
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous silica-protected silver nanoparticle disinfectant with controlled Ag+ ion release, efficient magnetic separation, and effective antibacterial activity†
Xiaoxin
Wang‡
 abc,
Wuzhu
Sun‡
dc,
Weiyi
Yang
c,
Shuang
Gao
e,
Caixia
Sun
fg and
Qi
Li
*c
abc,
Wuzhu
Sun‡
dc,
Weiyi
Yang
c,
Shuang
Gao
e,
Caixia
Sun
fg and
Qi
Li
*c
aCollege of Physics and Electronics, Dezhou University, Dezhou 253023, PR China
bShandong Key Laboratory of Biophysics, Dezhou University, Dezhou 253023, PR China
cEnvironment Functional Materials Division, Shenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, PR China. E-mail: qili@imr.ac.cn
dSchool of Materials Science and Engineering, Shandong University of Technology, Zibo 255000, PR China
eDivision of Energy and Environment, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, PR China
fKey Laboratory of New Metallic Functional Materials and Advanced Surface Engineering in Universities of Shandong, Qingdao Binhai University, Qingdao 266555, PR China
gSchool of Mechanical and Electronic Engineering, Qindao Binhai University, Qingdao 266555, PR China
First published on 15th November 2018
Abstract
Ag is the most effective metal disinfectant against pathogenic microorganisms and thus, various approaches have been exploited to enhance the dispersity and control the release of Ag+ ions from Ag nanoparticles. In this study, a superparamagnetic Fe3O4@SiO2@Ag@porous SiO2 disinfectant with a double-layer core–shell structure was developed. Its superparamagnetic Fe3O4 nanosphere core ensured its good dispersity in water and allowed its easy magnetic separation after treatment. Its dense SiO2 inner shell protected the Fe3O4 nanosphere core and allowed a good loading of Ag nanoparticles. Its mesoporous SiO2 outer layer effectively protected the Ag nanoparticles from detachment, and its mesoporous channels resulted in lower silver oxidation and dissolution for the controlled release of Ag+ ions. Thus, a highly efficient, silver-based disinfectant was developed, as demonstrated by its effective disinfection of Escherichia coli bacteria with good recycle performance, while the silver concentration in the treated water met the MCL of silver for drinking water.
Introduction
Water pollution is a critical issue, which has caused a serious water crisis worldwide.1,2 Various types of water pollutants exist in the aqueous environment, including inorganics, organics, and microorganisms, which make water treatment a complicated process.3 Among them, microbial contamination has received extensive attention due to the following reasons. First, water is a common medium for pathogenic microorganisms to spread diseases. For instance, Salmonella had been reported to spread through water, endangering human health.4 Second, biofilms are usually formed on industrial water pipes, which block and corrode them, thus affecting normal industrial production. Third, biological toxins released by microorganisms in their metabolic processes add complexity for water treatment. For example, cyanotoxins are secondary metabolites of blue algae, which must be considered when treating blue algae pollution.5 Hence, effective approaches need to be developed to solve microbial water pollution.Various antibacterial materials had been developed, including natural, organic, and inorganic antibacterial materials.6 Chitosan and sorbic acid are typical natural antibacterial agents, which are safe, environmentally benign, and have good disinfection capability.7 However, their long-term storage is a problem, and they cannot be used under high temperature. Organic antibacterial agents have a good sterilization effect, fast sterilization rate, and relatively low cost.8 However, their drawbacks are also prominent because they are toxic and easily cause microbial resistance. Sometimes, they may lead to the formation of carcinogenic disinfection by-products that are harmful to human health.9 Compared with their natural and organic counterparts, inorganic antibacterial materials exhibit a wide antimicrobial spectrum, excellent chemical and thermal stability, nontoxicity, and no microbial resistance.10,11 Among them, silver (including silver ions and silver-based compounds) is considered as the most effective metallic inorganic antibacterial candidate,12 which has long been used in dental resin composites and various consumer products.13 Although the antibacterial mechanism of silver is still debatable, the prevailing view is that ionic silver can strongly interact with the thiol groups of vital enzymes inside microorganisms and inactivate them.14 Subsequently, ionic silver can diffuse back into the environment and inactivate more microorganisms periodically. Thus, silver exhibits strong antibacterial activity for a wide range of microorganisms with long duration and safety.15
With the advantages of low volume, large specific surface area and high activity, silver nanoparticles are considered to be the most effective metal disinfectant against bacteria, viruses and other eukaryotic microorganisms due to their constant release of silver ions through slow oxidation.13,16 Various approaches have been developed for the fabrication of stable and well-dispersed silver nanoparticles, including chemical reduction, reverse micelle, sol–gel, and precipitation.16–18 Furthermore, to avoid their strong aggregation tendency and enhance their effectiveness for potential applications, various types of carriers were investigated for silver nanoparticles, including glass fibre,19 rice husk ash20 and Fe3O4@carbon.21 They were also integrated with multiple types of filters,22 fibre membranes23 and composites24 to improve the water purification efficiency. However, silver nanoparticles impregnated into carriers were prone to detach and could enter the aqueous environment, while colloidal silver nanoparticles showed toxicity toward mammalian cells.25,26 Furthermore, their fast dissolution rate shortened their life-time, and could easily induce higher silver ion concentrations over the WHO recommended maximum contaminant level limit (MCL = 0.05 mg L−1) for drinking water.27
To overcome these drawbacks, research efforts have been focused on protecting the efficient ingredient of silver nanoparticles from detachment to maintain the appropriate silver ion content for water purification. For example, the Ag@Fe2O3–GO nanocomposites synthesized by Gao et al. showed good long-term antibacterial property against both Gram-negative and Gram-positive bacteria due to the lower oxidation rate of Ag nanoparticles in the presence of GO.28 Liong et al. enclosed silver nanoparticles in a mesostructured silica shell, which slowed down the oxidation rate of the silver nanoparticles and displayed long-term antibacterial properties.17 Besides, carbon29 and titania30 shells were also adopted to protect the inner silver nanoparticles. Although the detachment of silver nanoparticles could be suppressed by these protection shells, the silver ion concentration in the treated water was not generally modulated in these studies, while it was difficult to separate these core–shell structure disinfectants from the treated water bodies to avoid potential environmental hazards.
