
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGalactose:PEGamine coated gold nanoparticles adhere to filopodia and cause extrinsic apoptosis†
Konstantina
Tzelepi
a,
Cristina
Espinosa Garcia
b,
Phil
Williams
b and
Jon
Golding
 *a
*a
aSchool of Life, Health and Chemical Sciences, The Open University, Walton Hall, Milton Keynes, MK7 6AA, UK. E-mail: Jon.Golding@open.ac.uk; Tel: +44 (0)1908 653748
bMidatech Pharma, 65 Innovation Drive, Milton Park, Abingdon, OX14 4RQ, UK
First published on 12th November 2018
Abstract
Ultra-small gold nanoparticles, surface functionalised with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio of a thiolated α-galactose derivative and a thiolated hexaethylene glycol amine, are toxic to HSC-3 oral squamous carcinoma cells. Differences in nanoparticle toxicity were found to be related to the synthesis duration, with 1 h reaction nanoparticles being less toxic than 5 h reaction nanoparticles. The ligand density decreased with longer reaction time, although the size, charge and ligand ratio remained similar. The concentration of sodium borohydride in the reaction decreased logarithmically over 5 h but remained within a concentration range sufficient to desorb weakly bound ligands, possibly explaining the observed gradual decrease in ligand density. Nanoparticle toxicity was abrogated by inhibition of either caspase 3/7 or caspase 8, but not by inhibition of caspase 9, consistent with extrinsic apoptosis. Electron microscopic analysis of cellular uptake demonstrated predominantly cytoplasmic localization. However, when energy-dependent transport was inhibited, by lowering the temperature to 4 °C, a remarkable adhesion of nanoparticles to filopodia was observed. Inhibition of filopodial assembly with a fascin inhibitor prevented nanoparticle adhesion to HSC-3 cells at 4 °C, while fascin inhibition at 37 °C resulted in less cytoplasmic uptake. More adhesion to HSC-3 filopodia was seen with the higher toxicity 5 h reaction time nanoparticles than with the 1 h nanoparticles. By including two further cell types (HaCaT keratinocytes and hCMEC/D3 endothelial cells), a pattern of increasing toxicity with filopodial binding of 5 h reaction nanoparticles became apparent.
:50 ratio of a thiolated α-galactose derivative and a thiolated hexaethylene glycol amine, are toxic to HSC-3 oral squamous carcinoma cells. Differences in nanoparticle toxicity were found to be related to the synthesis duration, with 1 h reaction nanoparticles being less toxic than 5 h reaction nanoparticles. The ligand density decreased with longer reaction time, although the size, charge and ligand ratio remained similar. The concentration of sodium borohydride in the reaction decreased logarithmically over 5 h but remained within a concentration range sufficient to desorb weakly bound ligands, possibly explaining the observed gradual decrease in ligand density. Nanoparticle toxicity was abrogated by inhibition of either caspase 3/7 or caspase 8, but not by inhibition of caspase 9, consistent with extrinsic apoptosis. Electron microscopic analysis of cellular uptake demonstrated predominantly cytoplasmic localization. However, when energy-dependent transport was inhibited, by lowering the temperature to 4 °C, a remarkable adhesion of nanoparticles to filopodia was observed. Inhibition of filopodial assembly with a fascin inhibitor prevented nanoparticle adhesion to HSC-3 cells at 4 °C, while fascin inhibition at 37 °C resulted in less cytoplasmic uptake. More adhesion to HSC-3 filopodia was seen with the higher toxicity 5 h reaction time nanoparticles than with the 1 h nanoparticles. By including two further cell types (HaCaT keratinocytes and hCMEC/D3 endothelial cells), a pattern of increasing toxicity with filopodial binding of 5 h reaction nanoparticles became apparent.
Introduction
Gold nanoparticles (AuNPs) hold enormous promise for delivering a variety of cargoes into cells. However, the details of how different AuNPs interact with and enter cells are still being elucidated.1Moreover, some AuNPs exhibit cytotoxicity that does not always correlate directly with AuNP uptake.2–4 This cytotoxicity can be useful when targeting cancer cells, but disastrous when targeting delicate or diseased cells to improve their survival or function. The physico-chemical parameters that can affect AuNP uptake and toxicity (without any targeting peptide or antibody ligands) include: nanoparticle size,5 charge6 and the nature and density of any coating polymers7–10 (reviewed by Yah3). Exactly how nanoparticles damage cells remains unclear, but can involve changes to biochemical stress signalling pathways11 and damage to organelles such as mitochondria, lysosomes, and DNA within the nucleus.12
We have previously shown that AuNPs coated with a 50:50 ratio of α-galactose:PEGamine ligands are efficiently taken up into kidney and brain endothelial cells, predominantly by energy-dependent mechanisms.13 These AuNPs show selective toxicity toward HSC-3 skin cancer cells, compared to a normal keratinocyte cell line (HaCaT) and this toxicity was caspase 3/7-dependent and could be prevented by antioxidants.2
In order to examine the interaction and toxicity of α-galactose:PEGamine AuNPs with HSC-3 cancer cells in more detail, we analysed AuNP distribution on the cell surface, and within the cytoplasm and nucleus. Two types of AuNPs were tested, which differed only in their synthesis time. One was reacted with sodium borohydride for 1 h (short synthesis) and the other was reacted for 5 h (long synthesis). We find that the shorter synthesis time AuNPs show less binding to filopodia and are less toxic.
Materials and methods
Nanoparticle synthesis
2-Thioethyl α-D-galactopyranoside as the disulphide dimer (α-galactose) and 1-thiohexaethylene glycol-17-ammonium acetate (PEG-amine) (both from GalChimia, Spain) were mixed together in a 50:50 molar ratio with HAuCl4 in Milli-Q water with a 3-fold molar excess of total ligands to gold and adjusted to pH 12 with 2 M NaOH. The mixture was stirred constantly and reduced by adding NaBH4 to 90 mM and samples removed after either 1 h or 5 h. AuNPs were then purified from unreacted ligands and reactants via five rounds of spin washing with excess Milli-Q water using an Amicon 10 kDa ultrafiltration device. AuNPs were resuspended in water to 2 mg ml−1 [Au] and stored in amber vials at 4 °C. Note that all AuNP concentrations stated in this work are based on gold content.
Sodium borohydride assay
The hydrolytic loss of NaBH4 during the AuNP synthesis reaction was monitored by the 2,4,6-trinitrobenzenesulfonic acid (TNBS) assay.14 Milli-Q water was adjusted to pH 12 with 0.2 M NaOH. To a 10 ml portion of this, NaBH4 was added to achieve a final concentration of 90 mM. To a 5 ml portion of the resulting 90 mM NaBH4 solution, 15 mg HAuCl4 and 39 mg 5,5-dithio-bis-(2-nitrobenzoic acid) were simultaneously added, corresponding to the molarities of gold and ligands used in a standard AuNP synthesis reaction (note that we had to replace the α-galactose and PEG-amine disulphide ligands with another disulphide, since TNBS reacts with primary amines). Twenty microlitre aliquots of this reaction mixture were removed at intervals and added to 180 μl of pH 12 water in 96-well plastic plates. Finally, 20 μl aliquots of the diluted samples were added to 200 μl aliquots of freshly prepared 2 mM TNBS in pH 12 water. After 15 min, the absorbance of the TNBS solution was measured at 470 nm using a FLUOstar Optima plate reader (BMG Labtech) and calibrated using a doubling dilution series of the 90 mM NaBH4 solution, treated in an identical way to the AuNP reaction.Nanoparticle physico-chemical characterization
000× magnification. Core sizes were calculated from TEM images using the automated particle analysis feature of ImageJ software and are reported as the mean and median values.
