic deposition process in the vicinity of the reduced cofactor, it is expected that the metal nanoclusters formed through this process will become electrically wired to the redox active sites. Thus, combined with the electrical contact observed in Fig. S4† between the metal cluster and the GC collector (via the MPCNP matrix), a conductive path for direct electron transfer (DET) bioelectrocatalysis is allegedly formed in the system. To test this assumption, we conducted a series of measurements monitoring the amperometric responses of the PtNC@GOx/MPCNP electrodes grown for different times during exposure to variable concentrations of glucose. Fig. 2(A) exemplifies the cyclic voltammograms obtained with a 60 minutes-grown PtNC@GOx/MPCNP electrode. As can be seen, bioelectrocatalytic currents starting at an onset voltage of E > −0.08 V vs. Ag/AgCl are observed and increase as the concentration of the substrate in the system is gradually elevated. Near the onset of catalysis, a small shoulder-like peak, extending up to ca. 0.0 V, is also detected. We will shortly discuss the possible origins of this peak. Fig. 2(B) depicts the bioelectrocatalytic responses of the PtNC@GOx/MPCNP electrode, curve (a), as compared to a set of control electrode assemblies, all tested in the presence of 60 mM glucose in the electrolyte. The bioelectrocatalytic oxidation current is attributed to the direct electron transfer process on the electrode described in eqn (5), regenerating the cofactor of GOx for continuous enzymatic glucose oxidation.
| FADH2 → FAD + 2H+ + 2e− (on the electrode) | (5) |
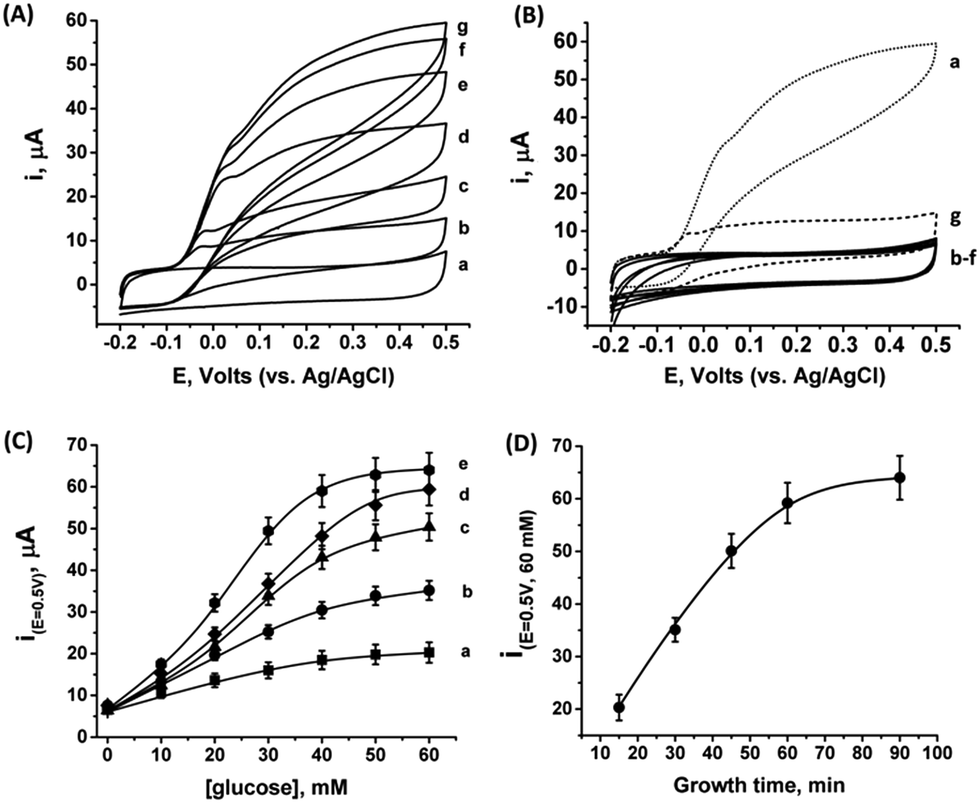 | ||
| Fig. 2 (A) Bioelectrocatalytic currents recorded on a 60 minutes inside-out-grown PtNC@GOx/MPCNP assembly in the presence of: (a) 0, (b) 10, (c) 20, (d) 30, (e) 40, (f) 50, and (g) 60 mM glucose. (B) Cyclic voltammograms showing the current responses obtained upon testing different electrode assemblies in the presence of 60 mM glucose: (a) 60 minutes grown PtNC@GOx/MPCNPs. Curves (b)–(d) demonstrate attempts to synthesize the PtNC@GOx/MPCNPs while excluding either glucose, curve (b), or PtCl62− ions from the pores, curve (c), or by replacing GOx with albumin, curve (d). (e) Attempt to synthesize the PtNC@GOx/MPCNP assembly in an O2-saturated electrolyte. (f) Electrochemically grown PtNCs inside albumin-capped MPCNPs. Curve (g) corresponds to a MPCNP-modified electrode on which solution phase-synthesized PtNC@GOx was deposited and crosslinked. (C) Calibration curves corresponding to the DET electrocatalytic currents obtained, in the presence of the indicated glucose concentrations at E = 0.5 V vs. Ag/AgCl, by different PtNC@GOx/MPCNP assemblies grown for: (a) 15, (b) 30, (c) 45, (d) 60, and (e) 90 minutes. (D) Dependence of the bioelectrocatalytic currents at E = 0.5 V vs. Ag/AgCl and in the presence of 60 mM glucose on the growth time-interval. All measurements were performed in a HEPES buffer (0.1 M, pH = 7.0) at a scan rate of 10 mV s−1. Error bars correspond to a set of 4 experiments. | ||
The diminished catalytic responses evidenced by curves (b)–(f) indicate that only the combined presence of glucose, PtCl62− ions, and glucose oxidase during the synthetic stage yields the catalytic PtNC implanted assembly necessary for the DET bioelectrocatalysis. Synthetic attempts to grow the cluster under saturated O2 conditions further resulted in a diminished current response due to the competing effect of the oxygen acceptor over the electrons provided by the reduced FADH2 cofactor at the synthesis stage. This led to the destructive pathway for the formation of Pt0 atoms as described in eqn (3). The minor, sole contribution of PtNCs to the electrocatalysis was also demonstrated by the application of a negative potential to reduce platinum ions entrapped in the pores by the electrochemically inert enzyme albumin. The lack of an amperometric response by the PtNCs themselves indicates that the bioelectrocatalysis, in contrast to electrocatalysis, is the primary source for the catalytic glucose oxidation in our system. The use of an electrochemical PtNCs synthesis will be further explored later, in conjunction with GOx, as a comparative methodology to the enzymatic growth. Curve (g) in Fig. 2(B) depicts an additional experiment according to which a PtNC was implanted in GOx in solution without the confinement of the pores.52 Following the solution phase growth of PtNC@GOx hybrids, they were allowed to adsorb on a MPCNP-modified GC surface, and chemically crosslinked to form a stable structure. It should be noted that the content of the active GOx in both the pore confined and surface adsorbed PtNC@GOx/MPCNP systems was fairly similar and showed, upon a spectroscopic assay, less than 11% variability. The significantly lower bioelectrocatalytic current revealed for the adsorbed PtNC@GOx system highlights the more intimate (superior electrical) Pt/C contact and/or the larger Pt/C contact surface area generated by the pore-confined surface-localized growth of the clusters.
