
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerating the water splitting kinetics of CoP microcubes anchored on a graphene electrocatalyst by Mn incorporation†
Xun
Xu
ab,
Hanfeng
Liang
 *c,
Guisheng
Tang
a,
Yingling
Hong
a,
Yaqiang
Xie
a,
Zhengbing
Qi
d,
Binbin
Xu
a and
Zhoucheng
Wang
*a
*c,
Guisheng
Tang
a,
Yingling
Hong
a,
Yaqiang
Xie
a,
Zhengbing
Qi
d,
Binbin
Xu
a and
Zhoucheng
Wang
*a
aCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: zcwang@xmu.edu.cn
bSchool of Materials and Mechanical & Electrical Engineering, Jiangxi Science and Technology Normal University, Nanchang 330013, China
cMaterials Science and Engineering, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia. E-mail: hanfeng.liang@kaust.edu.sa
dCollege of Materials Science and Engineering, Xiamen University of Technology, Xiamen 361024, China
First published on 21st November 2018
Abstract
CoP is considered as an efficient electrocatalyst for the hydrogen evolution reaction (HER) in acidic electrolytes but its performance in alkaline solutions is generally poor because of its slow reaction kinetics, which further limits its application in overall water splitting. Herein, we demonstrate a strategy to greatly accelerate its HER and OER kinetics in alkaline solutions through Mn incorporation. Ternary MnxCo1−xP microcubes with a tunable Mn/Co ratio strongly anchored on rGO were synthesized using Prussian blue analogues as precursors. The synergy between the high activity of MnxCo1−xP microcubes and the good conductivity of rGO leads to the superior performance of the hybrid toward water splitting in 1 M KOH. The optimized Mn0.6Co0.4P–rGO electrocatalyst shows high activity and stability towards both the HER and OER with low overpotentials of 54 and 250 mV at 10 mA cm−2, respectively. Furthermore, the water electrolyzer using Mn0.6Co0.4P–rGO as both the cathode and anode only requires a cell voltage as low as 1.55 V to reach a current density of 10 mA cm−2, making Mn0.6Co0.4P–rGO a competitive and cost-effective electrocatalyst for water splitting.
Hydrogen, as a clean energy carrier, has the potential to replace fossil fuels and fulfill the large future energy demand.1 Water splitting powered by renewable electricity is one of the most attractive methods for high purity hydrogen production. However, efficient water splitting requires highly efficient and robust electrocatalysts that can sufficiently overcome the sluggish kinetics of the two half reactions, namely the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER).2–5 Currently, noble metal (such as Pt, Ir and Ru)-based compounds are identified as the most active electrocatalysts, while their high cost and scarcity prevent them from being applied on a large scale.3–6 Consequently, major efforts have been devoted toward developing non-precious metal electrocatalysts with high activity (e.g., perovskite oxides and transition metal oxides/hydroxides for the OER and nitrides, chalcogenides, carbides, and phosphides for the HER), especially those that can catalyze both the HER and OER.2–26 Among them, transition-metal phosphides (TMPs) have been extensively explored due to their low cost and high activity.14–18,27–31 TMPs were first known as HER catalysts and have lately shown their efficacy for the OER, though real active materials are the surface (hydro)oxides in situ formed in the OER process.32
CoP, as a representative TMP material, has shown high electrocatalytic activity towards the HER in acidic electrolytes.33 However, its HER activity in alkaline solutions is poor, because of its weak ability to promote water dissociation. Incorporation with a second metal could potentially improve the alkaline HER performance due to the synergy between them. For example, transition metal (such as Fe, Zn, and Al)-doped CoP has shown enhanced performance.34–37 We noted that the Mn tetramer complex has shown reasonable catalytic activity towards water oxidation in photosystem II,38 suggesting relatively facile water dissociation kinetics. We therefore proposed that Mn incorporation into CoP could greatly enhance the HER activity in alkaline solutions. On the other hand, the Co might also promote the OER on the Mn oxide. This is possible as already confirmed in the case of CaMn2O4, where Ca incorporation improves catalytic rates.38 In fact, MnCoP particles have already been synthesized and used for OER electrocatalysis.39 However, they suffer from a significant performance decay with ∼25% activity loss within 15 h, possibly because of the surface oxidation and active mass leaching. Mn-doped CoP has also been explored but the amount of Mn dopant is generally quite low.40 Further optimization is therefore necessary to stabilize the catalyst and thus to achieve stable performance.
Decorating electrocatalysts with conductive graphene supports could be an efficient approach to improve the catalytic performance. Graphene can improve the interaction between electrocatalysts and electrode supports, which facilitates charge transport.41,42 In addition, delicate nanostructuring and compositional tuning can further boost the activity. In this context, Prussian blue analogues (PBAs) with uniform sizes, various compositions, and diverse morphologies and architectures can serve as ideal precursors to prepare electrocatalysts.43 The converted materials generally inherit the morphology and structure (tunable composition) of the PBAs and thus offer an excellent platform to study the impacts of the composition and structure on the catalytic activity. Although a number of metal oxides, sulfides, and selenides have been synthesized using PBAs as templates,44–46 the preparation of MnxCo1−xP, however, has rarely been reported.
Herein we report the synthesis of MnxCo1−xP microcubes anchored on reduced graphene oxide (rGO) from a MnCo PBA precursor and further their application in water splitting electrocatalysis. We show that Mn incorporation greatly enhances the reaction kinetics of CoP towards both the OER and HER in alkaline solutions. The PBA precursor allows us to easily tune the Mn/Co ratio in the product, and the optimized Mn0.6Co0.4P–rGO hybrid catalyst exhibits superior catalytic activity with overpotentials of 250 and 54 mV for the OER and HER in 1 M KOH along with outstanding stability, respectively. More importantly, the Mn0.6Co0.4P–rGO can also drive the overall water splitting electrocatalysis with a low cell voltage of 1.55 V at 10 mA cm−2 and remarkable durability, outperforming the Pt/C‖RuO2 couple at high current densities.