000×g for 1 min to remove any insoluble material and analysed by 1H-NMR at 500 MHz (Avance III HD, Bruker), using MestReNova software. The defining protons for the α-galactose and PEGamine ligands were identified to resonate at 4.95 ppm and 2.75 ppm, respectively. These correspond to the single anomeric proton of α-galactose (NMR doublet) and the two CH2 protons proximal to the terminal NH2 group in the PEG-amine linker (NMR triplet).
:gold (S:Au) ratio is based on the assumption that each ligand on the AuNP surface carries a single S atom, while Au atoms constitute the AuNP core. The geometry of AuNPs is assumed to be spherical.
Cell culture
HSC-3 squamous carcinoma cells were purchased from the American Type Culture Collection. HaCaT cells were a gift from Erik Walbeem, Erasmus Medical Centre, Rotterdam. Cells were cultured in low glucose (1 g l−1) DMEM media (Gibco) supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin/streptomycin in a humidified incubator at 37 °C with 5% CO2. Cells were grown in T-75 culture flasks to about 80% confluency before passage.Clonogenic assay
Cells were seeded at 300 cells per well in 24-well plates and allowed to adhere overnight. The next day, cells were exposed to AuNPs for 3 h and then washed in fresh medium. We previously demonstrated that 3 h incubation was optimal for clonogenic assay and uptake studies using these types of sugar:PEGamine AuNPs.2 Cells were left to grow for 6 days to form colonies and were then stained and fixed with 2% methylene blue in 50% ethanol. Experiments were repeated three times, each with technical triplicates (total n = 9). Images of stained plates were captured using a G:Box Chemi XX6 gel documentation system with GeneSys v1.4.6 software (Syngene). Colonies containing >50 cells were quantified using GeneTools v.4.03 software (Syngene) and expressed as percent of controls. Automated counts were verified by eye. IC50 values were estimated from logarithmic plots of AuNP concentration versus percent of control colonies.Cellular uptake and distribution
Cellular uptake was quantified by ICP-MS and TEM.000 cells per well overnight in 24-well plates. At this density, most cells were isolated from their neighbours. Cells were then incubated with AuNPs for 3 h at a final concentration of 10 μg ml−1, in triplicate. Cells were washed 3 times in medium, trypsinised and spin washed twice at 2000 rpm in 10 ml PBS. The number of cells in each sample was counted using a haemocytometer prior to the final spin and the cell pellet was dissolved in 1 ml 2% nitric acid for 2 days. Samples were analysed using a Perkin-Elmer NexION 300x ICP-MS with NexION software version 1.4. The gold amount was calculated against a standard curve of gold chloride plus washed cells. The amount of gold per cell was calculated for each sample by dividing the measured gold amount by the cell number.
000 cells per well were seeded onto 12-well transwell inserts overnight and then incubated with AuNPs for 3 h at a concentration of 10 μg ml−1. Experiments with HSC-3 cells were repeated three times, each with technical duplicates (total n = 6). Experiments with HaCaT cells were technical triplicates. Cells were fixed and AuNPs were silver enhanced for 45 min at room temperature (R-Gent, Aurion, Netherlands) and processed for imaging according to Gromnicova.15 Ultrathin sections of cells were visualized on a JEOL model 1010 EM instrument operated at an accelerating voltage of 80 kV at ×15000 and ×50000 magnifications. Only isolated cells were counted. It should be noted that silver enhancement increases the size of the nanoparticles, making them easier to detect in cell sections.
Nanoparticle TEM counts on cells
A systematic sampling method was used to evaluate nanoparticle counts in cells. This method was based on acquiring 25 TEM images at the same magnification and same settings across every sample at regular intervals, which included every fifth field of view of a cell. Each image was loaded into ImageJ software (NIH), where the visible areas of the nucleus and cytoplasm, and the length of the cell surface were measured, and the numbers of nanoparticles in each of these three regions were manually counted. Data are expressed as nanoparticles per micron2 for nuclear and cytoplasmic nanoparticle counts, and as nanoparticles per micron for cell surface nanoparticle counts.Fascin inhibition
The fascin inhibitor fascin-G2 (Xcessbio, M60269-2s) was used to inhibit filopodia formation. Cells were incubated with 50 μM fascin-G2 for 2 h at 37 °C. Then cells were incubated with 10 μg ml−1 AuNPs in 50 μM fascin-G2 for a further 3 h, either at 37 °C or at 4 °C. Cells were then washed, fixed and prepared for TEM analysis.Caspase inhibition
Cells were set up for clonogenic assay as described. Cells were pre-incubated with either 70 nM caspase 8 inhibitor I (Merck, 218773) or 50 nM caspase 9 inhibitor II (Merck, 218776) or caspase 3/7 inhibitor (Abcam, ab120382) for 1 h prior to addition of 10 μg ml−1 AuNPs and incubation for 3 h. 10 μM Antimycin A (Abcam, ab141904) was used as a positive control for apoptosis and 50 ng ml−1 Apo2 ligand/TRAIL as a positive control for inducing extrinsic apoptosis. Clonogenic assays were analysed after 6 days, as described. Experiments were repeated three times, each with technical triplicates (total n = 9).Statistical analyses
Data were analysed using GraphPad 6.0 Prism software. For cell work, multiple comparisons between many groups were performed by one-way ANOVA with Tukey's multiple comparison post-test; while comparison of the mean values between two groups was made by multiple two-tailed t-tests with a Sidak–Bonferroni post-test. Comparison of S:Au ratios, O:Au ratios and C:Au ratios were made using paired two-tailed t-tests.
Results and discussion
Physico-chemical and solution stability characterisation
TEM analysis revealed the short synthesis (1 h) and long synthesis (5 h) AuNPs to be spherical with mean core diameters of 1.6 ± 0.8 nm and 1.5 ± 0.9 nm, respectively (mean ± SD; ESI Fig. 1†). Consistent with this, the UV-vis spectra revealed no plasmon band, indicating a core diameter smaller than 2 nm (ref. 16) (ESI Fig. 2A†). DLS analysis showed a hydrodynamic diameter of 4.6 ± 0.9 nm for short synthesis AuNPs and 3.7 ± 1.0 nm for long synthesis AuNPs (mean ± SD; ESI Fig. 2B†). The zeta potential of the AuNPs at pH 7 was positive at 43.8 ± 16.4 mV for short synthesis AuNPs (with some split peaks) and 41.8 ± 10.6 mV for long synthesis AuNPs (mean ± SD; ESI Fig. 2C†). FPLC revealed no tendency to aggregate in PBS, with a single elution peak at 17.97 ml for short synthesis AuNPs and at 17.99 ml for long synthesis AuNPs (ESI Fig. 2D†).