By testing the PtNC@GOx/MPCNP assemblies grown for different time intervals, we assessed the size-dependent effects on the bioelectrocatalysis. Fig. 2(C) depicts the calibration curves corresponding to the different PtNC@GOx/MPCNP electrodes which were grown for variable time-intervals (raw data presented in Fig. S6, ESI†). These results imply that even in the presence of the smallest Pt nanoclusters obtainable under the preparative constrictions, particles associated with tgrowth = 15 minutes, a genuine DET current is obtained. Moreover, a definitive increase in the DET bioelectrocatalysis is clearly evident for electrodes consisting of clusters grown for longer times. This is further visualized by the dependence of bioelectrocatalytic saturation currents on the growth time shown in Fig. 2(D) and the simultaneous increase in the turnover rate of bioelectrocatalysis, Table S1.† As mentioned, the DET bioelectrocatalysis is facilitated via the existence of an electronically conductive path between the FAD and the GC collector which are wired through the PtNC implants and the MPCNP scaffold. Looking at the catalytic current responses obtained by the clusters grown for different times, Fig. S6,† we realize that the bioelectrocatalytic onset potential, ca. E = −0.08 V vs. Ag/AgCl, remained constant, showing no variation with the growth time. Further attempts to scan the potential from considerably more negative potentials revealed a weak response of the FAD cofactor near its expected thermodynamic potential in pH = 7.0, 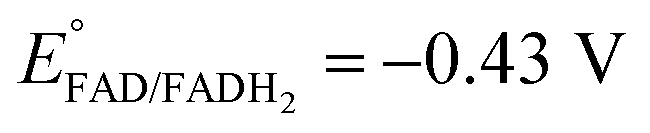 vs. Ag/AgCl, which was more clearly observed in the differential pulse voltammograms, DPV, shown in Fig. S7,† curve (a). The DPV response was found to be insensitive to the glucose concentration in the system. These observations indicate that while the cofactor is indeed electrically wired to the surface, no direct bioelectrocatalysis occurs in the absence of an applied overpotential, suggesting the existence of a short tunneling gap at the FAD/PtNC terminal. Furthermore, the invariability in the onset potential of the electrocatalytic currents with the growth time implies that for all clusters the FAD-PtNC distance remained the same. Combined with the amperometric observations from the H2SO4 scanning, Fig. S4,† we conclude that the PtNCs reach out from the interior of the enzyme to yield a physical contact with the interior of the MPCNPs. These concepts are visualized in Scheme 1(A). It should be noted that the observations are in agreement with the guidelines suggested by different recent reports for a genuine DET in GOx-based bioelectronic systems.61,62 Since we observe in our system (i) direct bioelectrocatalytic oxidation currents that increase with the levels of the substrate, (ii) no indirect catalytic reduction currents that might arise due to contaminants, (iii) a relatively high Michaelis constant, KM ∼ 20 mM, and (iv) specificity to glucose and no responses in its absence, we conclude that a real DET prevails in our case.
vs. Ag/AgCl, which was more clearly observed in the differential pulse voltammograms, DPV, shown in Fig. S7,† curve (a). The DPV response was found to be insensitive to the glucose concentration in the system. These observations indicate that while the cofactor is indeed electrically wired to the surface, no direct bioelectrocatalysis occurs in the absence of an applied overpotential, suggesting the existence of a short tunneling gap at the FAD/PtNC terminal. Furthermore, the invariability in the onset potential of the electrocatalytic currents with the growth time implies that for all clusters the FAD-PtNC distance remained the same. Combined with the amperometric observations from the H2SO4 scanning, Fig. S4,† we conclude that the PtNCs reach out from the interior of the enzyme to yield a physical contact with the interior of the MPCNPs. These concepts are visualized in Scheme 1(A). It should be noted that the observations are in agreement with the guidelines suggested by different recent reports for a genuine DET in GOx-based bioelectronic systems.61,62 Since we observe in our system (i) direct bioelectrocatalytic oxidation currents that increase with the levels of the substrate, (ii) no indirect catalytic reduction currents that might arise due to contaminants, (iii) a relatively high Michaelis constant, KM ∼ 20 mM, and (iv) specificity to glucose and no responses in its absence, we conclude that a real DET prevails in our case.