The preparation process of the MnxCo1−xP–rGO hybrid is illustrated in Fig. 1a (also see Methods in the ESI† for details). MnCo PBA microcubes (Fig. 1b) were first in situ grown on graphene oxide (GO) nanosheets. The resulting product was then collected from the solution and consequently freeze dried to form MnCo PBA–GO. Then MnCo PBA–GO was converted into MnxCo1−xP–rGO by a gas-phase reaction with PH3 (generated by sodium hypophosphite) at 400 °C. The composition of the MnxCo1−xP–rGO can be readily tuned by adjusting the ratio of Mn/Co in the PBA precursor, and the optimal composition was found to be Mn0.6Co0.4P–rGO. The morphologies of the MnCo PBA–GO and Mn0.6Co0.4P–rGO were revealed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The MnCo PBA microcubes with an average size of about 800 nm were homogeneously anchored on GO sheets (Fig. 1c–e and S1†). After thermal phosphorization, the MnCo PBA was converted into Mn0.6Co0.4P with a well-retained cubic structure (Fig. 1f–h and S1†). Meanwhile, GO was reduced to rGO because of the reducibility of PH3 gas. For comparison, SEM images of CoP–rGO, Mn0.2Co0.8P–rGO, and Mn0.4Co0.6P–rGO are also shown (Fig. S2†). The results show that the morphology and particle size of MnxCo1−xP–rGO are similar despite their different Mn/Co ratios. It is noteworthy that the Mn0.6Co0.4P microcubes are still evenly anchored on rGO sheets after thermal conversion, suggesting the strong binding between Mn0.6Co0.4P and rGO. Such geometric confinement of nanoparticles within rGO layers is expected to reinforce their interfacial contact and to prevent the Mn0.6Co0.4P from structural collapse and aggregation during electrocatalysis, which ensures high stability.6,21 In comparison, the Mn0.6Co0.4P microcubes without rGO are obviously more aggregated (Fig. S3†). The high-resolution TEM (HRTEM) image of Mn0.6Co0.4P is shown in the inset of Fig. 1h. The lattice fringe spacing of the particle was measured to be 0.28 nm, corresponding to the (110) plane of CoP. The energy-dispersive X-ray (EDX) spectrum reveals the coexistence of Mn, Co, and P and C (Fig. S4†). The corresponding TEM-EDX elemental mapping images of Mn0.6Co0.4P–rGO are shown in Fig. 1i. The Mn, Co and P are homogeneously distributed in a microcube, whereas the C outer shell layer is clearly identified, confirming that the Mn0.6Co0.4P is indeed anchored on rGO sheets. The atomic ratio of Mn, Co, and P of the sample is 0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4:1 as determined by inductively coupled plasma optical emission (ICP-OES) analysis (Table S1†).
:0.4:1 as determined by inductively coupled plasma optical emission (ICP-OES) analysis (Table S1†).
 | ||
| Fig. 1 Morphological characterization of the MnCo PBA–GO and Mn0.6Co0.4P–rGO: (a) schematic illustration of the synthesis process of the Mn0.6Co0.4P–rGO hybrid. (b) SEM image of MnCo PBA microcubes. (c) SEM and (d and e) TEM images of MnCo PBA–GO. (f) SEM (g and h) TEM images and (i) HAADF image and the corresponding elemental maps of Mn0.6Co0.4P–rGO. The inset of (h) shows the HRTEM image of Mn0.6Co0.4P–rGO. | ||
We then carried out X-ray diffraction (XRD) measurements to identify the detailed structure of the as-prepared samples. The XRD results confirm the phase of the MnCo PBA (JCPDS #51-1898) precursor, and the converted product Mn0.6Co0.4P adopts the structure of CoP (JCPDS #29-0497) (Fig. 2a). We further performed the X-ray photoelectron spectroscopy (XPS) measurements to probe the surface composition and chemical valence states of the Mn0.6Co0.4P–rGO. The Mn 2p3/2 region shows a singlet peak at 641.0 eV that can be ascribed to oxidized Mn species (Fig. 2b), indicating that the surface was oxidized because of the high oxophilicity of Mn.30,31,47 The Co 2p3/2 region (Fig. 2c) shows two main peaks at 778.7 and 781.4 eV that are attributed to Co species in phosphides and the oxidized species, respectively.48–50 As for the P 2p region (Fig. 2d), the peaks at 130.2 and 131.1 eV can be assigned to the P 2p doublet, consistent with those observed in metal phosphides.16,49–52 The peak near 133.8 eV is attributed to the oxidized phosphorus in phosphates, indicating that the surface of Mn0.6Co0.4P is oxidized, which is commonly seen in metal phosphides under air exposure.31,50,52 The XPS results confirm the successful conversion of MnCo PBA to Mn0.6Co0.4P. Furthermore, the XPS survey spectrum also suggests the presence of N (Fig. S5†), indicating that N might be doped into rGO during the thermal conversion process.