Herein, a double-layer core–shell structure was designed to create a novel silver-based disinfectant, which consisted of a superparamagnetic Fe3O4 nanosphere core, dense SiO2 inner shell, Ag nanoparticles, and mesoporous SiO2 outer shell. In this double-layer core–shell structure, the superparamagnetic Fe3O4 nanoparticle core endowed the disinfectant with good dispersion/redispersion in water when no external magnetic field was applied, which could then be easily separated magnetically from treated water bodies when an external magnetic field was applied to avoid it entering the aqueous environment. The dense SiO2 inner shell could protect the Fe3O4 nanosphere core and provide an easy attachment to silver species for a good loading of disinfectant Ag nanoparticles. The mesoporous SiO2 outer layer could protect the Ag nanoparticles from detachment during the disinfection operation, and the relatively long diffusion route along its mesoporous channels for silver ions could be beneficial for lower silver oxidation and dissolution for the controlled release of silver ions and long-term antibacterial performance. Thus, a highly efficient silver-based disinfectant was developed with good stability, while the silver concentration in the treated water met the WHO recommended maximum contaminant level limit (MCL = 0.05 mg L−1) for drinking water.
Experimental
Chemicals
All chemicals were of analytical grade and used as received without further purification. Ferric chloride hexahydrate, ethylene glycol, disodium ethylenediamine tetraacetate, sodium acetate, sodium citrate tribasic dihydrate, ethanol, ammonium hydroxide aqueous solution (28%), tetraethyl orthosilicate (TEOS), silver nitrate, glucose, stannous chloride and hydrochloric acid (37%) were purchased from Sinopharm Chemical Reagent Corporation (Shanghai, P. R. China). Polyvinylpyrrolidone (Mw ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was obtained from Aladdin Industrial Corporation (Shanghai, P. R. China). Deionized (DI) water was produced by a water purifier system.
000) was obtained from Aladdin Industrial Corporation (Shanghai, P. R. China). Deionized (DI) water was produced by a water purifier system.
Synthesis of monodispersed Fe3O4 nanospheres
Monodispersed Fe3O4 nanospheres (denoted as F) were prepared via a modified method reported by Lin et al.31 In a typical process, 2.72 g FeCl3·6H2O and 0.136 g EDTA-2Na were first dissolved in 80 mL ethylene glycol with vigorous stirring. After 30 min, 2.4 g NaAc was added to the above solution and it was further stirred for another 30 min. Next, the mixture solution was ultrasonically treated for 60 min and then sealed in a 100 mL autoclave. The autoclave was heated to and maintained at 200 °C for 10 h. After the autoclave cooled to room temperature, the black brown precipitate was collected, rinsed with ethanol and DI water three times, respectively, and then dispersed in 200 mL DI water. To improve the dispersibility of the Fe3O4 nanosphere core, 0.5 g sodium citrate tribasic dihydrate was added to the suspension and it was ultrasonically treated for 10 min. Finally, the product was collected from the water via magnetic separation, rinsed with DI water twice, and dried in vacuum oven at 60 °C for 12 h to obtain monodispersed Fe3O4 nanospheres.Synthesis of Fe3O4@SiO2 core–shell structures
Fe3O4@SiO2 core–shell structures (denoted as FS) were prepared through a modified Stöber process.32 In a typical process, 0.6 g Fe3O4 nanoparticle cores were dispersed in a solution of 300 mL ethanol, 80 mL DI water and 20 mL aqueous ammonia (28%) via ultrasonic treatment. After 10 min, a solution of 2 mL TEOS and 20 mL ethanol was added to it dropwise under vigorous stirring. After 6 h, the product was magnetically separated, washed with ethanol and DI water several times, and then dried in a vacuum oven at 60 °C for 12 h to obtain the final product.Synthesis of Fe3O4@SiO2@Ag core–shell structures
Fe3O4@SiO2@Ag (denoted as FSA) core–shell structures were synthesized using an improved seed-mediated growth method reported by Liu et al.33 In a typical process, an Sn(II) solution was prepared by dissolving 0.3 g SnCl2 in 10 mL DI water containing 0.1 mL concentrated HCl. 0.1 g FS sample was dispersed in the above SnCl2 solution via ultrasonication for 20 min. Then, the Sn2+-functionalised FS sample was separated magnetically to remove unabsorbed Sn2+, and then re-dispersed in water. The Sn2+-functionalised FS sample was added to 10 mL 0.35 M ammonia silver nitrate solution under ultrasonication for 20 min to anchor the Ag seeds on the surface of the SiO2 shell. Next, the rinsed silver nuclei-decorated FS sample was dispersed in 100 mL DI water, and sodium citrate tribasic dehydrate (0.2 g), excess glucose (0.667 g), and 1 mL 11.83 mmol L−1 ammonia silver nitrate solution were added to the suspension in sequence. After 1 h reduction at room temperature, FSA core–shell structures were obtained. The product was rinsed using DI water three times and then dried in a vacuum oven at 60 °C overnight.Synthesis of dense silica layer-protected Fe3O4@SiO2@Ag double-layer core–shell structures
Fe3O4@SiO2@Ag@d-SiO2 (denoted as FSAS) double-layer core–shell structures were prepared through a modified Stöber process.32 Typically, 2 mL NH3·H2O (28 wt%) was injected into a suspension of 0.1 g FSA sample, 20 mL DI water and 80 mL ethanol under moderate mechanical stirring. Then, 100 μL TEOS was added into the above suspension, and it was mechanically stirred for 6 h. The resulting product was collected by magnetic separation, washed three times with ethanol and water, respectively, to remove excess NH3·H2O, and dried in a vacuum oven at 60 °C for 12 h to obtain the FSAS sample.Synthesis of porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures
A modified surface-protected etching process34 was adopted to transform the dense silica outer shell into a mesoporous shell to allow the release of silver ions. First, 0.2 g FSAS sample was heated in 40 mL water at 95 °C in the presence of 2 g PVP (MW ∼ 10000) for 3 h. Next, the product (denoted as FSAS-P3) was collected by magnetic separation, rinsed with DI water several times, and re-dispersed in 90 mL DI water. Then, 10 mL NaOH (4 M) was added in with rapid mechanical stirring for the alkaline etching. After the etching process, the sample was collected by magnetic separation, washed three times with ethanol and water, respectively, and dried at 60 °C for 12 h to obtain the final product. A series of etching times was adopted at 0 min, 30 min, 60 min, 90 min, and 120 min, and the obtained samples were denoted as FSAS-P3-0, FSAS-P3-30, FSAS-P3-60, FSAS-P3-90 and FSAS-P3-120, respectively.