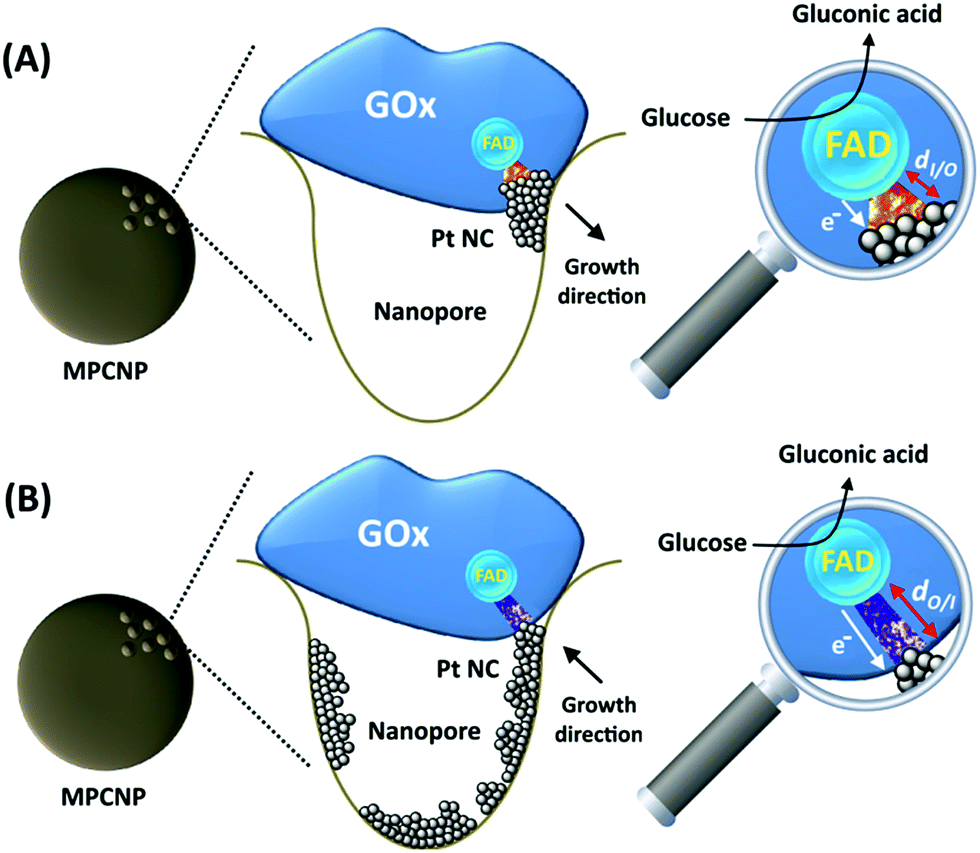 | ||
| Scheme 1 Illustrations corresponding to the suggested configurations of (A) the inside-out enzymatically synthesized PtNC@GOx/MPCNP electrode and (B) the outside-in electrochemically synthesized PtNC-GOx/MPCNP electrode. | ||
While the physical contact between the PtNCs and the carbonaceous matrix eliminates in all cases the need for a further tunneling of the biocatalytic electrons at the Pt/carbon terminal, no increase in the overpotential is required upon using the smaller cluster electrodes. A related observation focused on the effect of pH on the catalysis, Fig. S7(B).† In this experiment the bioelectrocatalytic response of the 60 minutes grown PtNC@GOx/MPCNP electrode was tested in the presence of glucose at three pHs. Upon gradually increasing the pH from 7.0 to 9.0, we observed a ca. −30 mV pH−1 potential shift in the onset of catalysis. The potential shift correlates with the 2e− transfer associated with the oxidation of glucose on one of the two pH-sensitive FAD cofactor units associated with the dimeric enzyme. Considering the spatial separation between the subunits of the enzyme,63 this description correlates (not to scale) with the wiring scheme illustrated in Scheme 1(A). It should also be noted that the intensities of the catalytic saturation currents evident in the figure fit the pH-induced redistribution of the [FADH2]/[FAD+] units on the surface. Based on these observations, we attempt to explain the appearance of the additional amperometric response that is clearly seen from tgrowth ≥ 30 minutes in Fig. S6.† The response, in the region of E = −0.1 to 0.0 V, is observed to be affected both by the presence of the glucose substrate and also by pH, and thus may be linked to the catalytic process. The direct dependence of the onset voltage associated with the peak on the pH, Fig. S7(B),† leads us to assume that this additional pre-catalytic signal originates from a co-effect of the pH-sensitive FAD and the PtNCs on the glucose oxidation. Such an effect may involve an initial adsorption stage of the glucose substrate onto the PtNC surface which is competitive with the presence of protons in the electrolyte. Interestingly, this is also reflected by the inverted dependence of the catalytic peak current intensity, at E > 0.1 V, on the pH, Fig. S7(B).† As the surface area of the PtNCs is increased by prolonging the enzymatic deposition intervals, more substrate molecules are adsorbed on the Pt nanoclusters and become activated for oxidation by the electrically wired FAD center of the GOx. This explains the relative increase in the size-dependent bioelectrocatalytic responses shown in Fig. 2(C) and (D). The kinetic aspects of the proposed mechanism, supporting these observations, are summarized in Scheme S1.† To further validate the assumptions we have also performed a comparative analysis, testing the current responses of both the pre-catalytic and the catalysis peaks in the presence of various concentrations of glucose, fructose, and their combinations, Fig. S8.† The results indicate that the presence of fructose, a weak interferent saccharide for GOx,64 in the system hinders the development of both the pre-catalytic and catalytic peaks, supporting the claim for an adsorption stage of glucose on the PtNCs prior to the bioelectrocatalytic process.
The enzymatically implanted PtNC@GOx/MPCNP electrode was then compared to another catalytic assembly showing a pore-confined generation of PtNCs in MPCNPs and communicating direct electron transfer from GOx to a GC collector, Scheme 1(B). In contrast to the enzymatic synthesis approach which promotes the inside-out growth of the PtNCs from the vicinity of the enzymatic cofactor towards the MPCNP matrix, here the nanoclusters are electrochemically deposited inside the pores through the application of a reductive potential. By modifying a previously reported study,50 we entrapped PtCl62− ions in GOx-capped MPCNPs and reduced them directly to metallic NCs through the application of a potential pulse, in accord with eqn (6).
| PtCl62− + 4e− → Pt0 + 6Cl− (on electrode) | (6) |
Following the outside-in electrochemical synthesis of the NCs, the content of the active GOx in the assembly was spectroscopically assayed and was found to be similar, variance ±1.8 × 10−11 mol mg−1 MPCNPs, to the case of the enzymatically inside-out-grown PtNC@GOx/MPCNPs. The similarity in the active content of the biocatalyst in the two configurations suggests that the GOx units were not unequally deformed throughout the different preparative stages. A ca. 60% higher content of Pt was also coulometrically revealed in the case of the outside-in electrochemically synthesized assembly. Fig. 3(A) compares the cyclic voltammograms obtained for the electrochemically grown, curves (a and a′) and enzymatically implanted, curves (b and b′) assemblies, in the presence and the absence of 40 mM glucose. While the electrochemically outside-in-grown PtNC-GOx/MPCNP assembly shows a DET bioelectroatalysis starting from an onset potential of ca. 0.2 V, the overpotential required for glucose oxidation on the enzymatically synthesized inside-out-grown PtNC@GOx/MPCNP matrix is more than 200 mV lower. A comparative testing of various concentrations of the substrate on both electrodes is shown in the calibration curves presented in Fig. 3(B), indicating a 5-fold difference in the current responses measured at E = 0.5 V vs. Ag/AgCl in the higher glucose concentration regime. Due to a lack of sufficient overpotential to drive the electron transfer in the outside-in-grown system, this ratio is sharply increased at scan potentials closer to the onset potential of bioelectrocatalysis of this assembly. From the saturation currents and the surface coverage values estimated for GOx in each of the cases, turnover rates of ca. 2580 e− s−1 and 855 e− s−1 were estimated for the enzymatically and electrochemically synthesized electrodes, respectively, see the Experimental section for further details.