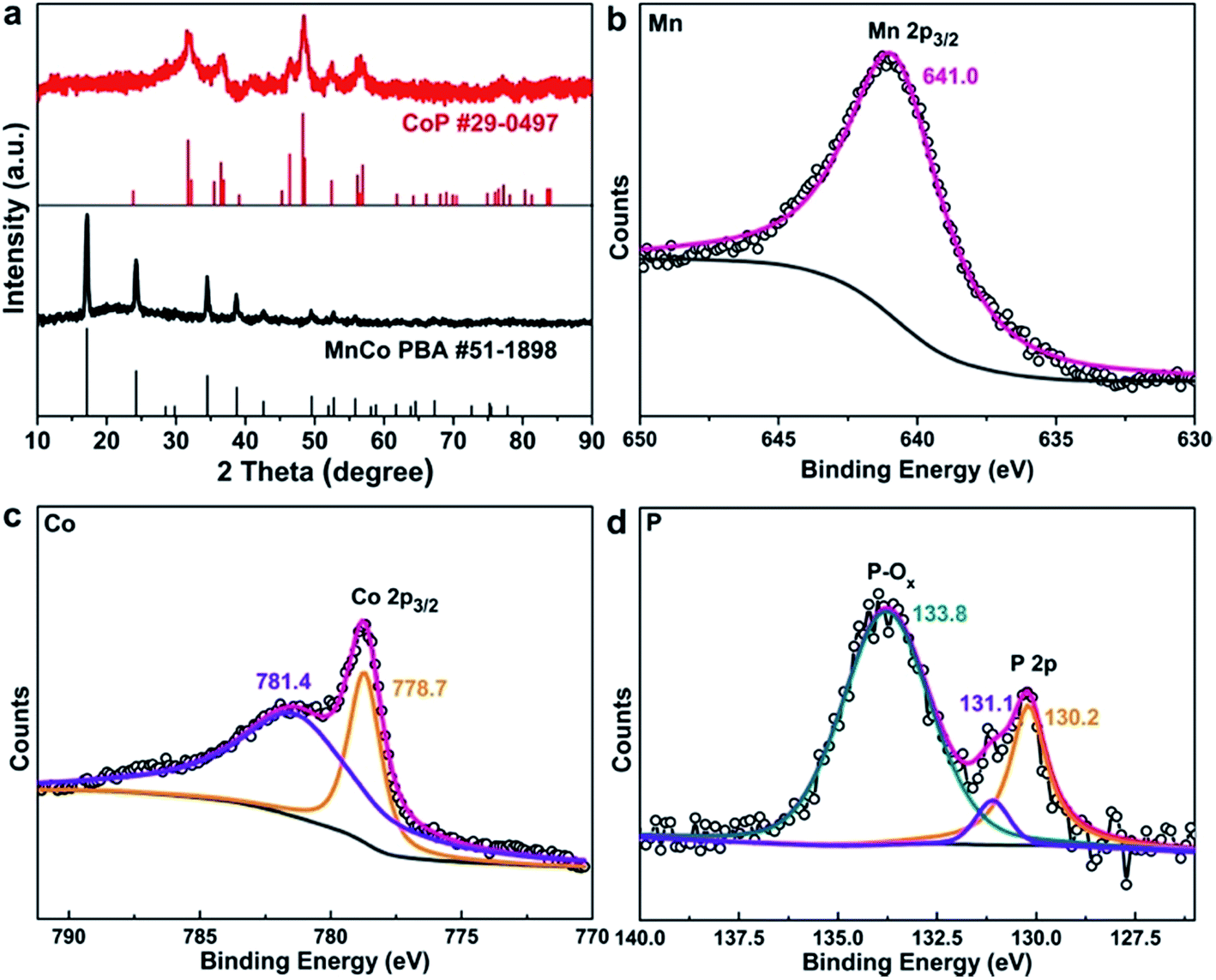 | ||
| Fig. 2 Structural characterization of the Mn0.6Co0.4P–rGO hybrid: (a) XRD patterns of MnCo PBA–GO and Mn0.6Co0.4P–rGO. (b) Mn 2p3/2, (c) Co 2p3/2, and (d) P 2p high resolution XPS spectra of the Mn0.6Co0.4P–rGO. | ||
We first assessed the catalytic OER activity of the MnxCo1−xP–rGO (x = 0.6, 0.4, 0.2), CoP–rGO, RuO2, and rGO in 1 M KOH using a standard three-electrode system (see Methods for details†). The polarization curves of these catalysts were obtained from linear sweep voltammograms (LSV) at a scan rate of 1 mV s−1. As shown in Fig. 3a, the phosphide catalysts show significantly higher activity than the rGO support, suggesting that the main OER contribution comes from the materials themselves. The pristine CoP–rGO already shows a low overpotential of 330 mV at a current density of 10 mA cm−2; the performance can be further boosted upon Mn incorporation, which confirms the efficacy of our strategy. Upon the introduction of a small amount of Mn (x = 0.2), though the overpotential of Mn0.2Co0.8P–rGO only decreases by 2 mV at 10 mA cm−2, the performance at high currents is largely improved, suggesting promoted reaction kinetics. Further enhancement is observed when more Mn is incorporated into CoP. For example, the overpotentials at 10 mA cm−2 further decrease to 290 and 250 mV on Mn0.4Co0.6P–rGO and Mn0.6Co0.4P–rGO, respectively. Note that the latter even outperforms the state-of-the-art catalyst, namely RuO2 (270 mV). These overpotentials compare favorably or at least comparable to those of recently reported high-performance earth-abundant OER catalysts (Table S2†).48,53–55 Further increasing the Mn ratio (x > 0.6) might result in even better activity; unfortunately we failed to synthesize the corresponding MnCo PBA precursor with a high Mn ratio, possibly because the high Mn ratio may cause structural instability. The OER performance of Mn0.6Co0.4P microcubes is also measured to investigate the synergistic effect between the Mn0.6Co0.4P and rGO (Fig. S6†). The results showed that the Mn0.6Co0.4P–rGO hybrid is much more active than Mn0.6Co0.4P microcubes, suggesting that the synergy between Mn0.6Co0.4P and rGO could greatly boost the OER activity. The direct integration of Mn0.6Co0.4P on rGO nanosheets ensures efficient charge transport in the hybrid, which can be confirmed by electrochemical impedance spectroscopy (EIS) studies (Fig. S7†). To further evaluate the catalytic kinetics of these catalysts, the corresponding Tafel plots were investigated. As shown in Fig. 3b, the Tafel slope of the CoP–rGO catalyst is 110 mV dec−1. This value decreases to 88, 84, and 65 mV dec−1 on Mn0.2Co0.8P–rGO, Mn0.4Co0.6P–rGO, and Mn0.6Co0.4P–rGO, indicating that the OER kinetics is promoted upon Mn incorporation. To better understand the intrinsic activity, we calculated the turnover frequency (TOF) of the catalyst (Fig. S8†), and the results show that the Mn0.6Co0.4P–rGO possesses the highest TOF value. The superior OER activity can be attributed to the synergy between Mn and Co and the strong binding with rGO that improves the conductivity and helps to stabilize the structure. We further carried out a simple cyclic voltammetry (CV) measurement50 to determine the electrochemically active surface area (ECSA) of the samples (Fig. 3d, S9 and S10†) and found that the Mn0.6Co0.4P–rGO possesses the highest double layer capacitance (33.3 mF cm−2) among the investigated samples, which is nearly 1.9 and 2.7 times those of the Mn0.6Co0.4P (17.4 mF cm−2) and CoP–rGO (12.3 mF cm−2), respectively, revealing more exposed active sites of Mn0.6Co0.4P–rGO.56
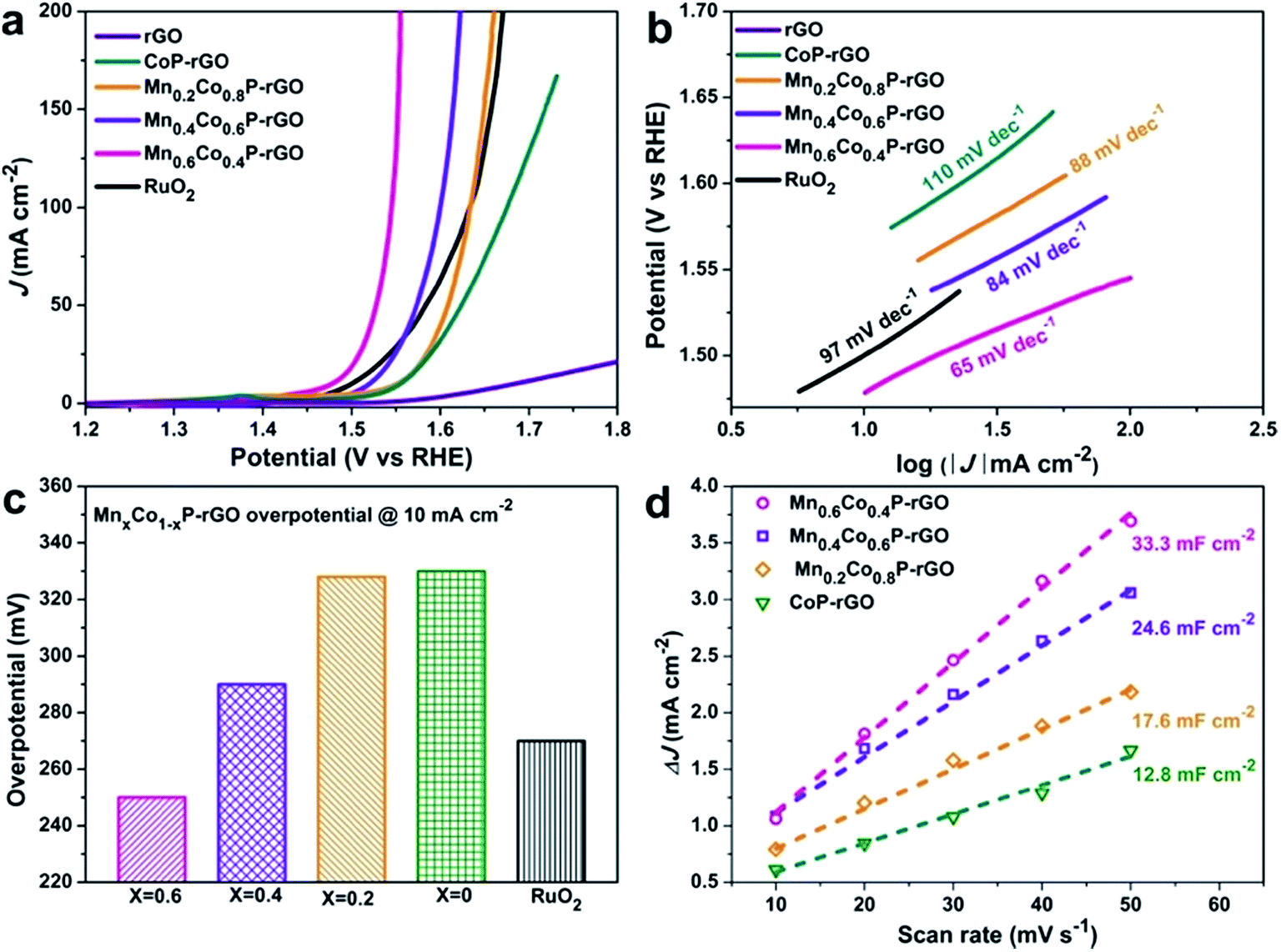 | ||
| Fig. 3 Electrocatalytic OER performance of MnxCo1−xP–rGO (x = 0.6, 0.4, 0.2, 0) loaded on carbon paper in 1 M KOH: (a) IR-corrected polarization curves of the MnxCo1−xP–rGO, rGO, and RuO2. (b) The corresponding Tafel plots. (c) Overpotentials required at J = 10 mA cm−2. (d) Plots showing the extraction of the double-layer capacitances that allow the estimation of the electrochemically active surface area (ECSA). | ||
Aside from the high OER activity, the operating stability is another indicator to evaluate a practical catalyst. The stability of the Mn0.6Co0.4P–rGO catalyst was tested by the controlled potential electrolysis (CPE) and continuous cyclic voltammetry (CV) measurements (Fig. S11†). As shown in Fig. S11a,† a slight activity loss is observed after 5000 CV cycles for the Mn0.6Co0.4P–rGO catalyst. To further investigate the stability of the catalyst for the OER, we conducted the stability test by applying a static overpotential of 310 mV (1.54 V vs. RHE) continuously for 24 h. As shown in Fig. S11b,† the current response only decreases by about 5% after 24 h operation, suggesting the robustness of the Mn0.6Co0.4P–rGO catalyst. In an effort to understand the surface compositional change of the Mn0.6Co0.4P–rGO, we conducted the XPS analysis on the post-OER sample (Fig. S12†). In contrast to the pristine Mn0.6Co0.4P–rGO, the intensity of Co (778.7 eV) and P (130.2 eV) peaks that correspond to the species in phosphides decreases and the peak corresponding to the oxidized Mn shifts to a higher binding energy (from 641.0 to 642.6 eV). This result suggests that the surface of Mn0.6Co0.4P–rGO is further oxidized upon OER to generate (hydro)oxides. This phenomenon is commonly seen for metal phosphides after OER catalysis.30,31,51 TEM observations further reveal an amorphous layer that is formed on the surface of particles, which is likely the (hydro)oxides that are generated during the OER process (Fig. S13†).30,32 Together with the XPS data, it is clear that the surface of the catalyst has been oxidized into hydroxides or phosphates, which should be the real OER active sites.