1H-NMR demonstrated an α-galactose:PEGamine ligand ratio of 53:47 for short synthesis AuNPs and 51:49 for long synthesis AuNPs (ESI Fig. 3†).
SEM-EDS analysis demonstrated a significant ∼15% decrease in the S:Au ratio from 0.1013 ± 0.0011 for short synthesis AuNPs to 0.0860 ± 0.0018 for long synthesis AuNPs (mean ± SD, P = 0.0001, n = 5 areas analysed per sample; ESI Fig. 4†), consistent with a loss of sulphur-bearing ligands. The ratios of O:Au and C:Au in these analysed regions also significantly decreased with increasing AuNP synthesis time, consistent with a loss of ligands (O:Au 0.1720 ± 0.0035 for short synthesis AuNPs to 0.1414 ± 0.0025 for long synthesis AuNPs; mean ± SD, P = 0.0001, n = 5. C:Au 0.3365 ± 0.0091 for short synthesis AuNPs to 0.2819 ± 0.0114 for long synthesis AuNPs; mean ± SD, P = 0.0006, n = 5. ESI Fig. 4†).
HPLC analysis similarly demonstrated a loss of ligands from the AuNPs with increasing reaction time (ESI Fig. 5†). Thus, short synthesis AuNPs had 40.8 ligands per 100 Au atoms, while long synthesis AuNPs had 34.8 ligands per 100 Au atoms (a decrease of ∼15%, consistent with SEM-EDS). A previous study has determined that 1.52 nm core diameter AuNPs contain an average of 116 Au atoms.17 This is within the size range of our AuNPs, as determined by TEM. Therefore, the HPLC values of ligands per 100 Au atoms approximate the actual number of ligands per AuNP. Interestingly, our data are similar to those of Jadzinsky et al.,18 in which AuNPs comprising 102 gold atoms were found to be surrounded by 44 thiol (p-mercaptobenzoic acid) ligands.
In agreement with our 1H-NMR data, the HPLC data demonstrated that the α-galactose:PEGamine ligand ratio remained stable during AuNP synthesis (53:47 for short synthesis AuNPs and 52:48 for long synthesis AuNPs; ESI Fig. 5†).
How are ligands being removed from AuNPs during synthesis? Above a concentration of 24 mM, NaBH4 has been shown to strip all ligands from readily formed >10 nm AuNPs within 10 min, while at lower NaBH4 concentrations, desorption becomes less effective (<30% desorption using 8 mM NaBH4 and no desorption using 0.5 mM NaBH4).19 No similar study has examined the kinetics of ligand desorption during AuNP formation. Two ways in which NaBH4 could be quenched in the reaction are via hydrolysis or reaction with disulphide bonds. The former possibility is unlikely, since although NaBH4 is hydrolysed within seconds at low pH, it is stable for days at high pH and our reaction pH remains alkaline, falling from pH 12 to pH 11.5 in 5 h. The second possibility is that a portion of the NaBH4 is lost as it reduces the gold salt and the disulphide bonds of the ligands. We therefore measured the NaBH4 concentration during AuNP synthesis and found that it decreased logarithmically from 90 mM before adding the reactants to 6 mM after 5 h. Importantly, the NaBH4 concentration fell below 24 mM within 35–40 min (ESI Fig. 6†). However, the same reaction in the absence of disulphides did not demonstrate any dramatic decrease in NaBH4 concentration for 5 h (not shown). Thus, in our usual synthesis reaction, a low concentration of NaBH4 persists in solution for several hours and this could set up a dynamic equilibrium between ligand desorption and resorption that favours a slow overall desorption of the more weakly bound ligands from the AuNPs.
Clonogenic assay
The cytotoxic/cytostatic potential of acute (3 h) exposure of HSC-3 cells to AuNPs was determined by clonogenic assay and showed a dose-dependent reduction in cell colonies. Short synthesis AuNPs had an IC50 of around 8 μg ml−1, while long synthesis AuNPs had an IC50 of around 3.5 μg ml−1 (Fig. 1). | ||
| Fig. 1 Clonogenic assay of short synthesis (1 h) and long synthesis (5 h) AuNPs on HSC-3 cells following 3 h acute exposure. All data are shown as mean ± SEM. ** = P < 0.01, *** = P < 0.001, n = 9. | ||
Cellular uptake and distribution of AuNPs
Total cellular uptake of AuNPs, which includes AuNPs both on and within cells, was determined by ICP-MS on low-density cell cultures (to permit the unimpeded extension of filopodia around each cell). Cells were found to accumulate similar amounts of each type of AuNP within 3 h (4.8 ± 1.8 pg per cell for short synthesis AuNPs and 6.8 ± 2.2 pg per cell for long synthesis AuNPs; two-tailed t-test P = 0.63).To examine the details of AuNP cellular uptake, we used a well-established quantitative TEM method.15,20–22 To discriminate between energy-dependent and passive transport, cell uptake studies were performed at 37 °C or 4 °C, respectively. Following 3 h incubation at 37 °C, AuNPs were found predominantly within the cytoplasm, with no significant differences between the accumulation of short synthesis or long synthesis AuNPs (Fig. 2), consistent with the ICP-MS data. However, when incubated instead at 4 °C, there was a dramatic shift in the distribution of both AuNPs to the cell surface, with significantly more long synthesis AuNPs being associated with the cell surface than short synthesis AuNPs (Fig. 3). At 4 °C AuNPs were not randomly distributed on the cell surface, but instead were preferentially associated with filopodia (88.9% (3385/3808) of long synthesis AuNPs and 82.5% (2254/2732) of short synthesis AuNPs at the cell surface were associated with filopodia; Fig. 3B and C).