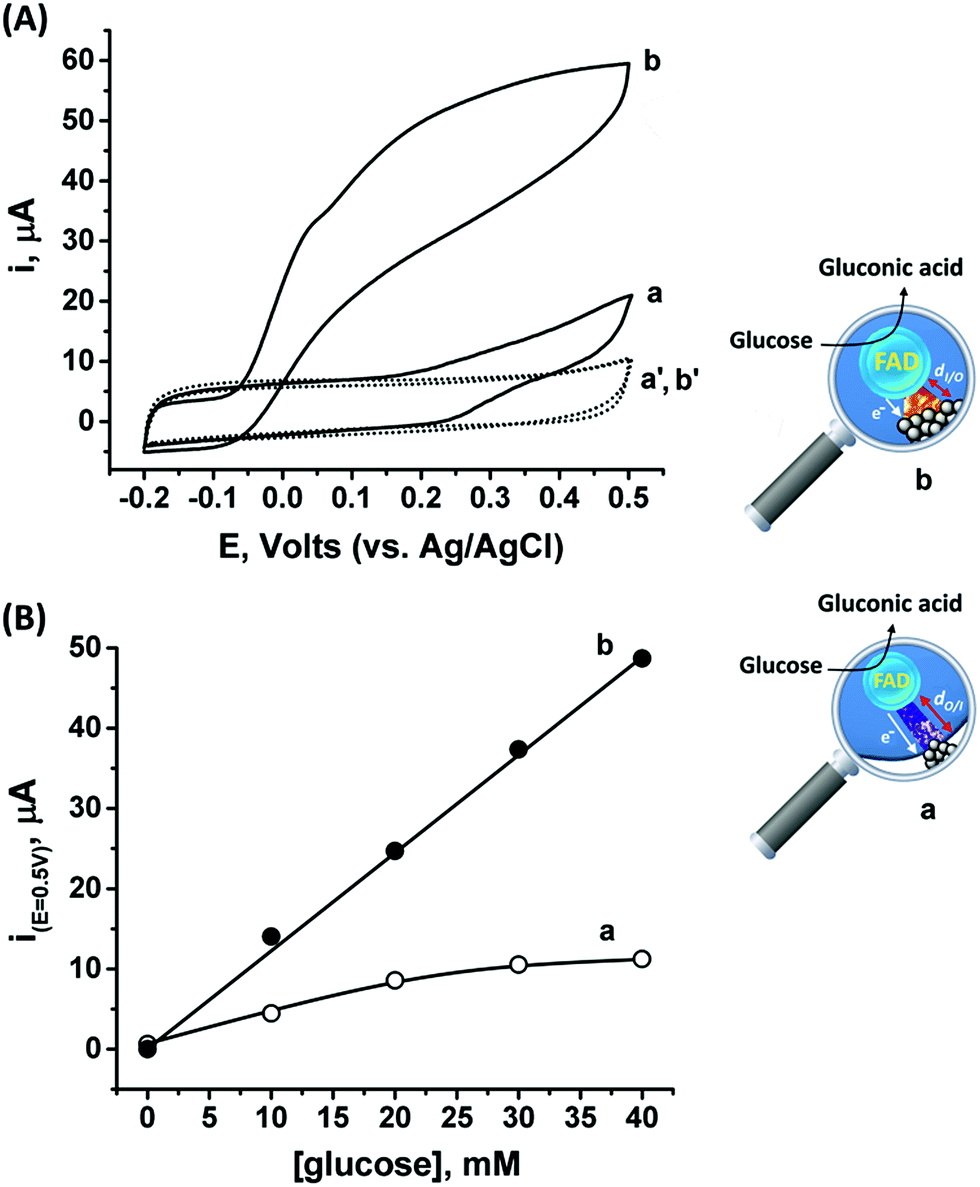 | ||
| Fig. 3 (A) Cyclic voltammograms corresponding to bioelectrocatalytic currents measured on (a) and (b) the electrochemically outside-in-synthesized PtNC-GOx/MPCNP electrode and the enzymatically inside-out-synthesized PtNC@GOx/MPCNP electrode, respectively, in the presence of 40 mM glucose, and (a′) and (b′) the electrodes in the absence of glucose. (B) Calibration curves comparing the bioelectrocatalytic currents, at E = 0.5 V vs. Ag/AgCl, generated by (a) the outside-in electrochemically synthesized PtNC-GOx/MPCNP electrode and (b) the inside-out enzymatically synthesized PtNC@GOx/MPCNP electrode in the presence of variable concentrations of glucose. Capacitance currents were subtracted from all curves. Measurements were performed in a HEPES buffer (0.1 M, pH = 7.0) at a scan rate of 10 mV s−1. | ||
The differences in the DET bioelectrocatalysis performance and their correlation with the preparative procedures are explained on the grounds of the relative distances between the wired FAD center of the GOx and the PtNCs, reflecting the directionality of the two synthetic routes. As the inside-out enzymatic synthesis of the metallic clusters is believed to occur from the electrostatically controlled minimal tunneling gap separating the cluster from the FAD site, a shorter FAD/PtNC distance is expected in this case as compared to the outside-in electrochemical growth. The latter synthetic process is initiated at the interior of the carbon pore allowing the Pt nanocluster to extend, among other possible directions, towards the FAD. As the rate of electron transfer correlates with the gap distances between the cofactor and the nanocluster, in accord with Marcus theory,65 a favorable wiring is expected for the enzymatically inside-out-grown PtNC@GOx/MPCNP assembly. A further support for these observations is revealed by the DPV scanning of the electrochemically synthesized assembly around the redox region of the FAD, Fig. S7(A),† curve (b). As can be seen, while the electrochemically synthesized electrode also demonstrates a direct electronic access to the redox responsive cofactor, the signal obtained in this case is considerably smaller as compared to the enzymatically grown PtNC@GOx/MPCNP assembly. The smaller response is further associated with a positive voltage shift of ca. 35 mV from the thermodynamic potential. While this shift is smaller than the difference in the overpotential for bioelectrocatalysis observed for the two assemblies, it provides an additional indication for the larger gap existing between the PtNCs and the redox cofactor in the electrochemically grown system, a barrier that evidently affects the shuttling of the bioelectrocatalyzed electrons.