Material characterization
The crystal structures of the samples were analysed via X-ray diffraction (XRD) using a D/MAX-2004 X-ray powder diffractometer (Rigaku Corporation, Tokyo, Japan) with Ni-filtered Cu Kα radiation (0.15418 nm) at 56 kV and 182 mA. TEM images were obtained on a JEOL-2100 transmission electron microscope (JEOL, Tokyo, Japan). The specific surface areas of the samples were measured on an Autosorb-1 automatic Surface Area and Pore Size Analyzer (Quantachrome Instruments, Boynton Beach, FL, USA) and calculated using the standard Brunauer–Emmett–Teller (BET) method. The mesopore size distributions were obtained using the Barrett–Joyner–Halenda (BJH) method and the desorption branch data from the nitrogen adsorption isotherms. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra of the catalysts were recorded on a Nicolet 6700 FTIR spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) in the wavelength range of 4000–650 cm−1. UV-vis measurement was carried out on a UV-2550 spectrophotometer (Shimadzu Corporation, Kyoto, Japan). An Agilent 240FS atomic absorption spectrometer (AAS) was used to measure the silver ion content in solution. Magnetic measurements were conducted on a vibrating sample magnetometer (VSM) standard option in a physical property measurement system (PPMS) equipped with a superconducting magnet with a maximum magnetic field of 14 T.E. coli disinfection experiment
The standard plate counting method was adopted to test the disinfection capability of our samples, and a model microorganism of wild-type Escherichia coli (E. coli) bacteria (ATCC 15597, American Type Culture Collection, VA, USA) was used for the disinfection experiment. In a typical disinfection test, the initial concentration of E. coli cells was ∼107 cfu mL−1, and the sample dosage was 0.05 mg (sample)/mL (E. coli cell suspension). This sample dosage was chosen based on the superior disinfection performance of silver nanoparticles, which was obtained as the optimized value after multiple pre-experiments. The E. coli cell density was determined using the value of logNt, whereNt was the number of viable cells in terms of colony-forming units (cfu mL−1) determined by the plate count method. Fig. S1† shows the optical images of the plates used for plate counting in a typical E. coli disinfection experiment, which clearly demonstrated that the colony-forming units continuously decreased with an increase in treatment time. Recycling and reuse disinfection experiments were also conducted to examine the long-term disinfection capability of our sample using the FSAS-P3-90 sample as a representative. For each run, the treatment time was chosen as 20 min according to the previous disinfection experiment result. After the disinfection experiment was finished in the prior run, the used FSAS-P3-90 sample was magnetically separated, collected, rinsed with ethanol and water, respectively, and then reused for the next run with a fresh E. coli cell suspension.
Results and discussion
Creation of porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structure
Fig. 1 shows a schematic of the synthesis of the porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structure from an Fe3O4 nanoparticle core, and the XRD patterns of the samples obtained in each step are shown in Fig. 2. For all the samples, the diffraction peaks of the orthorhombic phase of Fe3O4 (JCPDS card no. 75–0449) could be easily identified. After the loading of Ag nanoparticles, the diffraction peaks of the metallic Ag phase (JCPDS card no. 87–0720) could also be easily identified (see XRD patterns of N3–N8). No diffraction peaks belonging to SiO2 could be identified after the introduction of the silica shells (see XRD patterns of N2–N8), which is attributed to their amorphous nature. The existence of silica layers was verified by FTIR spectroscopy, as shown in Fig. S2 in the ESI.