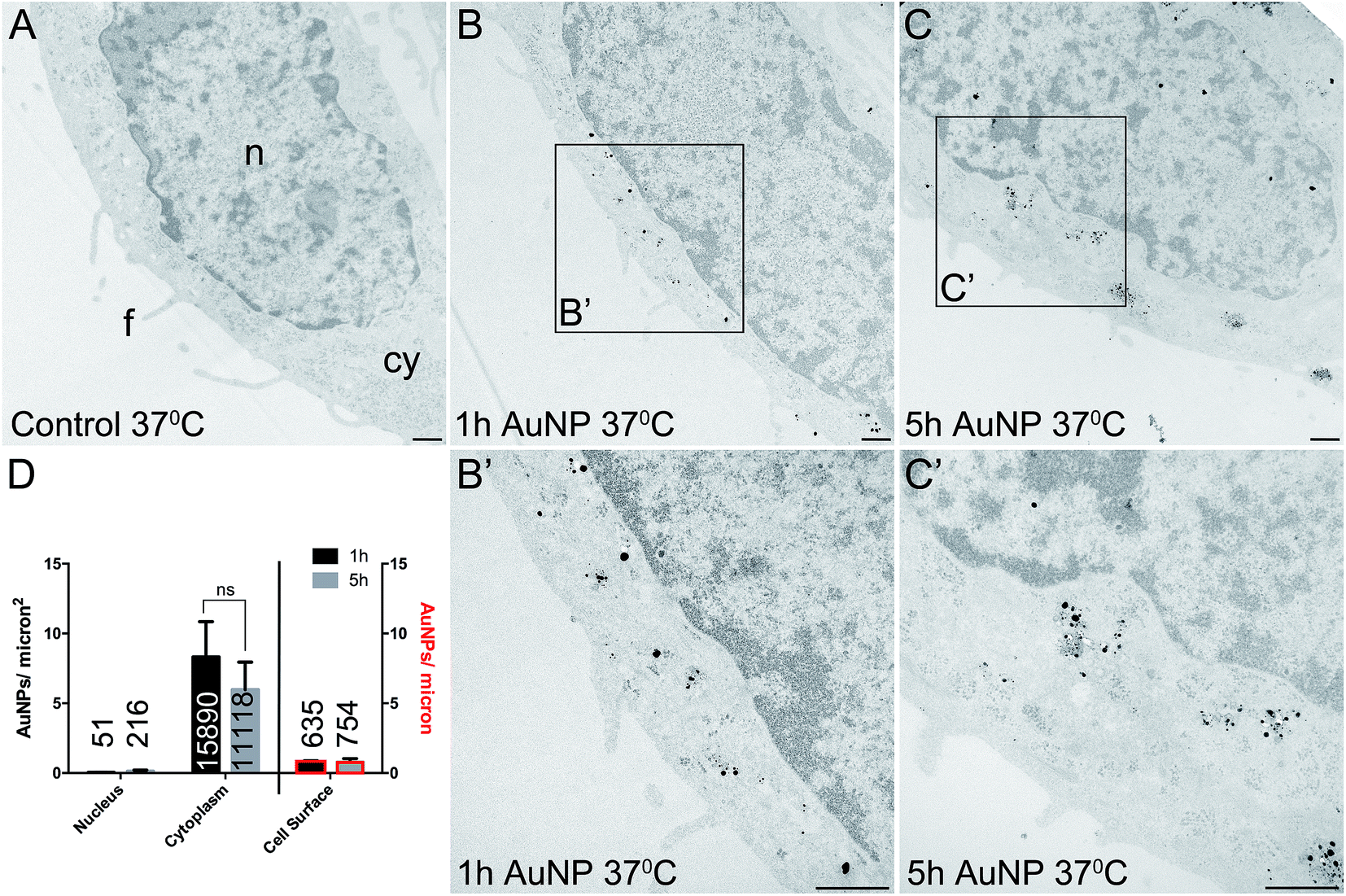 | ||
| Fig. 2 TEM images of silver enhanced HSC-3 cells after acute 3 h exposure to 10 μg ml−1 AuNPs at 37 °C. (A) No AuNP control. (B) Short synthesis (1 h) AuNPs. (C) Long synthesis (5 h) AuNPs. (B′) and (C′) show magnified portions of the boxed regions of (B) and (C), respectively. n = nucleus, cy = cytoplasm, f = filopodia. Scale bars = 500 nm. (D) Quantitation of AuNP counts per micron2 for the nucleus and cytoplasm (left axis) and AuNP counts per micron for the cell surface (right axis, bars with red border). Total numbers of AuNPs counted per category are shown on bars. All data are shown as mean ± SEM, n = 6. | ||
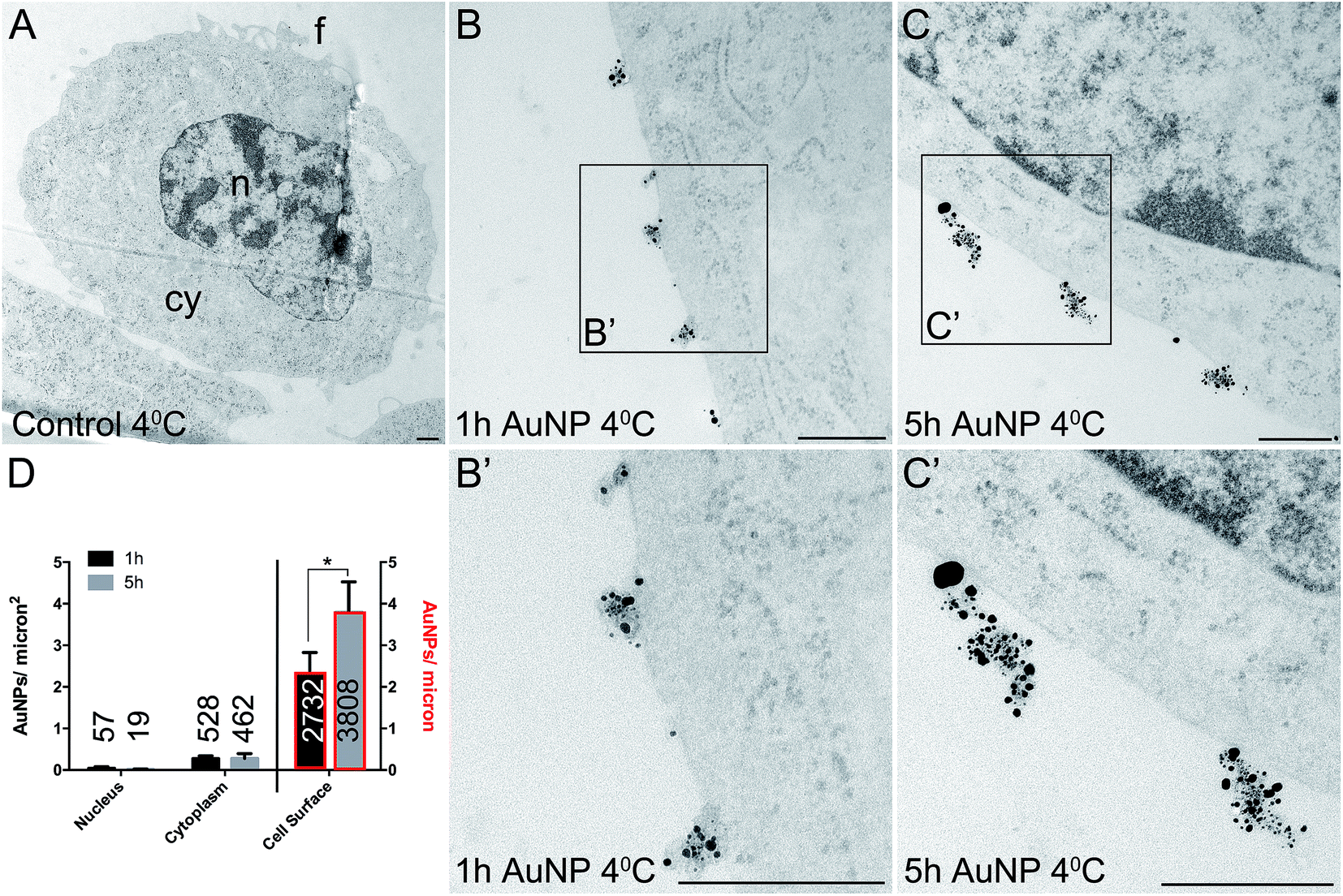 | ||
| Fig. 3 TEM images of silver enhanced HSC-3 cells after acute 3 h exposure to 10 μg ml−1 AuNPs at 4 °C. (A) No AuNP control. (B) Short synthesis (1 h) AuNPs. (C) Long synthesis (5 h) AuNPs. (B′) and (C′) show magnified portions of the boxed regions of (B) and (C), respectively. n = nucleus, cy = cytoplasm, f = filopodia. Scale bars = 500 nm. (D) Quantitation of AuNP counts per micron2 for the nucleus and cytoplasm (left axis) and AuNP counts per micron for the cell surface (right axis, bars with red border). Total numbers of AuNPs counted per category are shown on bars. All data are shown as mean ± SEM. * = P < 0.05, n = 6. | ||
Previous studies have shown that varying the ligand density affects nanoparticle cellular accumulation, although there is no consensus on whether low or high ligand density is optimal.7–10 Moreover, the precise distribution of ligands on the nanoparticle surface may be crucial, since computer simulations indicate that a homogeneous ligand distribution favours cellular uptake, while the presence of ligand-free areas prevents cellular uptake.23 However, in this study, AuNP intracellular accumulation was not significantly affected by ligand density, although binding to the cell surface was better with lower ligand density AuNPs.