As the favorable characteristics of the inside-out enzymatically implanted PtNCs@GOx system may potentially be implemented for the sensing of glucose, we have also decided to test its applicability for the generation of electrical power through the construction of an all-DET biofuel cell. To this end, we employed a cathode consisting of directly adsorbed bilirubin oxidase, BOD, on MPCNPs, operating in the absence of any mediating relay units, Fig. S9(A).† The content of the BOD on the surface corresponded to 4.6 × 10−11 mol mg−1 MPCNPs, with an enzymatic activity reaching ca. 92% of the solubilized free biocatalyst. Similarly to other members of the multicopper oxidase family, BOD has been previously shown to support DET bioelectrocatalysis upon adsorption on different carbonaceous nanostructured substances, supposedly due to interactions between its hydrophobic pocket engulfing the redox active T1 blue copper site and the matrices.45,46 Following the adsorption of the enzyme on the MPCNP matrix and its chemical crosslinking, the BOD/MPCNPs demonstrated, in the presence of the O2 substrate, Fig. S9(B),† a DET bioelectrocathodic current that follows the reaction described in eqn (7).
| O2 + 4H+ + 4e− → 2H2O | (7) |
The onset of the electrocatalytic response is clearly evident at E = 0.50 V vs. Ag/AgCl. As the turnover rate in the case of the PtNC@GOx/MPCNP assembly, 2580 e− s−1, is over 4 times higher than the electron exchange rate between GOx and its native O2 acceptor,66 the anodic response is expected to be inert to the presence of the O2 oxidizer required for the operation of the BOD/MPCNP cathode. We have further verified this experimentally and observed only a minor, less than a 5%, decrease in the bioelectrocatalytic current upon testing the PtNC@GOx/MPCNP assembly for glucose oxidation under O2-saturated conditions. Encouraged by these results, we combined the anodic inside-out-grown PtNC@GOx/MPCNP and cathodic BOD/MPCNP assemblies into a mediatorless, all-DET biofuel cell. Fig. 4(A) and (B), curves (a), respectively depict the discharge voltage and power output obtained by the PtNC@GOx/MPCNPs//BOD/MPCNP cell upon its discharge over variable external resistances. An open circuit potential of nearly 600 mV is evident, reflecting the difference between the onset potential for catalysis by the two electrode assemblies. The discharge was accompanied by a maximal power output of 45 μW cm−2, peaking at a current density of 105 μA cm−2. These discharge characteristics are comparable to those of other biofuel cells based on mesoporous carbon substances,67–71 and especially to those of systems which employ DET pathways, Table S2.† It should be noted that no further optimization was performed prior to the discharge, suggesting that the performance can be further elevated for example by tuning the discharge temperature or by synthetically increasing the PtNC size, as well as by other means occasionally reported to enhance the charge transfer in biofuel cells, such as introducing convection to the cell. To highlight the inherent advantages of the enzymatically implanted metallic NC methodology, we have further compared this biofuel cell to two related assemblies. In one, curves (b), the anode consisted of the electrochemically outside-in-synthesized PtNCs-GOx/MPCNPs, while in the other, curves (c), the anode employed ferrocenemethanol, FcMe, as a diffusional relay unit mediating the enzymatic charge transfer in the system. The latter configuration, depicted in Fig. S10(A),† was based on the entrapment of FcMe species in the GOx-capped MPCNPs, and yielded the bioelectrocatalytic currents shown in Fig. S10(B).† During the GOx-catalyzed oxidation of glucose, the electrode potential is scanned positively to generate ferrocenyl cation ions FcMe+ which, following their pore confined diffusion to the enzymatic redox site, become reduced by the FADH2 cofactor.72–74 A regeneration of GOx activity is thus accompanied by the FcMe/FcMe+-mediated bioelectrocatalytic oxidative current according to eqn (8) and (9) that follow eqn (1).
| Glucose + FcMe+ → gluconic acid + FcMe (on GOx) | (8) |
| FcMe → FcMe+ + e− (on electrode) | (9) |
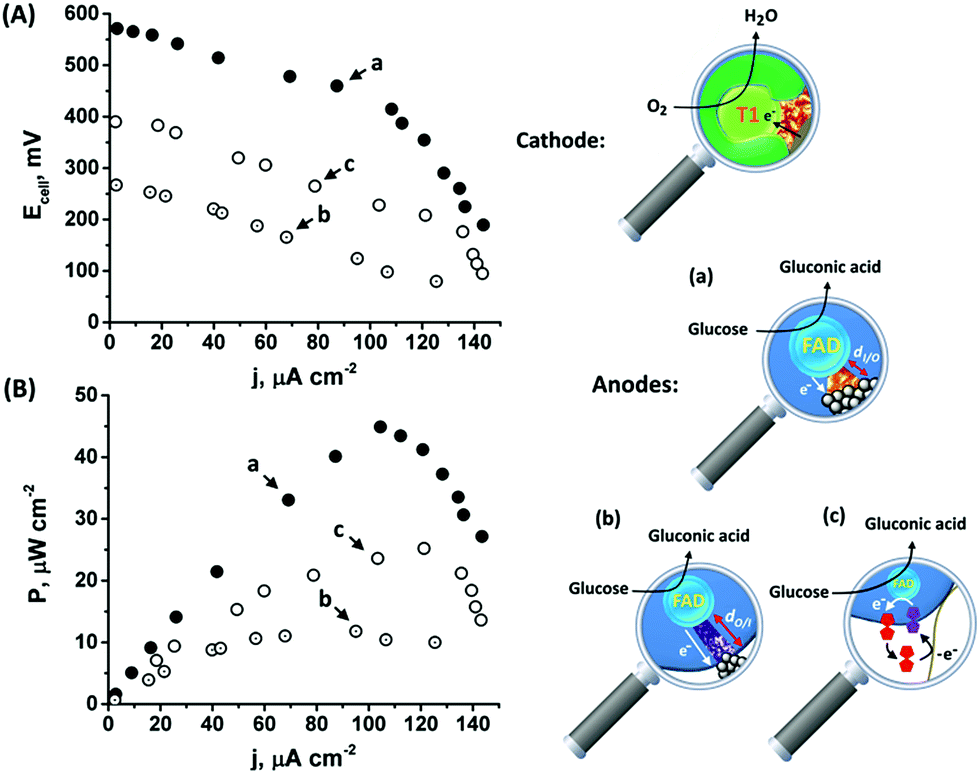 | ||
| Fig. 4 (A) Polarization curves corresponding to the discharge of (a) the inside-out enzymatically synthesized, 90 minutes grown, PtNC@GOx/MPCNPs//BOD/MPCNP biofuel cell, (b) the outside-in electrochemically synthesized PtNC-GOx/MPCNPs//BOD/MPCNP biofuel cell, and (c) the FcMe-entrapped, GOx-capped MPCNPs//BOD/MPCNP biofuel cell. (B) The power outputs associated with the discharge of the cells presented in (A). The cells were discharged versus different constant external resistances in an O2-saturated HEPES buffer (0.1 M, pH = 7.0) containing 60 mM glucose. | ||
The comparative biofuel cell discharges indicate that the superb electrical contact originating from the inside-out synthetic approach leads to a favorable performance in terms of discharge potential and power outputs as compared to both the outside-in and the pore-confined diffusion methodologies. It should be noted that a few GOx mediators exhibiting redox potentials more negative than FcMe do exist,75,76 yet are unsuitable to act inside the pores due to size and solubility limitations. Furthermore, as the relay molecules are freely dissolved in the pore confined electrolyte, they tend to leak out from the matrix with time, thus effecting the long-term operation of the biofuel cell. Upon testing the time-dependent production of power from the cells, both the DET enzymatic and electrochemically synthesized PtNC systems were able to sustain up to 80% of the initial maximal power for several days, while the voltage/power characteristics of the FcMe-based cell decayed to less than 50% within a single day of continuous operation.