† After the introduction of the silica shells, two characteristic peaks appeared at ∼1103 cm−1 and ∼947 cm−1, which were assigned to the Si–O–Si bond and the Si–OH bond, respectively, and verified the existence of the SiO2 shells.35 From Fig. S2,† the obvious COO− symmetric stretching vibration at ∼1410 cm−1 and asymmetric stretching vibration at ∼1610 cm−1 could be observed for all the samples, which was due to the use of citric acid in the synthesis of the Fe3O4 nanoparticle core and the loading of Ag nanoparticles.31,36 The XRD analysis results demonstrated that the silica coating and the followed etching process did not change the crystal structures of Fe3O4 nanoparticle core and Ag nanoparticles because their XRD diffraction patterns remained almost unchanged after these processes. The average crystallite sizes of the Fe3O4 and Ag nanoparticles were determined to be ∼10 nm using the Scherrer formula and their strongest XRD peaks.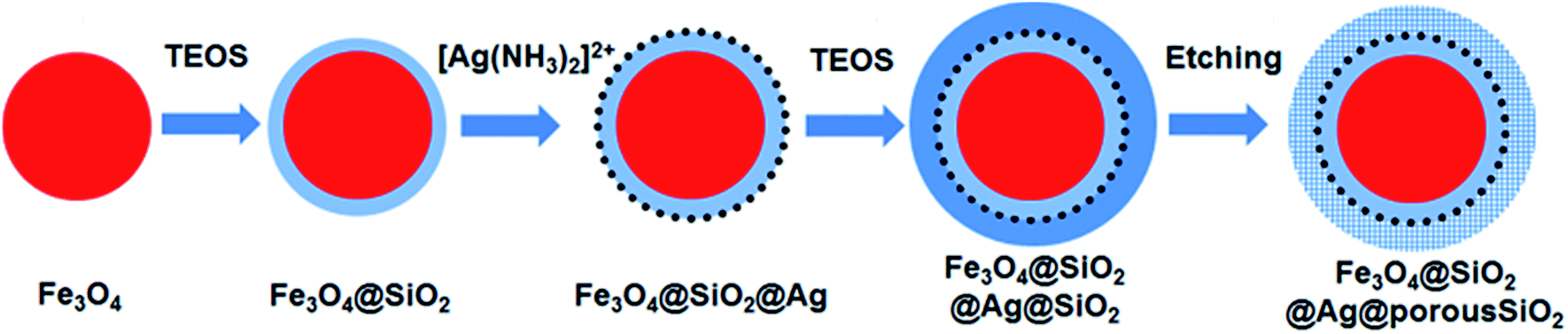 | ||
| Fig. 1 Schematic illustration of the synthesis of the mesoporous silica-protected Fe3O4@SiO2@Ag double layer core–shell structure. | ||
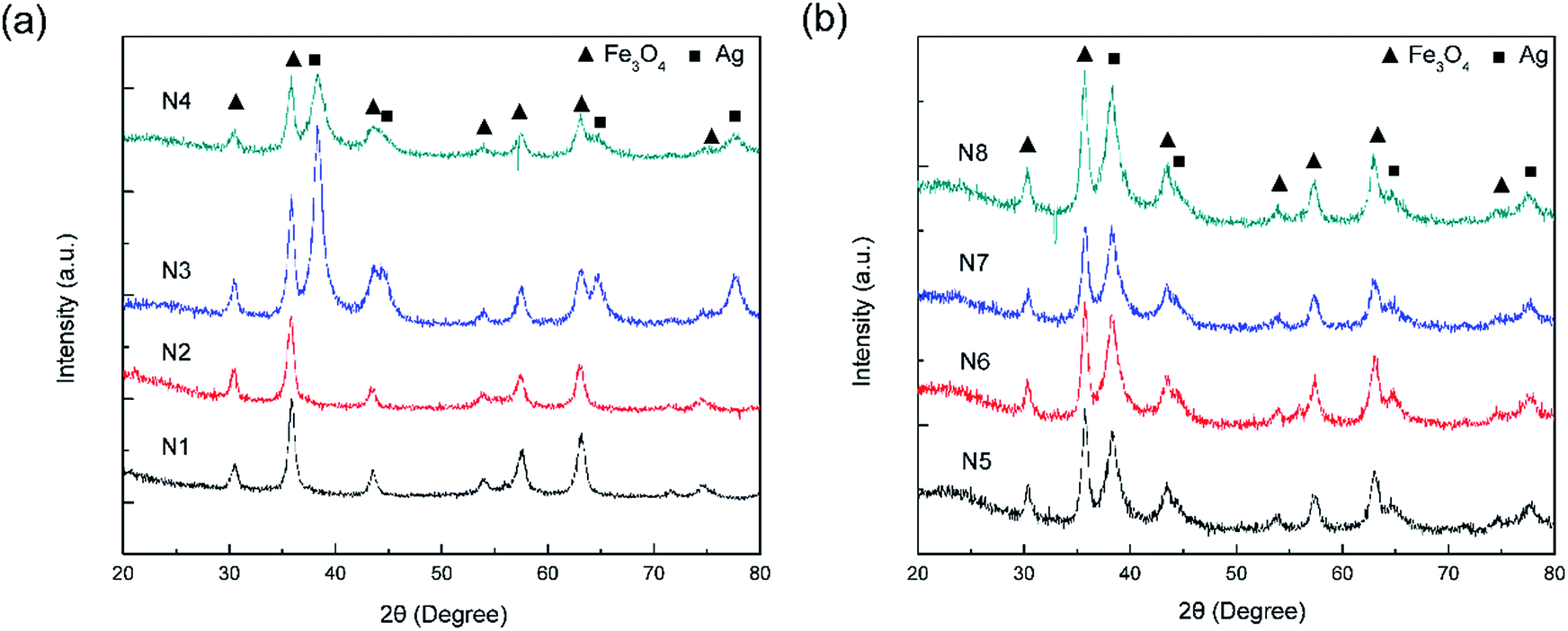 | ||
| Fig. 2 (a) XRD patterns of the F (N1), FS (N2), FSA (N3), and FSAS (N4) samples. (b) XRD patterns of the FSAS-P3-0 (N5), FSAS-P3-30 (N6), FSAS-P3-60 (N7), and FSAS-P3-90 (N8) samples. | ||
Fig. 3a–d show the TEM images of the F, FS, FSA, and FSAS samples, respectively. Fig. 3a shows that the monodispersed Fe3O4 nanospheres were composed of aggregated fine Fe3O4 nanoparticles, and their average diameter was ∼200 nm. The introduced citrate during the synthetic process was critical to enhance their dispersity due to its adsorption on the Fe3O4 nanospheres to increase the electrostatic repulsion between them.36 When no citrate was introduced, the good dispersion of the Fe3O4 nanospheres was found to last only several hours. After the modified Stöber process, a thin silica shell was coated on the surface of the Fe3O4 nanosphere core (Fig. 3b). The thickness of the silica shell was ∼20 nm thick, and it had a dense structure. Thus, it could provide protection to the Fe3O4 nanosphere core from interaction with substances in water bodies to avoid secondary pollution and keep its superparamagnetic behaviour. Furthermore, rich surface hydroxyl (–OH) groups existed on the surface of the SiO2 shell and their negative charges could facilitate the adsorption of Sn2+ ions by electrostatic attraction.37
 | ||
| Fig. 3 TEM images of the sample at the different synthesis stages: (a) F, (b) FS, (c) FSA, and (d) FSAS. | ||
After the Sn2+-functionalised FS sample emerged in the silver ammonia solution, its colour changed from bright orange to dark grey immediately, indicating the formation of metal silver nuclei on the silica shell surface through the reduction of silver ammonia by Sn2+. Under the reducing environment created by glucose, Ag atoms subsequently grew on these Ag nuclei to finally form uniformly distributed silver nanoparticles on the SiO2 shell surface, as demonstrated in Fig. 3c. The average diameter of these silver nanoparticles was ∼10 nm, which is in accordance with the XRD crystal size analysis result. A similar core–shell structure of Fe3O4@SiO2@Ag was synthesized by Fu et al.,16 which exhibited efficient antibacterial activity and reusability. However, the detachment and excessive dissolution (exceeding MCL of 0.05 g L−1) issues hindered its practical applications. In our approach, a protective SiO2 outer shell was introduced to prevent the detachment of Ag nanoparticles due to the robust coating procedure based on a room temperature sol–gel process and readiness for the surface protecting etching process.32,38Fig. 3d shows the TEM image of the FSAS sample. The thickness of its SiO2 outer layer was ∼50 nm, which had a dense structure.