To confirm whether AuNPs were selectively binding to filopodia, HSC-3 cells were pre-incubated with fascin-G2, an inhibitor of the actin bundling ability of fascin, that thereby destabilizes filopodia and prevents their formation. Fascin inhibition resulted in 85% fewer filopodia per micron of the cell surface (compare Fig. 4A and D with Fig. 4B, C, E and F).
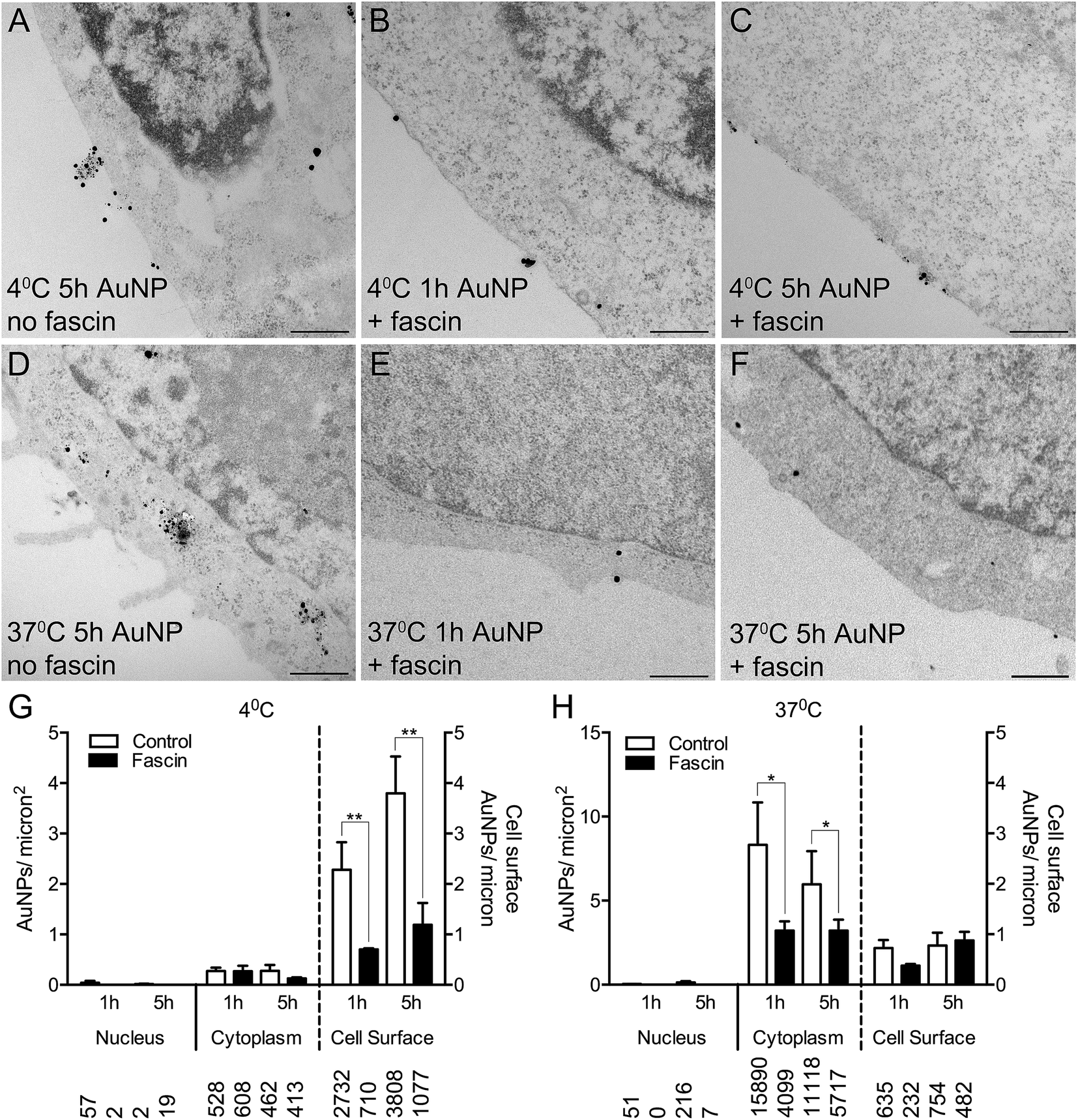 | ||
| Fig. 4 (A–F) TEM images of silver enhanced HSC-3 cells incubated for 3 h with 10 μg ml−1 AuNPs at 4 °C in (A) the absence or (B, C) the presence of a fascin inhibitor or at 37 °C in (D) the absence or (E, F) the presence of a fascin inhibitor. Scale bars = 500 nm. (G, H) Quantitation of AuNP counts per micron2 for the nucleus and cytoplasm (left axis) and AuNP counts per micron for the cell surface (right axis) with or without a fascin inhibitor at either (G) 4 °C or (H) 37 °C. Total numbers of AuNPs counted per category are shown below bars. All data are shown as mean ± SEM. * = P < 0.05, ** = P < 0.01, n = 6. | ||
Following fascin inhibition, cells loaded with short synthesis or long synthesis AuNPs at 4 °C demonstrated a significant three-fold reduction of AuNPs at the cell surface (Fig. 4G), while cells loaded with short synthesis or long synthesis AuNPs at 37 °C demonstrated significantly fewer AuNPs within the cytoplasm (Fig. 4H).