Experimental
Materials
N-methyl-2-pyrrolidone (NMP), Nafion™, perfluorinated resin solution (5% wt), mesoporous carbon nanoparticles (MPCNPs, d < 500 nm, average pore size 6.4 nm), glucose oxidase (GOx, Type-VII, from Aspergillus niger, 100000 units per g), horseradish peroxidase (HRP, Type VI-A), albumin (from human serum), bilirubin oxidase (BOD, from Myrothecium verrucaria), potassium hexachloroplatinate, glucose, fructose, polyvinylidene fluoride (PVDF), and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS2−) were purchased from Sigma.
Electrode preparation
A slurry was prepared by mixing 5 mg MPCNPs in 0.5 mL of NMP containing 5 mM K2PtCl6 and 15 mM glucose. The slurry was sonicated for 10 minutes using an ultrasonic bath. 20 μL NMP containing 10% (w/w) PVDF was added to the slurry, and the resulting suspension was stirred for an additional 20 minutes. Glassy carbon, GC, electrodes (d = 3 mm, from ALS company) were successively polished with 1.0 and 0.3 μm alumina powder, and were further sonicated in water and ethanol for 15 minutes. Four μL of the MPCNP suspension were then placed on top of the clean GC surface and allowed to dry at room temperature, yielding a non-functionalized “Type A” electrode. Similarly, “Type B” electrodes lacking the glucose substrate were prepared.The enzymatically inside-out-grown PtNC@GOx/MPCNP electrodes were prepared by rinsing the “Type A” electrodes in water to remove any remains of surface-bound excess of glucose and hexachloroplatinate, followed by drying in nitrogen for one hour. Subsequently, 6 μL of HEPES buffer solution (0.1 M, pH 7.0) containing 0.1 mg mL−1 GOx were deposited on the surface, and the electrode was allowed to dry in an enclosed chamber exposed to a constant flow rate of N2 for a fixed time interval that determines the duration of the enzymatic synthesis. The enzyme was crosslinked for 15 minutes using 2 μL of an aqueous solution containing 0.5 mg mL−1 bis(sulfosuccinimidyl) suberate, BS,3 and then covered by 3 μL perfluorinated Nafion™ solution (0.5% wt). The resulting modified electrodes were dried at room temperature and thoroughly rinsed in buffer solution to remove any remains of non-reacted precursors from the system.
The electrochemically outside-in-grown PtNC-GOx/MPCNP assembly was prepared by covering the “Type B” electrode with 6 μL of GOx, 0.1 mg mL−1, which was followed by drying, BS3-crosslinking, application of Nafion™, redrying, and washing as described above. The modified electrode was then transferred to a three-electrode electrochemical cell connected to a potentiostat, and a reduction potential corresponding to E = −0.9 V vs. Ag/AgCl was applied for 10 seconds on its surface in 0.1 M HEPES buffer solution to reduce the metal ions at the pores. It should be noted that attempts to increase the deposition time indicated no significant effect on the electrode's bioelectrocatalytic performance. Following the electrodeposition process the electrode was washed thoroughly and allowed to dry at room temperature.
For the experiments employing a solution phase synthesis of PtNC@GOx assemblies, 3 mL of N2-purged HEPES buffer solution (100 mM, pH 7.0) containing 5 mg mL−1 GOx, 5 mM K2PtCl6 and 20 mM glucose were gently stirred under N2 for 60 minutes. The resulting PtNC@GOx hybrids were purified through centrifugation using an Amicon Ultra 50 KD filter.
Methods and instrumentation
Electrochemical measurements were performed using a PalmSens 4 potentiostat. A Ag/AgCl and a carbon rod (d = 5 mm) were used as reference and counter electrodes, respectively. UV/visible spectroscopic measurements were performed on a Smart Spec Plus spectrophotometer by Bio-Rad, using a quartz cuvette. STEM images were taken on a Hitachi HD-2700 using a Z-contrast detector. The STEM grids were loaded with aqueous dispersions of the active materials that had been mechanically removed from the electrode surfaces and vigorously stirred. Particle size distribution was derived using ImageJ software analysis on the STEM images. The biofuel cells were discharged against variable external resistances using a Keithley 2000 multimeter.The estimation of the surface bound GOx was based on a spectroscopic assay performed in a HEPES (0.1 M, pH = 7.0) solution. Different amounts of GOx were interacted with 25 mM glucose in the presence of 20 nM HRP and 3.5 mM ABTS2− under aerated conditions. Upon the GOx-catalyzed reduction of O2 to H2O2, the peroxide acted as a substrate for the HRP, which catalyzed its further reduction concurrently with oxidizing ABTS2− to the colorful ABTS˙− species (λmax = 422 nm). The measurements allowed the derivation of a calibration curve correlating the absorbance of ABTS˙− to the tested GOx concentrations. By similarly assaying the absorbance of the surface bound GOx associated with the different assemblies and using the derived calibration curve, we estimated the surface coverage of GOx on the electrode. It should be noted that the assay assumes that the solubilized GOx activity does not change upon adsorption to the MPCNP matrix.