To allow the oxidation of Ag nanoparticles and the subsequent release of Ag+ ions for effective disinfection, a surface protecting etching method34,39 was adopted to transform the dense SiO2 outer shell into a mesoporous structure. Fig. 4a–d shows the TEM images of the FSAS samples after the water boiling process with the surface protecting etching times of 0 min, 30 min, 60 min, and 90 min, respectively. For the sample treated only by boiling water, a difference in contrast could be observed at the SiO2 outer layer edge (∼20 nm from the outside to the inside along the radial direction), as shown in the TEM image in Fig. 4a, which can be attributed to the dissolution and precipitation equilibrium between the Si–O–Si network and the monomer Si(OH)4 during the long time heating.40,41 Some micropores may have formed at the surface layer of the SiO2 outer shell, but no obvious penetrating pores were observed. During the water boiling process, strong hydrogen bonds could be formed between PVP and silica to increase the stability of the silica shell in NaOH solution for the following controllable surface protecting etching process to create a mesoporous structure.34 Without the protection of PVP, the silica shell could be etched away rapidly.42 After 30 min etching, the SiO2 outer shell surface became rough and the observed contrast difference in Fig. 4a became weaker, as shown in Fig. 4b. After 60 min etching, the SiO2 outer shell thickness decreased to ∼45 nm (5 nm smaller than the original shell thickness) due to the continuous alkaline etching and its structure became mesoporous (see Fig. 4c). When the alkaline etching time reached 90 min, the SiO2 outer shell thickness decreased to ∼35 nm and its mesoporous structure became more obvious (see Fig. 4d). Thus, porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures were successfully created. With the prolonged etching time, the SiO2 outer layer could be completely removed as demonstrated in the TEM image of the FSAS sample after 120 min etching, as shown in Fig. S3 in the ESI,† which then lost the protection of the Ag nanoparticles.
 | ||
| Fig. 4 TEM images of FSAS samples with a series of etching times of 0 min, 30 min, 60 min, and 90 min: (a) FSAS-P3-0, (b) FSAS-P3-30, (c) FSAS-P3-60, and (d) FSAS-P3-90 (Note, the inset images show the edges of the samples with a higher magnification). | ||
Mesoporous structure of the protective SiO2 outer shells with different etching times
The TEM observation clearly demonstrated that the surface protecting etching method successfully converted the dense SiO2 outer shell into a mesoporous structure. N2 adsorption/desorption isotherm studies were conducted to further investigate the SiO2 outer shell structure of the etched samples. Fig. S4 and S5 in the ESI† show the isotherms and BJH pore distributions of the samples. The BET specific surface areas of the FSAS-P3-0, FSAS-P3-30, FSAS-P3-60, and FSAS-P3-90 samples were calculated to be ∼11.43, 83.34, 90.37 and 113.9 m2 g−1, respectively, based on their N2 adsorption/desorption isotherms. For the FSAS-P3-0 sample, no obvious pore structure was observed (see Fig. S5a†), while the alkaline etched samples all demonstrated obvious mesoporous structures, which is in accordance with their significantly increased BET specific surface areas compared with that of the FSAS-P3-0 sample without alkaline etching. The average pore sizes were found to be ∼1.93, 2.05, and 2.06 nm for the FSAS-P3-30, FSAS-P3-60, and FSAS-P3-90 samples, respectively. These results clearly demonstrated that the BET specific surface area and pore size increased with an increase in alkaline etching time, and the resulting mesoporous structures could provide channels in the protective outer shell to allow the Ag nanoparticles to interact with the aqueous environment. The resultant Ag+ ions could diffuse from these channels gradually into the water body for a long-term disinfection capability. Fig. S6† shows the optical images of the F, FS, FSA, FSAS, and FSAS-P3-90 samples. Sample F had a dark brown color due to its Fe3O4 nature, while sample FS was mostly brown after the formation of the SiO2 layer on the Fe3O4 core. Sample FSA had a dark grey color because silver nanoparticles were deposited on its surface, while the color of sample FSAS was slightly lighter after coating with the second SiO2 layer. The surface protecting etching process did not change the sample color obviously, as demonstrated by sample FSAS-P3-90 as a representative.Effective protection of silver nanoparticles from detachment