AuNP interactions with other cell lines
In order to determine if there were any obvious trends in the way that long synthesis AuNPs interact with cell membranes, we examined two additional cell types: hCMEC/D3 human endothelial cells and HaCaT human keratinocytes.First, we examined whether incubation at 4 °C resulted in an inhibition of cellular uptake and a redistribution of AuNPs to the cell surface. As shown in Fig. 4, the uptake of 10 μg ml−1 long synthesis AuNPs into HSC-3 cells is inhibited by 95% if the incubation is performed at 4 °C (mean cytoplasmic AuNPs: 5.97 ± 1.97/μm−2 at 37 °C versus 0.28 ± 0.12/μm−2 at 4 °C, Fig. 4G and H). The accumulation of 10 μg ml−1 long synthesis AuNPs into HaCaT cells was similarly inhibited by 94% when the incubation was performed at 4 °C (mean cytoplasmic AuNPs: 7.39 ± 1.02/μm−2 at 37 °C versus 0.43 ± 0.12/μm−2 at 4 °C; ESI Fig. 7A and B†). Finally, under conditions comparable to those used here, we previously reported that the accumulation of 8 μg ml−1 long synthesis AuNPs into hCMEC/D3 cells for 3 h is inhibited by 98% if the incubation is instead performed at 4 °C.13
Next, we focussed on the proportion of long synthesis AuNPs associated with filopodia on the cell surface at 4 °C. As mentioned above, HSC-3 cells exhibit 88.9% association of AuNPs with filopodia at 4 °C (3385 AuNPs on filopodia of 3808 total AuNPs on the cell surface). In contrast, HaCaT cells exhibit 52.5% association of AuNPs with filopodia at 4 °C (519 AuNPs on filopodia of 988 total AuNPs on the cell surface, ESI Fig. 7B†). A novel re-analysis of our existing TEM data for hCMEC/D3 cells at 4 °C (ref. 13) showed 11.9% association of AuNPs with filopodia (78 AuNPs on filopodia of 657 total AuNPs on the cell surface, ESI Fig. 7C and D†).
From this limited dataset, it is possible to see a trend whereby the proportion of AuNPs associated with filopodia on the cell surface correlates with toxicity. Thus, HSC-3: 88.9% filopodia binding, IC50 3.5 μg ml−1; HaCaT: 52.5% filopodia binding, IC50 10 μg ml−1 (ESI Fig. 7E†); and hCMEC/D3: 11.9% filopodia binding, IC50 > 75 μg ml−1 (note that hCMEC/D3 does not grow clonogenically). However, AuNPs were non-toxic at 75 μg ml−1 after continuous exposure for 48 h (ref. 13). These data suggest that filopodial binding may stimulate cell death receptor(s), and this possibility is further explored in the next section using HSC-3 cells.
This is the first unequivocal report of AuNPs interacting specifically with filopodia. On macrophages, bacterial particles are captured by the shaft of the filopodia and then directed towards the cell surface into phagocytic cups.24 Similarly, exosome nanovesicles attach to filopodia and become directed to endocytic hotspots at their base.25 AuNPs were only seen on filopodia at 4 °C when energy-dependent transport was inhibited; this suggests that at 37 °C they are normally rapidly cleared and/or endocytosed. However, we have no evidence that the extent of filopodial binding is a predictor of subsequent uptake: they could be independent.
We cannot yet definitively state whether AuNPs initially bind to pre-existing filopodia or whether AuNP binding to the cell surface locally stimulates filopodia formation. A study involving silica nanoparticles found that the nanoparticle aspect ratio affected filopodia formation and pinocytosis.26
One possibility is that these AuNPs are capable of binding to receptors expressed preferentially on filopodia, possibly in an analogous manner to the selective cancer cell targeting of some oncolytic viruses.27
Another possibility is that electrostatic interactions may occur between the positively charged AuNPs and the negatively charged cell membrane.28,29 Selectivity could arise due to variations in membrane potential, which can differ by up to 90 mV between different cell types.28 It is currently unknown whether the membrane potential of filopodia is more negative than that of the other regions of the cell membrane. However, the curvature of the filopodial shaft is sculpted from within by negative charges on the convex surface of small, ‘banana-shaped’ intracellular proteins, which repel and bend the adjacent negatively charged cell membrane.30 Speculatively, this may locally increase the negative charge on the filopodia surface. Consistent with this idea, positively charged 1.4 nm “Nanogold” AuNPs appear to bind to filopodia more strongly than the rest of the cell membrane on fixed COS7 cells.31 Although the magnification used in that SEM study was unable to resolve individual AuNPs. The exact nature of the ligand shell on nanogold is proprietary, but the manufacturer states there are 6 primary amine groups per AuNP.32 This differs from the ligand structure of our AuNPs, disfavouring (but not disproving) the idea of a common filopodial receptor for both types of AuNPs and instead favouring the idea of an electrostatic contribution to AuNP binding to cell surfaces, with the exact distribution of AuNPs on each cell surface being determined by charge heterogeneities.
Independent of AuNP charge, the energy-independent insertion of AuNPs coated in amphiphilic organic ligands into the cell membrane lipid bilayer occurs preferentially in regions of high cell membrane curvature33 and is dramatically improved when the core size is below 3 nm.34 Although these studies did not specifically examine filopodia, the tips and bases of filopodial protrusions and the small radius of the filopodial shaft all constitute highly curved regions of the cell membrane and could therefore act as preferential adhesion sites for nanoparticles capable of this kind of membrane insertion via ‘snorkeling’.35 In the present study, the hexaethylene glycol portion of the PEGamine ligand may be sufficiently amphiphilic to allow such an energy-independent interaction with the cell membrane. Further detailed studies will be required to compare the filopodial binding and cellular uptake of AuNPs using different ratios of α-galactose:PEGamine. Our previous study2 demonstrated differences in uptake (assessed by ICP-MS) into HSC-3 and HaCaT cells as the α-galactose:PEGamine ratio was varied. The highest uptake being seen with 50:50 ratio AuNPs (50:50 > 60:40 ≈ 40:60 ≈ 0:100 > 100:0). However, we do not yet have any TEM data regarding the filopodial interactions of these different α-galactose:PEGamine ratio AuNPs under energy-independent conditions, such as incubation at 4 °C, and we have found no similar TEM study in the literature.
AuNP toxicity is due to extrinsic apoptosis
These studies were done using the more toxic, long synthesis AuNPs. Co-incubation of HSC-3 cells with AuNPs and a caspase 3/7 inhibitor prevented AuNP-induced cell death in clonogenic assays, confirming apoptosis (Fig. 5A).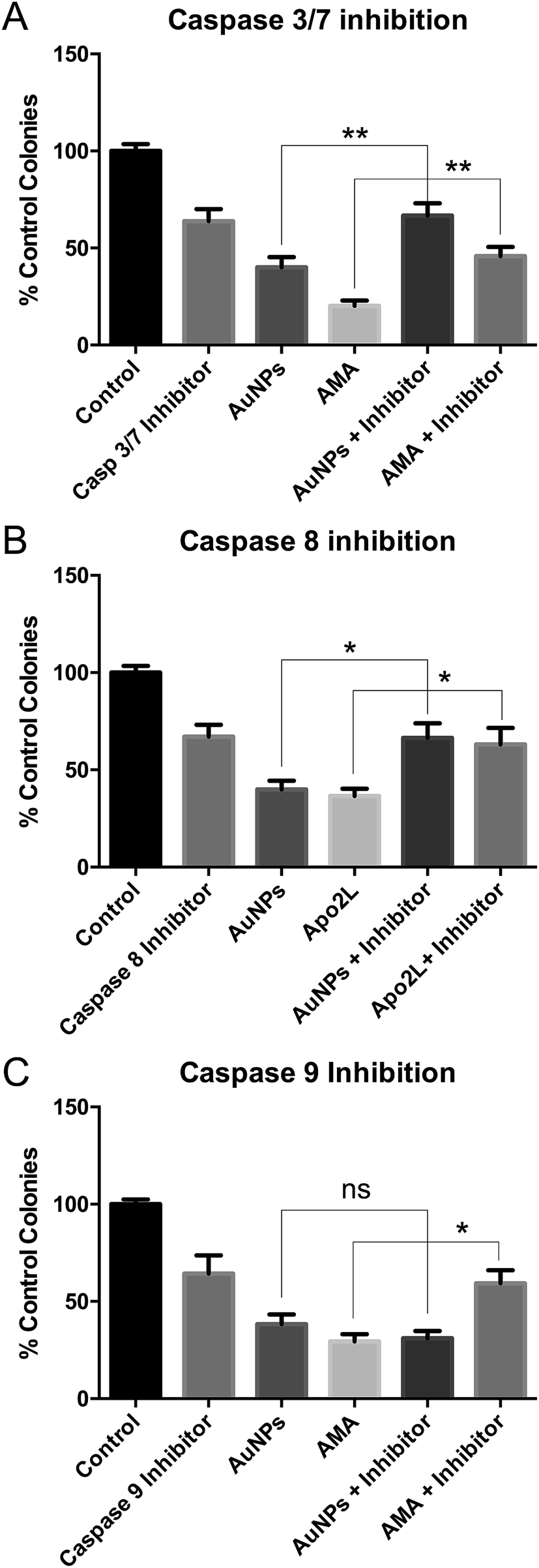 | ||
| Fig. 5 Clonogenic assays of HSC-3 cells treated with long synthesis time AuNPs or apoptosis-inducing drugs and inhibitors of (A) caspase 3/7 or (B) caspase 8 or (C) caspase 9. All are data shown as mean ± SEM. ns = not significant. * = P < 0.05, ** = P < 0.01, n = 9. | ||
To discriminate between classical caspase-dependent extrinsic and intrinsic apoptosis pathways, cells were co-incubated with AuNPs and either a caspase 8 inhibitor (apical caspase in the extrinsic pathway) (Fig. 5B) or a caspase 9 inhibitor (apical caspase in the intrinsic pathway) (Fig. 5C).36 Caspase 9 inhibition had no effect on AuNP toxicity, while caspase 8 inhibition prevented AuNP toxicity, suggesting extrinsic apoptosis. In each case, cell death due to positive control apoptosis-inducing agents was prevented by the caspase inhibitors (Fig. 5). Note that each caspase inhibitor alone is slightly toxic and rescue of AuNP or positive control drug toxicity brings the cell viability back to the inhibitor-only value.
Nanoparticle toxicity,37 specifically AuNP toxicity,38 is usually reported to occur via mitochondrial37 or endoplasmic reticulum stress,39 leading to cleavage of caspase 9.38,40 By contrast, nanoparticle-induced extrinsic apoptosis, due to caspase 8 cleavage, is rarely reported. Cleavage of caspase 8 and accumulation of FADD were seen in JB6 mouse skin cells within 1 h of adding tungsten carbide–cobalt nanoparticles,41 while cleavage of caspase 8 was seen in human keratinocyte HaCaT cells within 6 h of adding titanium dioxide nanoparticles.42 However, to our knowledge, extrinsic apoptosis has not been reported before with AuNPs. We do not claim that there is anything particularly unusual about our AuNPs that leads to extrinsic apoptosis, simply that most papers demonstrating AuNP-induced apoptosis only look at caspase 3 activation (reviewed in ref. 38).
It is tempting to link our observations of increased filopodial binding of AuNPs with extrinsic apoptosis. However, our data do not yet allow us to conclude that excessive filopodial binding causes apoptosis. Future work will determine whether these, currently independent, observations are linked.
For instance, it is not known whether any of the death receptors (TNFr, TRAMP, Fas, or DRs 4 to 6),43 which locally cleave and activate caspase 8, are concentrated within filopodia and could thereby become activated by excessive or prolonged AuNP binding. It is nonetheless intriguing that increased filopodial density is often reported in cancer cells,44 being important for tumour progression, migration and dissemination.44,45 Moreover, caspase 8 is associated with cell migration in cancer cells,46 being specifically associated with focal adhesion complexes,47 raising the possibility that caspase 8 may also be concentrated within filopodia.
Once inside cells, AuNPs can damage or impair the function of various organelles (reviewed in ref. 48), including lysosomes,49 mitochondria50 and the nucleus.12 AuNPs can also activate metal exposure and oxidative stress signalling pathways,11 resulting in autophagy.51
AuNP-induced cell death is therefore likely to be multifactorial, with distinct temporal sequences of events converging via several pathways to kill the cell.
Conclusions
We have identified that 50:50 α-galactose:PEGamine AuNPs accumulate to differing extents on the filopodia of various cell types when energy-dependent uptake is prevented. These data provide a fascinating insight into how AuNPs initially interact with cell membranes and how the extent of filopodial interaction may be a deciding factor in the initiation of extrinsic death pathways. It will now be important to survey a large number of normal and diseased cell types to determine whether there is a correlation between how AuNPs interact with cell membranes and resulting cell behaviours, not limited to cell death.
Nanoparticle synthesis time is rarely reported in the literature. Our observation that longer synthesis time AuNPs are more adherent to HSC-3 cell membranes and are more toxic than short synthesis AuNPs underscores that future publications would be wise to routinely quote synthesis time.
From a practical perspective, our data suggest that by varying the synthesis time, AuNPs could be engineered for either enhanced or reduced toxicity, depending on the desired application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Konstantina Tzelepi was funded by a joint grant from Midatech Pharma and the Open University. We thank Dr Radka Gromnicova and Gordon Imlach at the Open University for preparing sections for TEM and performing SEM-EDS, respectively. We additionally thank Dr Radka Gromnicova and Professor David Male at the Open University for sharing unpublished TEM images of hCMEC/D3 cells.References
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- S. Grellet, K. Tzelepi, M. Roskamp, P. Williams, A. Sharif, R. Slade-Carter, P. Goldie, N. Whilde, M. A. Smialek, N. J. Mason and J. P. Golding, PLoS One, 2017, 12, e0181103 CrossRef PubMed.
- C. S. Yah, Biomed. Res., 2013, 24, 400–413 CAS.
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- T. Lund, M. F. Callaghan, P. Williams, M. Turmaine, C. Bachmann, T. Rademacher, I. M. Roitt and R. Bayford, Biomaterials, 2011, 32, 9776–9784 CrossRef CAS PubMed.
- H. Liu, T. L. Doane, Y. Cheng, F. Lu, S. Srinivasan, J.-J. Zhu and C. Burda, Part. Part. Syst. Charact., 2015, 32, 197–204 CrossRef CAS.
- D. R. Elias, A. Poloukhtine, V. Popik and A. Tsourkas, Nanomedicine, 2013, 9, 194–201 CrossRef CAS PubMed.
- D. H. M. Dam, R. C. Lee and T. W. Odom, Nano Lett., 2014, 14, 2843–2848 CrossRef CAS PubMed.
- E. Bajak, M. Fabbri, J. Ponti, S. Gioria, I. Ojea-Jiménez, A. Collotta, V. Mariani, D. Gilliland, F. Rossi and L. Gribaldo, Toxicol. Lett., 2015, 233, 187–199 CrossRef CAS PubMed.
- M. Tsoli, H. Kuhn, W. Brandau, H. Esche and G. Schmid, Small, 2005, 1, 841–844 CrossRef CAS PubMed.