The enzymatic turnover rates of electron transfer were calculated,77 according to:
| ket = imax/nFAΓeff | (10) |
Conclusions
This study explores a new synthetic paradigm for the assembly of bioelectronic elements (biosensors and biofuel cell elements) which relies on enzymatic reduction of metal nanoclusters inside the confined mesopores of a carbonaceous matrix. The relatively close vicinity between the clusters grown and the FAD redox active center of the GOX on one hand and the direct electrical contact of the synthesized NCs with the interior of the pore on the other yield an effective DET pathway to electrobiocatalyze the oxidation of glucose. This was reflected by the uniquely high turnover rate of electrons, 2580 e− s−1, as well as the negative onset of oxidation, E = −0.08 V vs. Ag/AgCl. The inside-out implanted PtNC@GOx/MPCNP assembly was compared to another configuration facilitating DET in which PtNCs were electrically grown outside-in towards the cofactor inside the porous matrix. These two methods demonstrate elegant bidirectional synthetic routes to achieve resembling, yet different, direct electron transfer pathways for an enzyme immobilized onto a conductive matrix. The comparison highlighted the existence of a shorter gap between the PtNCs and the FAD center in the case of the enzymatic inside-out approach, which drives a more facile DET bioelectrocatalytic process. These features were further utilized in the construction of an all-DET biofuel cell employing a BOD-modified MPCNP cathode that supports the four electron direct reduction of oxygen. Operated with the PtNC@GOx/MPCNP anode, the integrated cell yielded a power output of 45 μW cm−2 prior to any optimization process and in the absence of the frequently used stirring conditions, optimized temperature, etc. Notably, this electrical power was obtained in the absence of potentially leaking diffusional mediators. It should also be emphasized that the novelty of the suggested methodology stems from the synthetic conjugation between enzymatic activity and nanopore “reactors”, and also from the observation that a single heterogeneous step may be utilized to effectively yield DET-based biosensing electrodes and biofuel cell elements. The conjugation between enzymes and nanopores may be further extended to activate other biocatalysts and harness their respective substrates in applications involving detection and/or energy production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Assistance by the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zürich is gratefully acknowledged.References
- C. D. Bostick, S. Mukhopadhyay, I. Pecht, M. Sheves, D. Cahen and D. Lederman, Rep. Prog. Phys., 2018, 81, 026601 CrossRef PubMed.
- M. Falk, Z. Blum and S. Shleev, Electrochim. Acta, 2012, 82, 191 CrossRef CAS.
- C. Gonzalez-Solino and M. Di Lorenzo, Biosensors, 2018, 8, 11 CrossRef PubMed.
- M. Torculas, J. Medina, W. Xue and X. Hu, ACS Biomater. Sci. Eng., 2016, 2, 1211 CrossRef CAS.
- M. Rasmussen, S. Abdellaoui and S. D. Minteer, Biosens. Bioelectron., 2016, 76, 91 CrossRef CAS PubMed.
- A. J. Bandodkar, J. Electrochem. Soc., 2017, 164, H3007 CrossRef CAS.
- S. Guo and E. Wang, Acc. Chem. Res., 2011, 44, 491 CrossRef CAS PubMed.
- S. Guo and E. Wang, Nano Today, 2011, 6, 240 CrossRef CAS.
- H. Li, S. Liu, Z. Dai, J. Bao and X. Yang, Sensors, 2009, 9, 8547 CrossRef CAS PubMed.
- R. D. Milton and S. D. Minteer, J. R. Soc., Interface, 2017, 14, 20170253 CrossRef PubMed.
- P. N. Bartlett, P. Tebbutt and R. G. Whitaker, Prog. React. Kinet., 1991, 16, 55 CAS.
- X. Q. Cui, C. M. Li, J. F. Zang and S. C. Yu, Biosens. Bioelectron., 2007, 22, 3288 CrossRef CAS PubMed.
- A. Konash and E. Magner, Anal. Chem., 2005, 77, 1647 CrossRef CAS PubMed.
- W. Schuhmann, T. J. Ohara, H.-L. Schmidt and A. Heller, J. Am. Chem. Soc., 1991, 113, 1394 CrossRef CAS.
- Y. Degani and A. Heller, J. Am. Chem. Soc., 1988, 110, 2615 CrossRef CAS.
- W. Schuhmann, Biosens. Bioelectron., 1995, 10, 181 CrossRef CAS.
- N. German, A. Ramanavicius, J. Voronovic and A. Ramanaviciene, Colloids Surf., A, 2012, 413, 224 CrossRef CAS.
- A. Ramanavicius, N. German and A. Ramanaviciene, J. Electrochem. Soc., 2017, 164, 45 CrossRef.
- N. German, A. Ramanavicius and A. Ramanaviciene, Electroanalysis, 2017, 29, 1267 CrossRef CAS.
- G. Bagdžiūnas, Š. Žukauskas and A. Ramanavičius, Biosens. Bioelectron., 2018, 102, 449 CrossRef PubMed.
- A. Heller, Curr. Opin. Chem. Biol., 2006, 10, 664 CrossRef CAS PubMed.
- M. T. Meredith, D.-Y. Kao, D. Hickey, D. W. Schmidtke and D. T. Glatzhofer, J. Electrochem. Soc., 2011, 158, B166 CrossRef CAS.
- K. Nguyen, Y. Holade and S. D. Minteer, ACS Catal., 2016, 6, 2603 CrossRef CAS.
- S. Liu, D. Leech and H. Ju, Anal. Lett., 2003, 36, 1 CrossRef CAS.
- N. D. Dimcheva and E. G. Horozova, Chem. Pap., 2015, 69, 17 CAS.
- J. M. Pingarrón, P. Yáñez-Sedeño and A. González-Cortés, Electrochim. Acta, 2008, 53, 5848 CrossRef.
- P. Si, S. Ding, J. Yuan, X. W. Lou and D. H. Kim, ACS Nano, 2011, 5, 7617 CrossRef CAS PubMed.
- D. Ivnitski, K. Artyushkova, R. A. Rincón, P. Atanassov, H. R. Luckarift and G. R. Johnson, Small, 2008, 4, 357 CrossRef CAS PubMed.
- Z. Y. Wang, S. N. Liu, P. Wu and C. X. Cai, Anal. Chem., 2009, 81, 1638 CrossRef CAS PubMed.
- C. X. Guo and C. M. Li, Phys. Chem. Chem. Phys., 2010, 12, 12153 RSC.
- Y. Oztekin, A. Ramanaviciene, Z. Yazicigil, A. O. Solak and A. Ramanavicius, Biosens. Bioelectron., 2011, 26, 2541 CrossRef CAS PubMed.
- G. Sanzó, C. Tortolini, R. Antiochia, G. Favero and F. Mazzei, J. Nanosci. Nanotechnol., 2015, 15, 3423 CrossRef.
- M. R. Tarasevich, V. A. Bogdanovskaya, N. M. Zagudaeva and A. V. Kapustin, Russ. J. Electrochem., 2002, 38, 335 CrossRef CAS.
- M. Wooten, S. Karra, M. Zhang and W. Gorski, Anal. Chem., 2014, 86, 752 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, Y. Cheng and S. P. Jiang, ACS Omega, 2018, 3, 667 CrossRef CAS PubMed.
- Y. Ogawa, S. Yoshino, T. Miyake and M. Nishizawa, Phys. Chem. Chem. Phys., 2014, 16, 13059 RSC.