We then evaluated the HER activity of the MnxCo1−xP–rGO loaded on carbon paper in 1 M KOH (see Methods for details†). For comparison, the HER activity of 20 wt% Pt/C (about 2.5 mg cm−2 mass loading) and rGO on carbon paper was also tested. As expected, the Pt/C catalyst exhibits the best performance with a near zero onset overpotential and achieves a current density of 10 mA cm−2 at an overpotential of 31 mV (Fig. 4a). Similar to what we observed in OER catalysis, the Mn0.6Co0.4P–rGO again shows enhanced catalytic activity towards the HER compared to CoP–rGO. The current density of 10 mA cm−2 is achieved at an overpotential as low as 54 mV, whereas larger overpotentials are required for Mn0.4Co0.6P–rGO (81 mV), Mn0.2Co0.8P–rGO (101 mV), and CoP–rGO (143 mV) catalysts (Fig. 4b). These potentials compare favorably to those of reported TMPs, chalcogenides, nitrides and other earth-abundant HER catalysts in alkaline electrolytes (Table S3†).56–63 The excellent activity of the Mn0.6Co0.4P–rGO is also confirmed by its relatively small Tafel slope of 63 mV dec−1, which is much smaller than those of Mn0.4Co0.6P–rGO (92 mV dec−1), Mn0.2Co0.8P–rGO (106 mV dec−1), and CoP–rGO (149 mV dec−1) catalysts (Fig. 4b), which again confirms the efficacy of using Mn to promote the water splitting kinetics of CoP. Besides, the HER performance of Mn0.6Co0.4P–rGO microcubes is also superior to that of Mn0.6Co0.4P microcubes due to the synergistic effect between Mn0.6Co0.4P and rGO (Fig. S6†). The Mn0.6Co0.4P–rGO also shows the highest TOF of 0.0698 s−1, which is much larger than those of Mn0.4Co0.6P–rGO, Mn0.2Co0.8P–rGO, and CoP–rGO catalysts (Fig. S8†). Moreover, the Mn0.6Co0.4P–rGO can also efficiently catalyze the HER in acidic and neutral electrolytes (Fig. S14†). The overpotentials at 10 mA cm−2 are 43 and 70 mV in 0.5 M H2SO4 and 1 M PBS, respectively. The Mn0.6Co0.4P–rGO electrode also exhibits good stability towards the HER (Fig. S11†). Only a slight performance decay was observed for the Mn0.6Co0.4P–rGO catalyst after 5000 CV cycles, suggesting its good stability under basic conditions. Indeed, the current density achieved at a given overpotential of 140 mV barely decreases after 24 h. The XPS analysis of the post-HER Mn0.6Co0.4P–rGO electrode reveals that the main composition remains unchanged. However, both the Mn 2p and Co 2p peaks slightly shift to higher binding energies (Fig. S12†), which could be due to the partial surface oxidation in strong basic solutions.7,12,16,63
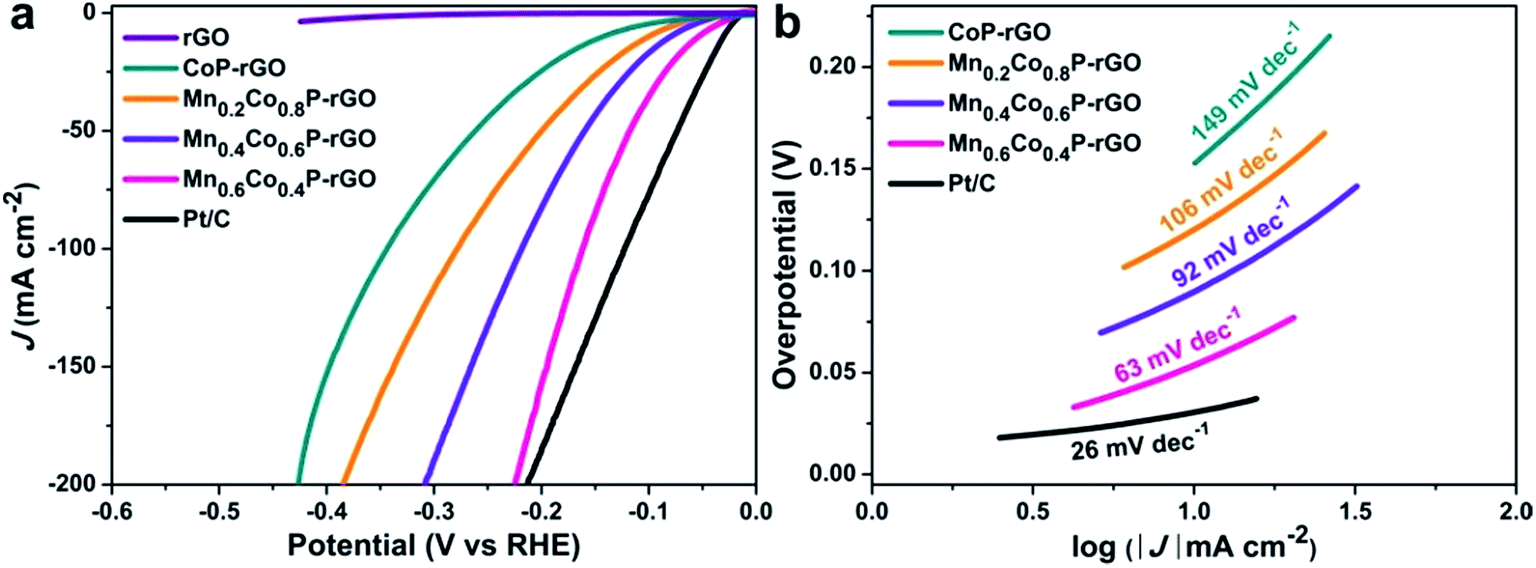 | ||
| Fig. 4 Electrocatalytic HER performance of MnxCo1−xP–rGO (x = 0.6, 0.4, 0.2, 0) loaded on carbon paper in 1 M KOH: (a) IR-corrected polarization curves of the MnxCo1−xP–rGO, rGO, and 20 wt% Pt/C. (b) The corresponding Tafel plots. | ||