- R. Gromnicova, M. Kaya, I. A. Romero, P. Williams, S. Satchell, B. Sharrack and D. Male, PLoS One, 2016, 11, e0161610 CrossRef PubMed.
- B. Elamin and G. E. Means, Anal. Chim. Acta, 1979, 107, 405–409 CrossRef CAS.
- R. Gromnicova, H. A. Davies, P. Sreekanthreddy, I. A. Romero, T. Lund, I. M. Roitt, J. B. Phillips and D. K. Male, PLoS One, 2013, 8, e81043 CrossRef PubMed.
- G. L. Nealon, B. Donnio, R. Greget, J.-P. Kappler, E. Terazzi and J.-L. Gallani, Nanoscale, 2012, 4, 5244 RSC.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- S. M. Ansar, F. S. Ameer, W. Hu, S. Zou, C. U. Pittman and D. Zhang, Nano Lett., 2013, 13, 1226–1229 CrossRef CAS PubMed.
- A. Elsaesser, C. A. Barnes, G. McKerr, A. Salvati, I. Lynch, K. A. Dawson and C. V. Howard, Nanomedicine, 2011, 6, 1189–1198 CrossRef CAS PubMed.
- A. P. Brown, R. M. D. Brydson and N. S. Hondow, J. Phys.: Conf. Ser., 2014, 522, 012058 CrossRef.
- B. Rothen-Rutishauser, D. A. Kuhn, Z. Ali, M. Gasser, F. Amin, W. J. Parak, D. Vanhecke, A. Fink, P. Gehr and C. Brandenberger, Nanomedicine, 2014, 9, 607–621 CrossRef CAS PubMed.
- V. Schubertová, F. J. Martinez-Veracoechea and R. Vácha, Soft Matter, 2015, 11, 2726–2730 RSC.
- M. Horsthemke, A. C. Bachg, K. Groll, S. Moyzio, B. Müther, S. A. Hemkemeyer, R. Wedlich-Söldner, M. Sixt, S. Tacke, M. Bähler and P. J. Hanley, J. Biol. Chem., 2017, 292, 7258–7273 CrossRef CAS PubMed.
- W. Heusermann, J. Hean, D. Trojer, E. Steib, S. von Bueren, A. Graff-Meyer, C. Genoud, K. Martin, N. Pizzato, J. Voshol, D. V. Morrissey, S. E. L. Andaloussi, M. J. Wood and N. C. Meisner-Kober, J. Cell Biol., 2016, 213, 173–184 CrossRef CAS PubMed.
- H. Meng, S. Yang, Z. Li, T. Xia, J. Chen, Z. Ji, H. Zhang, X. Wang, S. Lin, C. Huang, Z. H. Zhou, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4434–4447 CrossRef CAS PubMed.
- P. K. Singh, J. Doley, G. R. Kumar, A. P. Sahoo and A. K. Tiwari, Indian J. Med. Res., 2012, 136, 571–584 Search PubMed.
- M. Yang and W. J. Brackenbury, Front. Physiol., 2013, 4, 185 Search PubMed.
- E. C. Cho, J. Xie, P. A. Wurm and Y. Xia, Nano Lett., 2009, 9, 1080–1084 CrossRef CAS PubMed.
- S. H. Lee, F. Kerff, D. Chereau, F. Ferron, A. Klug and R. Dominguez, Structure, 2007, 15, 145–155 CrossRef PubMed.
- H. Nishiyama, K. Teramoto, M. Suga and C. Sato, Microsc. Res. Tech., 2014, 77, 153–160 CrossRef PubMed.
- Positively charged nanogold (product 2022) Nanoprobes.com, http://www.nanoprobes.com/instructions/Inf2022.html, accessed 20 June 2018.
- R. C. Van Lehn, M. Ricci, P. H. J. Silva, P. Andreozzi, J. Reguera, K. Voïtchovsky, F. Stellacci and A. Alexander-Katz, Nat. Commun., 2014, 5, 4482 CrossRef CAS PubMed.
- R. C. Van Lehn, P. U. Atukorale, R. P. Carney, Y.-S. Yang, F. Stellacci, D. J. Irvine and A. Alexander-Katz, Nano Lett., 2013, 13, 4060–4067 CrossRef CAS PubMed.
- A. K. Chamberlain, Y. Lee, S. Kim and J. U. Bowie, J. Mol. Biol., 2004, 339, 471–479 CrossRef CAS PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- D.-D. Ma and W.-X. Yang, Oncotarget, 2016, 7, 40882–40903 Search PubMed.
- H. Sun, J. Jia, C. Jiang and S. Zhai, Int. J. Mol. Sci., 2018, 19, 754 CrossRef PubMed.
- C. Noël, J.-C. Simard and D. Girard, Toxicol. Vitro, 2016, 31, 12–22 CrossRef PubMed.
- N. Morishima, K. Nakanishi, H. Takenouchi, T. Shibata and Y. Yasuhiko, J. Biol. Chem., 2002, 277, 34287–34294 CrossRef CAS PubMed.
- J. Zhao, J. Zhao, L. Bowman, L. Bowman, R. Magaye, R. Magaye, S. S. Leonard, S. S. Leonard, V. Castranova, V. Castranova, M. Ding and M. Ding, Int. J. Oncol., 2013, 42, 1349–1359 CrossRef CAS PubMed.
- C. Wright, A. K. V. Iyer, L. Wang, N. Wu, J. S. Yakisich, Y. Rojanasakul and N. Azad, Drug Chem. Toxicol., 2017, 40, 90–100 CrossRef CAS PubMed.
- H. Walczak, Cold Spring Harbor Perspect. Biol., 2013, 5, a008698 Search PubMed.
- A. Arjonen, R. Kaukonen and J. Ivaska, Cell Adhes. Migr., 2011, 5, 421–430 CrossRef PubMed.
- G. Jacquemet, H. Hamidi and J. Ivaska, Curr. Opin. Cell Biol., 2015, 36, 23–31 CrossRef CAS PubMed.
- R. P. Graf, N. Keller, S. Barbero and D. Stupack, Curr. Mol. Med., 2014, 14, 246–254 CrossRef CAS PubMed.
- S. Barbero, A. Mielgo, V. Torres, T. Teitz, D. J. Shields, D. Mikolon, M. Bogyo, D. Barilà, J. M. Lahti, D. Schlaepfer and D. G. Stupack, Cancer Res., 2009, 69, 3755–3763 CrossRef CAS PubMed.
- M. Kodiha, Y. M. Wang, E. Hutter, D. Maysinger and U. Stochaj, Theranostics, 2015, 5, 357–370 CrossRef PubMed.
- X. Ma, Y. Wu, S. Jin, Y. Tian, X. Zhang, Y. Zhao, L. Yu and X.-J. Liang, ACS Nano, 2011, 5, 8629–8639 CrossRef CAS PubMed.
- Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann, G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076 CrossRef CAS PubMed.
- J. J. Li, D. Hartono, C.-N. Ong, B.-H. Bay and L.-Y. L. Yung, Biomaterials, 2010, 31, 5996–6003 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00270c |
| This journal is © The Royal Society of Chemistry 2019 |