- R. Ortiz, H. Matsumura, F. Tasca, K. Zahma, M. Samejima, K. Igarashi, R. Ludwig and L. Gorton, Anal. Chem., 2012, 84, 10315 CrossRef CAS PubMed.
- K. Wang, H. Yang, L. Zhu, Z. Ma, S. Xing, Q. Lv, J. Liao, C. Liu and W. Xing, Electrochim. Acta, 2009, 54, 4626 CrossRef CAS.
- B. Liang, L. Fang, G. Yang, Y. Hu, X. Guo and X. Ye, Biosens. Bioelectron., 2013, 43, 131 CrossRef CAS PubMed.
- C. Shan, H. Yang, J. Song, D. Han, A. Ivaska and L. Niu, Anal. Chem., 2009, 81, 2378 CrossRef CAS PubMed.
- G. Fei, G.-H. Ma, P. Wang and Z.-G. Su, Encycl. Ind. Biotechnol., 2010, 2, 2086 Search PubMed.
- K. Komori, J. Huang, N. Mizushima, S. Ko, T. Tatsumaa and Y. Sakaiab, Phys. Chem. Chem. Phys., 2017, 19, 27795 RSC.
- K. Komori, T. Tatsuma and Y. Sakai, Langmuir, 2016, 32, 9163 CrossRef CAS PubMed.
- M. Zhao, Y. Gao, J. Sun and F. Gao, Anal. Chem., 2015, 87, 2615 CrossRef CAS PubMed.
- S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A. I. Yaropolov, J. W. Whittaker and L. Gorton, Biosens. Bioelectron., 2005, 20, 2517 CrossRef CAS PubMed.
- S. Cosnier, M. Holzinger and A. Le Goff, Front. Bioeng. Biotechnol., 2014, 2, 45 Search PubMed.
- L. Fruk, C. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 1550 CrossRef CAS PubMed.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877 CrossRef CAS PubMed.
- F. Patolsky, Y. Weizmann and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 2113 CrossRef CAS PubMed.
- A. Trifonov, R. Tel-Vered, M. Fadeev, A. Cecconello and I. Willner, Electroanalysis, 2015, 27, 2150 CrossRef CAS.
- D. Scott, M. Toney and M. Muzikár, J. Am. Chem. Soc., 2008, 230, 865 CrossRef PubMed.
- O. Yehezkeli, S. Raichling, R. Tel-Vered, E. Kesselman, D. Danino and I. Willner, J. Phys. Chem. Lett., 2010, 1, 2816 CrossRef CAS.
- D. Zhan, J. Velmurugan and M. V. Mirkin, J. Am. Chem. Soc., 2009, 131, 14756 CrossRef CAS PubMed.
- A. P. Yadav, A. Nishikata and T. Tsuru, Electrochim. Acta, 2007, 52, 7444 CrossRef CAS.
- V. Stamenkovic, N. M. Markovic and P. N. Ross Jr, J. Electroanal. Chem., 2001, 500, 44 CrossRef CAS.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098 CrossRef CAS PubMed.
- M. V. A. Martins, A. R. Pereira, R. A. S. Luz, R. M. Lost and F. N. Crespilho, Phys. Chem. Chem. Phys., 2014, 16, 17426 RSC.
- J. H. T. Luong, J. D. Glennon, A. Gedanken and S. K. Vashist, Microchim. Acta, 2017, 184, 369 CrossRef CAS.
- D. Petrović, D. Frank, S. C. L. Kamerlin, K. Hoffmann and B. Strodel, ACS Catal., 2017, 7, 6188 CrossRef PubMed.
- J. P. Roth and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 62 CrossRef CAS PubMed.
- G. S. Wilson, Biosens. Bioelectron., 2016, 82, vii CrossRef CAS PubMed.
- P. N. Bartlett and F. A. Al-Lolage, J. Electroanal. Chem., 2018, 819, 26 CrossRef CAS.
- G. Wohlfahrt, S. Witt, J. Hendle, D. Schomburg, H. Kalisz and H. J. Hecht, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 969 CrossRef CAS.
- K. Ponlakhet, M. Amatatongchai, W. Sroysee, P. Jarujamrusa and S. Chairama, Anal. Methods, 2016, 8, 8288 RSC.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CrossRef CAS.
- C. Bourdillon, C. Demaille, J. Gueris, J. Moiroux and J. M. Savéant, J. Am. Chem. Soc., 1993, 115, 12264 CrossRef CAS.
- K. Y. Kwon, J. Youn, J. H. Kim, Y. Park, C. Jeon, B. C. Kim, Y. Kwon, X. Zhao, P. Wang, B. I. Sang, J. Lee, H. G. Park, H. N. Chang, T. Hyeon, S. Ha, H.-T. Jung and J. Kim, Biosens. Bioelectron., 2010, 26, 655 CrossRef CAS PubMed.
- G. M. Olyveira, J. H. Kim, M. V. A. Martins, R. M. Lost, K. N. Chaudhari, J.-S. Yu and F. N. Crespilho, J. Nanosci. Nanotechnol., 2012, 12, 356 CrossRef CAS PubMed.
- C. X. Guo, F. P. Hu, X. W. Lou and C. M. Li, J. Power Sources, 2010, 195, 4090 CrossRef CAS.
- A. Korani, A. Salimi and B. Karimi, Electroanalysis, 2017, 29, 2646 CrossRef CAS.
- M. Zhou, L. Deng, D. Wen, L. Shang, L. Jin and S. Dong, Biosens. Bioelectron., 2009, 24, 2904 CrossRef CAS PubMed.
- A. Trifonov, K. Herkendell, R. Tel-Vered, O. Yehezkeli, M. Woerner and I. Willner, ACS Nano, 2013, 7, 113358 CrossRef PubMed.
- A. Trifonov, R. Tel-Vered, M. Fadeev and I. Willner, Adv. Energy Mater., 2015, 5, 1401853 CrossRef.
- K. Herkendell, R. Tel-Vered and A. Stemmer, Nanoscale, 2017, 9, 14118 RSC.
- H. Deng, A. K. L. Teo and Z. Gao, Sens. Actuators, B, 2014, 191, 522 CrossRef CAS.
- H. M. Bambhania, D. Chakraborty, H. Wen and S. C. Barton, J. Electrochem. Soc., 2017, 164, 232 CrossRef.
- B. Willner and I. Willner, in Bioelectronics – From Theory to Applications, ed. I. Willner and E. Katz, Wiley-VCH, Weinheim, 2005, ch. 3, pp. 35–97 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00177d |
| This journal is © The Royal Society of Chemistry 2019 |