Water splitting catalyzed by the same catalyst could simplify the water splitting system and reduce the operating cost. Encouraged by the excellent catalytic activity and robust stability toward both the OER (although the real active species is the surface hydroxides as demonstrated earlier) and HER, we then assembled a two-electrode water electrolyzer using the Mn0.6Co0.4P–rGO as both the cathode and anode for overall water splitting in 1 M KOH. The Pt/C‖RuO2 loaded on carbon paper was also tested for comparison. As shown in Fig. 5a, overall water-splitting using Mn0.6Co0.4P–rGO electrodes requires a low cell voltage of 1.55 V to drive 10 mA cm−2. This voltage is slightly lower than that of Pt/C‖RuO2 (1.54 V) and is among the lowest reported values (Table S4†). Note that the Mn0.6Co0.4P–rGO electrodes can drive high current densities (e.g., 100 mA cm−2) at cell voltages that are much lower than those of Pt/C‖RuO2, which is vital for industrial H2 production. Moreover, the Mn0.6Co0.4P–rGO exhibits great mechanical robustness and durability as demonstrated by the multi-step chronopotentiometric and chronopotentiometric measurements over 48 h (Fig. 5c and d). The voltage required to achieve 50 mA cm−2 barely changes after 48 h catalysis. The faradaic efficiency of MnCoP–rGO was calculated to be ∼97% by comparing the experimental H2/O2 production amounts with the theoretical values (Fig. 5b).
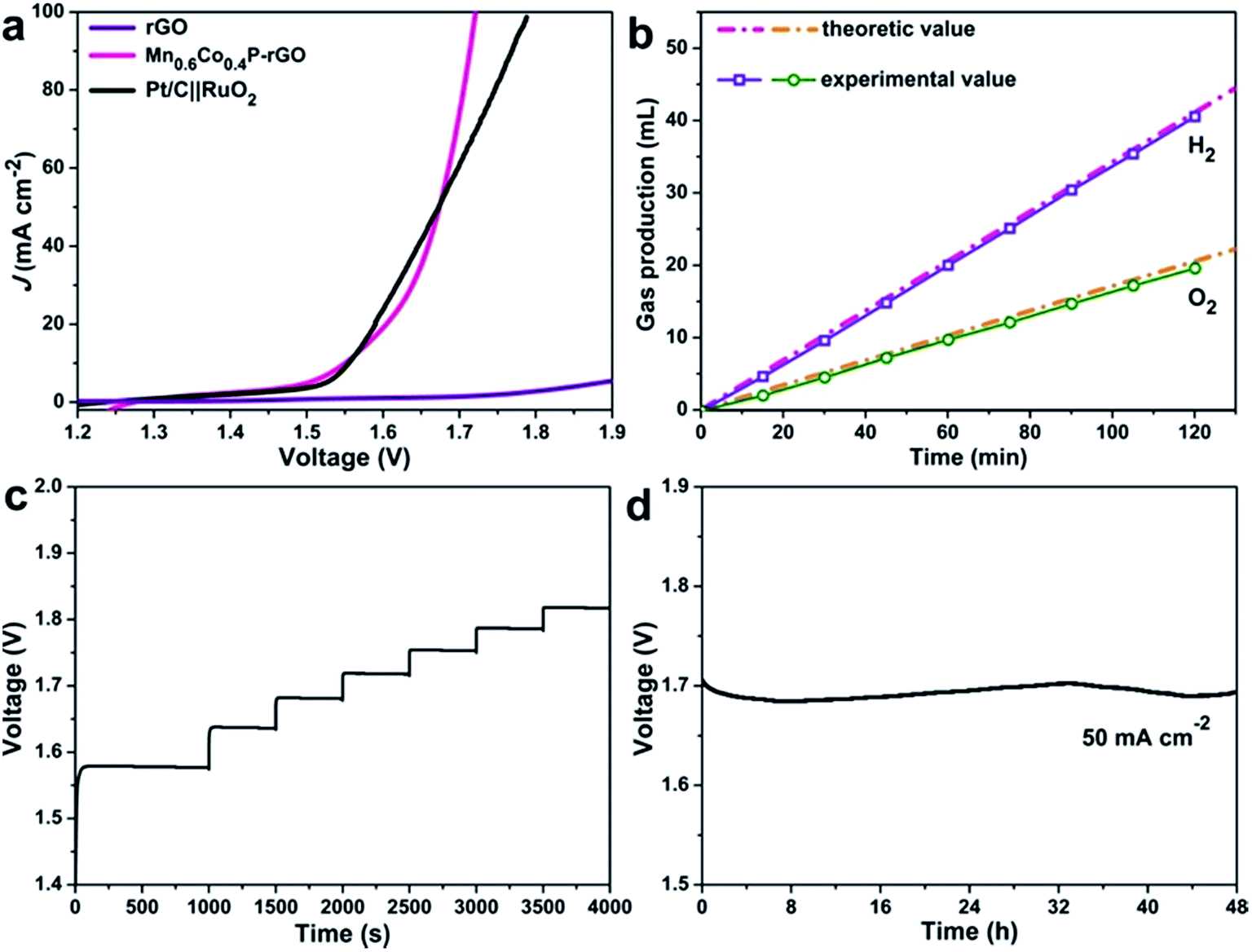 | ||
| Fig. 5 Mn0.6Co0.4P–rGO for overall water splitting electrocatalysis in 1 M KOH. (a) IR-corrected polarization curves. (b) H2 and O2 production catalyzed by Mn0.6Co0.4P–rGO in 1 M KOH at room temperature. The dash lines present the theoretic value H2 and O2 amounts expected for a 100% Faradaic efficiency. (c) The multi-step chronopotentiometric curve. (d) Long-term stability test carried out at 50 mA cm−2. | ||
In summary, we suggest that Mn incorporation can greatly promote the water splitting kinetics of CoP. By further anchoring the optimized Mn0.6Co0.4P on rGO, high electrocatalytic activity towards both the OER and HER in 1 M KOH is achieved. Specifically, the Mn0.6Co0.4P–rGO only requires overpotentials as low as 54 and 250 mV to drive HER and OER current densities of 10 mA cm−2, respectively. Moreover, the material can drive overall water splitting at a low cell voltage of 1.55 V (at 10 mA cm−2) with a nearly 100% faradaic efficiency and outstanding stability. Our work not only establishes the Mn0.6Co0.4P as a high-performance water splitting catalyst but also provides a general strategy to synthesize phosphide/graphene composites with delicate nanostructures and tunable compositions and further to significantly boost their performance in electrocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (51372212) for financial support.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- H. Wang, H. W. Lee, Y. Deng, Z. Lu, P. C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS.