The size of the mesopores (∼2 nm) in the protective SiO2 outer shell was much smaller than that of the Ag nanoparticles (∼10 nm). Thus, the Ag nanoparticles could not pass through the SiO2 outer shell, which provided the Ag nanoparticles the desired protection. The protection effect of Ag nanoparticles from detachment by the mesoporous SiO2 outer layer was examined via an Ag fall-off experiment. Typically, 0.1 g FSA sample and 0.1 g FSAS-P3-90 sample were dispersed in 100 mL PBS buffer separately in Erlenmeyer flasks, which were shaken vigorously on a shaker. Fig. 5 shows the optical absorbance spectra of the PBS buffer solutions, in which the dispersed FSA sample and FSAS-P3-90 sample were mechanically shaken for a series of time up to 72 h before they were magnetically separated. A large absorption peak was observed at ∼420 nm occurred for the FSA sample for the shaking time from 12 h up to 72 h, and its intensity constantly increased with an increase in the shaking time. This observed large absorption peak can be attributed to the plasmon resonance absorption of the Ag nanoparticles.43 This observation clearly suggested that the detachment of Ag nanoparticles occurred on the FSA sample during the shaking process and more and more Ag nanoparticles detached from the surface of the FAS sample with an extension in the shaking time. However, for the FSAS-P3-90 sample, no absorption peak at ∼420 nm was observed even after shaking for 72 h, suggesting that no Ag nanoparticle detachment occurred. Among the alkaline etched samples, the FSAS-P3-90 sample had the thinnest SiO2 outer layer with the largest mesopore size. Thus, the comparison results clearly verified that the mesoporous SiO2 outer layer successfully protected the Ag nanoparticles and prevented their detachment during the water disinfection process, which could avoid the dispersion of Ag nanoparticles into the aqueous environment and be crucial for their long-term disinfection performance.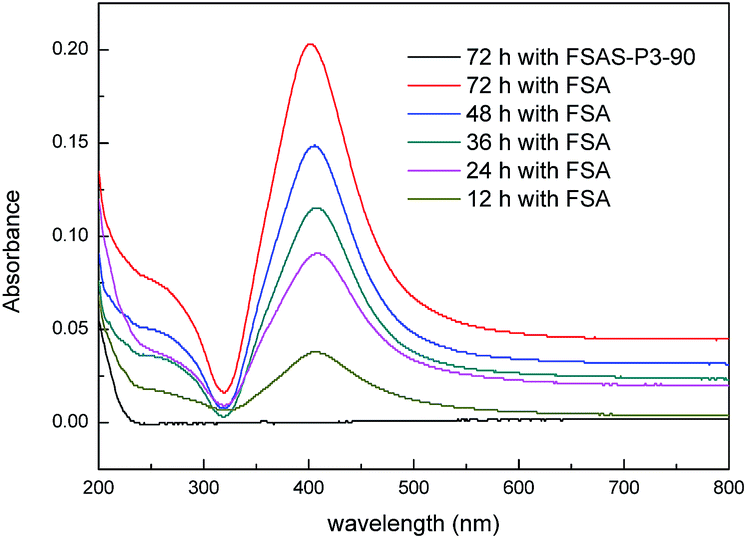 | ||
| Fig. 5 UV-vis absorbance spectra of PBS buffer solutions in the Ag fall-off experiments for the FSA and FSAS-P3-90 samples. | ||
Superparamagnetic behavior of porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures
Fig. 6 shows the magnetic field-dependent behaviors of the Fe3O4 nanosphere core and the FSAS-P3-90 sample as the representative of porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures at room temperature. Both of them showed typical superparamagnetic behavior, as demonstrated by the lack of a hysteretic lag in their loops.44 This can be attributed to the ultrafine size of the Fe3O4 nanoparticles that aggregated to form the Fe3O4 nanosphere core.45 Thus, their remanence and coercivity were both zero, which eliminated magnetic attraction between them and was beneficial for their dispersion in water bodies. Their saturation magnetization, Ms, was obtained by extrapolating their graphs of M vs. 1/H to 1/H → 0 (for H > 10 kOe). The Ms of the Fe3O4 nanosphere core was determined to be ∼72.3 emu g−1 and that for the FSAS-P3-90 sample was ∼24.9 emu g−1. The much lower Ms of the FSAS-P3-90 sample was due to the presence of non-magnetic Ag nanoparticles and SiO2 inner and outer shells. Nevertheless, it was strong enough for its rapid magnetic separation from water after treatment when an external magnetic field was applied. Thus, the superparamagnetic behavior and strong saturation magnetization of the FSAS-P3-90 sample could ensure both its good dispersion in water bodies and easy separation from treated water bodies, which are beneficial for its potential applications.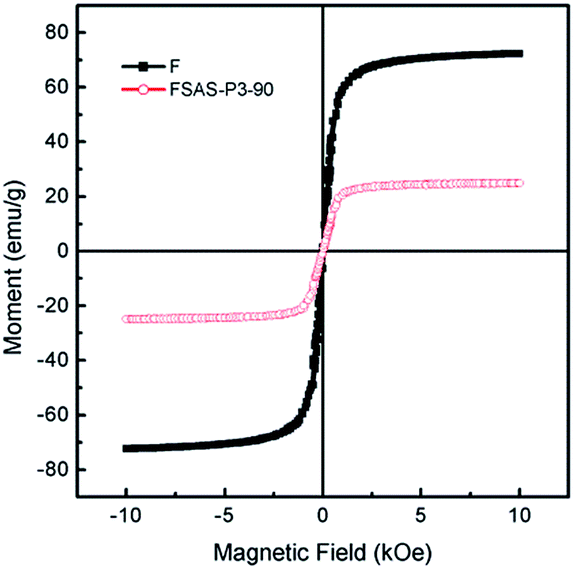 | ||
| Fig. 6 Room-temperature magnetization curves of the Fe3O4 nanosphere core and the FSAS-P3-90 sample. | ||
Disinfection activities of porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures
The disinfection performances of the porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures were investigated with E. coli bacterium as the model microorganism. Fig. 7a shows the cell density of E. coli bacteria treated with the FSAS-P3-0, FSAS-P3-30, FSAS-P3-60 and FSAS-P3-90 samples. In PBS buffer solution, the E. coli cells could remain active up to 2 weeks. However, with just a low disinfectant loading of 0.05 mg mL−1, the cell density of E. coli cells decreased sharply for the alkaline etched samples. Among them, the FSAS-P3-90 sample demonstrated the fastest disinfection performance. After only 17 min treatment, the E. coli cell density dropped 7 orders of magnitude, which suggested that all the E. coli cells in the treated solution (107 mfu mL−1) were sterilized. For the FSAS-P3-60 sample and the FSAS-P3-30 sample, the E. coli cell density dropped 7 orders of magnitude and over 5 orders of magnitude, respectively, after 20 min treatment. However, for the FSAS-P3-0 sample, the E. coli cell density dropped less than one order of magnitude after 20 min treatment. It is generally believed that ionic silver can strongly interact with the thiol groups of vital enzymes inside microorganisms and inactivate them.14 Thus, the disinfection of E. coli cells by our samples relied on the release of Ag+ ions into the aqueous environment. The disinfection experiment results for the different samples clearly indicated that the release of Ag+ ions was enhanced by a longer alkaline etching time because it reduced the thickness of the SiO2 outer shell and created more and larger mesopores, which were beneficial for the release of Ag+ ions.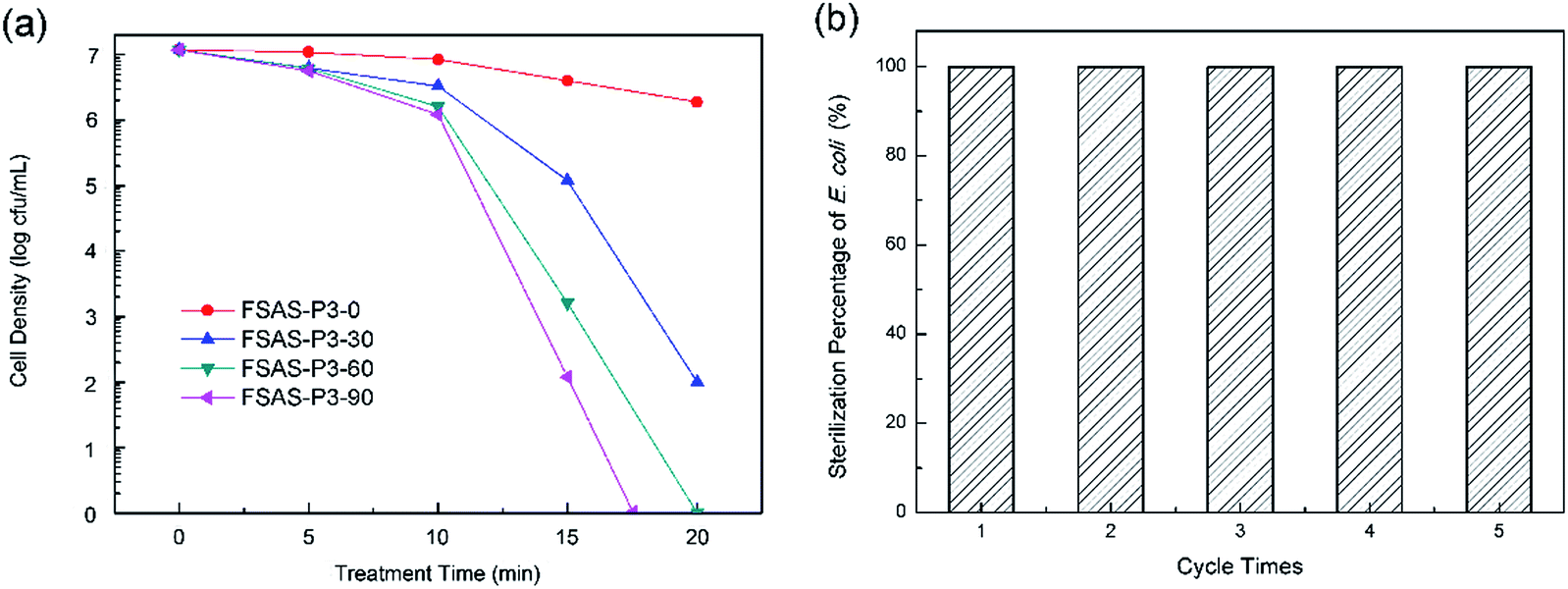 | ||