- J. Xie and Y. Xie, Chem.–Eur. J., 2015, 22, 3588–3598 CrossRef PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- W. Tang, J. Wang, L. Guo, X. Teng, T. J. Meyer and Z. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 41347–41353 CrossRef CAS PubMed.
- H. Shi, H. Liang, F. Ming and Z. Wang, Angew. Chem., Int. Ed. Engl., 2017, 56, 573–577 CrossRef CAS PubMed.
- C. Xiao, B. Zhang and D. Li, Electrochim. Acta, 2017, 242, 260–267 CrossRef CAS.
- F. Ming, H. Liang, H. Shi, X. Xu, G. Mei and Z. Wang, J. Mater. Chem. A, 2016, 4, 15148–15155 RSC.
- C. Wu and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 41314–41322 CrossRef CAS.
- F. Ming, H. Liang, H. Shi, G. Mei, X. Xu and Z. Wang, Electrochim. Acta, 2017, 250, 167–173 CrossRef CAS.
- Y. Ma, Z. He, Z. Wu, B. Zhang, Y. Zhang, S. Ding and C. Xiao, J. Mater. Chem. A, 2017, 5, 24850–24858 RSC.
- P. W. Menezes, A. Indra, C. Das, C. Walter, C. Göbel, V. Gutkin, D. Schmeiβer and M. Driess, ACS Catal., 2016, 7, 103–109 CrossRef.
- Z. Xing, Q. Liu, A. M. Asiri and X. Sun, ACS Catal., 2014, 5, 145–149 CrossRef.
- X. D. Wang, Y. Cao, Y. Teng, H. Y. Chen, Y. F. Xu and D. B. Kuang, ACS Appl. Mater. Interfaces, 2017, 9, 32812–32819 CrossRef CAS.
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlogl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 CrossRef CAS PubMed.
- P. Wang, F. Song, R. Amal, Y. H. Ng and X. Hu, ChemSusChem, 2016, 9, 472–477 CrossRef CAS.
- J. Wang, Y. Gao, D. Chen, J. Liu, Z. Zhang, Z. Shao and F. Ciucci, ACS Catal., 2017, 8, 364–371 CrossRef.
- Y. Zhang, H. Zhang, L. Fang, J. Deng and Y. Wang, Electrochim. Acta, 2017, 245, 32–40 CrossRef CAS.
- S. Zhang, X. Yu, F. Yan, C. Li, X. Zhang and Y. Chen, J. Mater. Chem. A, 2016, 4, 12046–12053 RSC.
- J. Ping, Y. Wang, Q. Lu, B. Chen, J. Chen, Y. Huang, Q. Ma, C. Tan, J. Yang, X. Cao, Z. Wang, J. Wu, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 7640–7645 CrossRef CAS PubMed.
- H. Liang, L. Li, F. Meng, L. Dang, J. Zhuo, A. Forticaux, Z. Wang and S. Jin, Chem. Mater., 2015, 27, 5702 CrossRef CAS.
- G. F. Chen, T. Y. Ma, Z. Q. Liu, N. Li, Y. Z. Su, K. Davey and S. Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314–3323 CrossRef CAS.
- C. Huang, T. Ouyang, Y. Zou, N. Li and Z.-Q. Liu, J. Mater. Chem. A, 2018, 6, 7420–7427 RSC.
- H. Cheng, Y.-Z. Su, P.-Y. Kuang, G.-F. Chen and Z.-Q. Liu, J. Mater. Chem. A, 2015, 3, 19314–19321 RSC.
- C. G. Read, J. F. Callejas, C. F. Holder and R. E. Schaak, ACS Appl. Mater. Interfaces, 2016, 8, 12798–12803 CrossRef CAS PubMed.
- Y. Yan, B. Y. Xia, X. Ge, Z. Liu, A. Fisher and X. Wang, Chem.–Eur. J., 2015, 21, 18062–18067 CrossRef CAS PubMed.
- T. Zhou, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, A. Borgna and R. Xu, ACS Catal., 2017, 7, 6000–6007 CrossRef CAS.
- D. Li, H. Baydoun, B. Kulikowski and S. L. Brock, Chem. Mater., 2017, 29, 3048–3054 CrossRef CAS.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- C. Tang, L. Gan, R. Zhang, W. Lu, X. Jiang, A. M. Asiri, X. Sun, J. Wang and L. Chen, Nano Lett., 2016, 16, 6617–6621 CrossRef CAS.
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Adv. Energy Mater., 2017, 7, 1700020 CrossRef.
- R. Zhang, C. Tang, R. Kong, G. Du, A. M. Asiri, L. Chen and X. Sun, Nanoscale, 2017, 9, 4793–4800 RSC.
- R. Zhang, X. Ren, S. Hao, R. Ge, Z. Liu, A. M. Asiri, L. Chen, Q. Zhang and X. Sun, J. Mater. Chem. A, 2018, 6, 1985–1990 RSC.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027 RSC.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS PubMed.
- T. Liu, X. Ma, D. Liu, S. Hao, G. Du, Y. Ma, A. M. Asiri, X. Sun and L. Chen, ACS Catal., 2016, 7, 98–102 CrossRef.
- Y. Liu, S. Liu, X. Lai, J. Miao, D. He, N. Li, F. Luo, Z. Shi and S. Liu, Adv. Funct. Mater., 2015, 25, 4480–4485 CrossRef CAS.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 RSC.
- X.-Y. Yu, Y. Feng, B. Guan, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 RSC.
- A. Kumar and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2017, 9, 41906–41915 CrossRef CAS.
- X. Y. Yu, Y. Feng, Y. Jeon, B. Guan, X. W. Lou and U. Paik, Adv. Mater., 2016, 28, 9006–9011 CrossRef CAS PubMed.
- X. Xu, H. Liang, F. Ming, Z. Qi, Y. Xie and Z. Wang, ACS Catal., 2017, 7, 6394–6399 CrossRef CAS.
- C.-H. Kuo, I. M. Mosa, A. S. Poyraz, S. Biswas, A. M. El-Sawy, W. Song, Z. Luo, S.-Y. Chen, J. F. Rusling, J. He and S. L. Suib, ACS Catal., 2015, 5, 1693–1699 CrossRef CAS.
- K. Liu, C. Zhang, Y. Sun, G. Zhang, X. Shen, F. Zou, H. Zhang, Z. Wu, E. C. Wegener, C. J. Taubert, J. T. Miller, Z. Peng and Y. Zhu, ACS Nano, 2017, 12, 158–167 CrossRef PubMed.
- X. Yang, A.-Y. Lu, Y. Zhu, M. N. Hedhili, S. Min, K.-W. Huang, Y. Han and L.-J. Li, Nano Energy, 2015, 15, 634–641 CrossRef CAS.
- L. Ai, Z. Niu and J. Jiang, Electrochim. Acta, 2017, 242, 355–363 CrossRef CAS.
- H. Liang, A. N. Gandi, C. Xia, M. N. Hedhili, D. H. Anjum, U. Schwingenschlögl and H. N. Alshareef, ACS Energy Lett., 2017, 2, 1035–1042 CrossRef.
- H. Liang, C. Xia, Q. Jiang, A. N. Gandi, U. Schwingenschlögl and H. N. Alshareef, Nano Energy, 2017, 35, 331–340 CrossRef CAS.
- H. Sun, X. Xu, Z. Yan, X. Chen, F. Cheng, P. S. Weiss and J. Chen, Chem. Mater., 2017, 29, 8539–8547 CrossRef CAS.
- K. Liang, L. Guo, K. Marcus, S. Zhang, Z. Yang, D. E. Perea, L. Zhou, Y. Du and Y. Yang, ACS Catal., 2017, 7, 8406–8412 CrossRef CAS.
- F. Xie, H. Wu, J. Mou, D. Lin, C. Xu, C. Wu and X. Sun, J. Catal., 2017, 356, 165–172 CrossRef CAS.
- M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jin, J. Am. Chem. Soc., 2014, 136, 10053–10061 CrossRef CAS PubMed.
- X. Zhang, Y. Han, L. Huang and S. Dong, ChemSusChem, 2016, 9, 3049–3053 CrossRef CAS.
- Y. Wang, B. Zhang, W. Pan, H. Ma and J. Zhang, ChemSusChem, 2017, 10, 4170–4177 CrossRef CAS.
- X. L. Wang, Y. J. Tang, W. Huang, C. H. Liu, L. Z. Dong, S. L. Li and Y. Q. Lan, ChemSusChem, 2017, 10, 2402–2407 CrossRef CAS.
- J. Wan, J. Wu, X. Gao, T. Li, Z. Hu, H. Yu and L. Huang, Adv. Funct. Mater., 2017, 27, 1703933 CrossRef.
- A. Irshad and N. Munichandraiah, ACS Appl. Mater. Interfaces, 2017, 9, 19746–19755 CrossRef CAS PubMed.
- T. Liu, Q. Liu, A. M. Asiri, Y. Luo and X. Sun, Chem. Commun., 2015, 51, 16683–16686 RSC.
- N. Jiang, B. You, M. Sheng and Y. Sun, ChemCatChem, 2016, 8, 106–112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00261d |
| This journal is © The Royal Society of Chemistry 2019 |