| Fig. 7 (a) E. coli cell density vs. treatment time by the FSAS-P3-0, FSAS-P3-30, FSAS-P3-60 and FSAS-P3-90 samples. (b) E. coli sterilization percentage for 5 consecutive runs of treatment by the FSAS-P3-90 sample. | ||
The long-term disinfection capability of the FSAS-P3-90 sample was examined via a recycling and reuse experiment. After each treatment for 20 min, the FSAS-P3-90 sample was magnetically separated, collected, rinsed, and then reused for the next run with a fresh E. coli cell suspension. Fig. 7b shows the E. coli sterilization percentage for 5 consecutive runs, which clearly demonstrated that the FSAS-P3-90 sample could completely sterilize all E. coli cells in every run although some sample loss was inevitable during the collection and rinsing process. This result suggested that our porous silica-protected Fe3O4@SiO2@Ag double-layer core–shell structures could be an effective disinfectant for water treatment in a long run.
The controllable Ag+ ion release within the MCL for drinking water
For drinking water disinfection, the MCL requirement (0.05 mg L−1) must be met to ensure its safety. To examine the Ag+ ion release with different protective SiO2 outer shells, the FSAS-P3-0, FSAS-P3-30, FSAS-P3-60, and FSAS-P3-90 samples were used, and the FSA sample without the protective SiO2 outer shell was also used as a comparison. 0.05 mg mL−1 sample was added to PBS buffer solution, mechanically shaken for 30 min, and then magnetically separated. Then, the Ag+ ion concentration in the solution was examined. Table 1 summaries the Ag+ ion concentration in the solution for the different samples. Without the protective SiO2 outer shell, the Ag+ ion concentration released by the FSA sample reached 0.232 mg L−1, which was four times higher than the MCL for silver in drinking water. However, for the SiO2 outer shell-protected samples, the Ag+ ion concentrations released by them were all below the MCL for silver in drinking water. Thus, they can be readily used for drinking water disinfection, and the relatively lower Ag+ ion concentration can also be beneficial for their long-term disinfection performance.| Sample | FSAS-P3-0 | FSAS-P3-30 | FSAS-P3-60 | FSAS-P3-90 | FSA |
| C Ag (mg L−1) | 0.0189 | 0.0401 | 0.0414 | 0.0485 | 0.232 |
Conclusions
In summary, a mesoporous SiO2 outer layer was coated on the Fe3O4@SiO2@Ag core shell structure to create a novel Ag nanoparticle-based disinfectant to solve the problems of Ag nanoparticle detachment and excessive Ag+ ion concentration over its MCL for drinking water associated with traditional silver-based disinfectants. In this double-layer core–shell structure, the superparamagnetic Fe3O4 nanosphere core provided an easy magnetic separation function to avoid its loss to the aqueous environment, its dense SiO2 inner shell protected the Fe3O4 nanosphere core and provided a good substrate to load Ag nanoparticles as the active component for disinfection, and its mesoporous SiO2 outer shell created by the protective etching process protected the Ag nanoparticles from detachment and also controlled the release of Ag+ ions from its mesoporous channels. These porous silica-protected Fe3O4@SiO2@Ag double layer core–shell structures exhibited an efficient disinfection effect, satisfactory MCL of Ag+ ions in drinking water, a stable and long-term disinfection performance, and easy magnetic separation capability from treated water bodies. Thus, they have the application potential as an efficient and safe disinfectant for drinking water disinfection with easy operation and low cost.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The experimental assistance on the wild type of Escherichia coli bacteria (ATCC 15597) culture by Ms Mian Song and Ms Shuang Jiao was greatly appreciated. This study was supported by the National Natural Science Foundation of China (Grant No. 51502305, 51672283 and 51602316), the Basic Science Innovation Program of Shenyang National Laboratory for Materials Science (Grant No. Y4N56R1161 and Y5N56F2161), the Natural Science Foundation of Shandong Province, China (Grant No. ZR2016FQ08 and ZR2017MEM017) and Scientific Research Foundation of DeZhou University (Grant No. 2016kjrc07).Notes and references
- E. J. Lee and K. J. Schwab, J. Water Health, 2005, 3, 109–127 CrossRef PubMed.
- L. Andrade, J. O'Dwyer, E. O'Neill and P. Hynds, Environ. Pollut., 2018, 236, 540–549 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 102, 307–315 Search PubMed.
- A. D. Wales, V. M. Allen and R. H. Davies, Foodborne Pathog. Dis., 2010, 7, 3–15 CrossRef CAS PubMed.
- X. Wang, J. Zhang, W. Sun, W. Yang, J. Cao, Q. Li, Y. Peng and J. K. Shang, Chem. Eng. J., 2015, 264, 437–444 CrossRef CAS.
- I. Sondi and B. Salopek-Sondi, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS PubMed.
- Z. Karahaliloglu, E. Kilicay and E. B. Denkbas, Artif. Cells, Nanomed., Biotechnol., 2017, 45, 1–14 CrossRef PubMed.
- B. Dizman, M. O. Elasri and L. J. Mathias, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5965–5973 CrossRef CAS.
- D. Zhang, G. Li and C. Y. Jimmy, J. Mater. Chem., 2010, 20, 4529–4536 RSC.
- R. Fagan, D. E. Mccormack, D. D. Dionysiou and S. C. Pillai, Mater. Sci. Semicond. Process., 2016, 42, 2–14 CrossRef CAS.
- L. Esteban-Tejeda, C. Prado, B. Cabal, J. Sanz, R. Torrecillas and J. S. Moya, PLoS One, 2015, 10, e0132709 CrossRef PubMed.
- H. H. Lara, E. N. Garzatreviño, L. Ixtepanturrent and D. K. Singh, J. Nanobiotechnol., 2011, 9, 30 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. Diendorf, M. Köller and M. Epple, Chem. Mater., 2010, 22, 4548–4554 CrossRef CAS.
- P. Dibrov, J. Dzioba, K. K. Gosink and C. C. Häse, Antimicrob. Agents Chemother., 2002, 46, 2668–2670 CrossRef CAS PubMed.
- Q. Feng, J. Wu, G. Chen, F. Cui, T. Kim and J. Kim, J. Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS PubMed.
- X. Wang, Y. Dai, J. l. Zou, L. Y. Meng, S. Ishikawa, S. Li, M. Abuobeidah and H. G. Fu, RSC Adv., 2013, 3, 11751–11758 RSC.
- M. Liong, B. France, K. A. Bradley and J. I. Zink, Adv. Mater., 2009, 21, 1684–1689 CrossRef CAS.
- J. Zhang, B. Han, J. Liu, X. Zhang, J. He, Z. Liu, T. Jiang and G. Yang, Chem.–Eur. J., 2015, 8, 3879–3883 CrossRef.
- G. Nangmenyi, X. Li, S. Mehrabi, E. Mintz and J. Economy, Mater. Lett., 2011, 65, 1191–1193 CrossRef CAS.
- D. He, A. Ikeda-Ohno, D. D. Boland and T. D. Waite, Environ. Sci. Technol., 2013, 47, 5276–5284 CrossRef CAS PubMed.
- Q. Yu, A. Fu, H. Li, H. Liu, R. Lv, J. Liu, P. Guo and X. S. Zhao, Colloids Surf., A, 2014, 457, 288–296 CrossRef CAS.
- V. A. Oyanedel-Craver and J. A. Smith, Environ. Sci. Technol., 2008, 42, 927–933 CrossRef CAS PubMed.
- W. L. Chou, D. G. Yu and M. C. Yang, Polym. Adv. Technol., 2005, 16, 600–607 CrossRef CAS.
- Y. Lv, L. Hong, W. Zhen, S. Liu, L. Hao, Y. Sang, D. Liu, J. Wang and R. I. Boughton, J. Membr. Sci., 2009, 331, 50–56 CrossRef CAS.
- S. George, S. Lin, Z. Ji, C. R. Thomas, L. J. Li, M. Mecklenburg, H. Meng, X. Wang, H. Zhang and T. Xia, ACS Nano, 2012, 6, 3745–3759 CrossRef CAS PubMed.
- E. Mcgillicuddy, I. Murray, S. Kavanagh, L. Morrison, A. Fogarty, M. Cormican, P. Dockery, M. Prendergast, N. Rowan and D. Morris, Sci. Total Environ., 2017, 575, 231–246 CrossRef CAS PubMed.
- World Health Organization (WHO), Guidelines for drinking-water quality, Geneva, 2011 Search PubMed.
- N. Gao, Y. Chen and J. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 11307–11314 CrossRef CAS PubMed.
- S. Sun, W. Wang, L. Zhang, M. Shang and L. Wang, Catal. Commun., 2009, 11, 290–293 CrossRef CAS.
- X. H. Yang, H. T. Fu, K. Wong, X. C. Jiang and A. B. Yu, Nanotechnology, 2013, 24, 415601 CrossRef CAS PubMed.
- M. Lin, H. Huang, Z. Liu, Y. Liu, J. Ge and Y. Fang, Langmuir, 2013, 29, 15433–15441 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- T. Liu, D. Li, D. Yang and M. Jiang, Chem. Commun., 2011, 47, 5169–5171 RSC.
- Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS PubMed.
- D. Kandpal, S. Kalele and S. Kulkarni, Pramana, 2007, 69, 277–283 CrossRef CAS.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- B. Lee, Z. Ma, Z. Zhang, C. Park and S. Dai, Microporous Mesoporous Mater., 2009, 122, 160–167 CrossRef CAS.
- A. K. Van Helden, J. W. Jansen and A. Vrij, J. Colloid Interface Sci., 1981, 81, 354–368 CrossRef CAS.
- J. Ge, Q. Zhang, T. Zhang and Y. Yin, Angew. Chem., 2008, 120, 9056–9060 CrossRef.
- G. B. Alexander, W. Heston and R. K. Iler, J. Phys. Chem., 1954, 58, 453–455 CrossRef CAS.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- R. A. Caruso and M. Antonietti, Chem. Mater., 2001, 13, 3272–3282 CrossRef CAS.
- A. K. Gade, P. Bonde, A. P. Ingle, P. D. Marcato, N. Durán and M. K. Rai, J. Biobased Mater. Bioenergy, 2008, 2, 243–247 CrossRef.
- X. Yu, S. Liu and J. Yu, Appl. Catal., B, 2011, 104, 12–20 CrossRef CAS.
- T. Hieu Ngo, D. L. Tran, H. M. Do, V. Hoang Tran, V. H. Le and X. Phuc Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2010, 1, 035001 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00275d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |