
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl of the compositions and morphologies of uranium oxide nanocrystals in the solution phase: multi-monomer growth and self-catalysis†
Qiang
Yan
 ab,
Yiwu
Mao
a,
Xiaosong
Zhou
a,
Jianhua
Liang
a,
Shuming
Peng
*a and
Minyou
Ye
*b
ab,
Yiwu
Mao
a,
Xiaosong
Zhou
a,
Jianhua
Liang
a,
Shuming
Peng
*a and
Minyou
Ye
*b
aInstitute of Nuclear Physics and Chemistry, China Academy of Engineering Physics, Mianyang, 621900, China. E-mail: pengshuming@caep.cn
bSchool of Physical Sciences, University of Science and Technology of China, Hefei, 230026, China. E-mail: yemy@ustc.edu.cn
First published on 11th February 2019
Abstract
The presence of mixed products and impurities, which always confuse researchers, are common during synthesizing nanomaterials. Even though many studies have been conducted with an objective to control the synthesis of nanomaterials, very few studies have investigated a mechanism to control the composition of nanomaterials. Various products include UO3·H2O, U3O8, UO2, and U4O9 were produced by simply adjusting the pH with ammonia. The morphology of UO2 and U3O8 are tunable. In this study, we suggest two mechanisms that can be used to control the nanomaterial composition. Various experiments have been conducted to understand the mechanism that controls the composition of nanomaterials. We indicate that a multi-monomer growth model can be used to control the uranium oxide composition. We have developed a new oxidation–reduction system using acetone, and this system is capable of controlling both the morphology and composition of uranium oxide micro/nanomaterials. Further, the presence of the self-catalysis mechanism can be used to regulate processes that control the monomer transformation. Thus, the results of this study can be applied to help in the construction of mixed-valence metal oxides.
Nanomaterials have attracted considerable attention due to their many unique and novel properties, such as optical, electronic, magnetic and catalytic properties. Nanomaterials have many potential innovative applications in various fields, as nanogenerators,1 catalysts,2,3 semiconductors,4–6 photoluminescent materials7,8 and biomedicines.9,10 These properties are closely related to the shape and size of the materials.
A number of studies have attempted to control the synthesis of nanomaterials, of which most have focused on the size and shape of the nanomaterials. Numerous theories11–16 have been proposed describing the control of the shapes or sizes of nanomaterials, such as the LaMer theory, agglomeration, aggregation, coalescence, orientation attachment (OA), the Kirkendall effect, self-assembly, and Ostwald ripening (OR). However, very few studies exist that discuss how to control nanomaterial composition. Thus, these mechanisms are proficient at describing the nucleation, growth process and shape changes of nanomaterials. However, they fail to explain the composition control and appearance of mixed products.
Impurities or mixtures are common in final products. For example, for Fe3O4/α-Fe2O3 composites,17 U3O8 appears in UO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and Cu in Cu2O.19 The reason for the appearance of impurities or a mixture in the final products still puzzles researchers.
18 and Cu in Cu2O.19 The reason for the appearance of impurities or a mixture in the final products still puzzles researchers.
Uranium is best known, and feared, for its involvement in nuclear energy. Currently, uranium has been gaining an increased amount of attention due to its exclusive combination of properties.20 Uranium oxides also have a number of important properties, as well as their potential application as semiconductors,21 and catalysts for short chain alkane destruction22 and volatile organic compound (VOCs) pollutants.2,23 On the other hand, uranium, which can form various oxides, is a multi-valence state metal. Four of the uranium oxides are stoichiometric thermodynamically stable phases, i.e. UO3, U3O8, U4O9, and UO2.24 The others are metastable stoichiometric phases, such as U2O5, which is only observable at elevated pressure, and U3O7, which appears only during UO2 oxidation at temperatures below 400 °C.25 Therefore, uranium oxides are ideal materials to investigate the mechanisms that control composition.
As is well known, the composition of micro/nanomaterials changes with the alteration of reaction times, temperature, pH, and chemical agent concentration during synthesis. Shi et al.26 produced UO2 nanospheres and U3O8 nanocuboids using a hydrothermal method. Wang et al.27 successfully synthesized ultrathin UO2, U3O7, and U3O8 nanomaterials using hydrothermal methods. However, the mechanism that plays an important role in forming the different valence state metal oxide products was unclear.
In this study, we successfully controlled both the morphology and shape of uranium oxide nanomaterials in aqueous phases. Based on the results, we suggest two mechanisms that are crucial in controlling nanomaterial composition.
The uranium oxide nanocrystals were synthesized using a hydrothermal method. Uranium acetate hydrate (UAH) (100 mg) was dissolved in a mixture of DI water (5 mL) and acetone (10 mL) in a capped glass bottle while being magnetically stirred. The ammonia (undiluted), ranging in volume from 25 μL to 1500 μL, was added to the solution to adjust the pH and stirred for 5 minutes. The entire mixture was then transferred to a 30 mL Teflon-lined stainless steel autoclave and sealed. The autoclave was maintained at 200 °C for 6 hours and cooled naturally to room temperature. The precipitates were centrifuged, and washed with ethanol and acetone. This procedure was repeated several times. Detailed experimental parameters are listed in the (ESI†).
X-ray powder diffraction (XRD) analyses indicate that the product compositions are different at various pH values (Fig. 1). The products include a mixture of UO3·H2O and U3O8 (pH = 4.76), pure U3O8 (pH = 7.83), a mixture of U3O8 and UO2 (pH = 8.25), pure UO2 (pH = 9.81) and pure U4O9 (pH = 10.66).
 | ||
| Fig. 1 XRD patterns of the products at various pH values (symbols: ● = U4O9, ♦ = UO2, ♣= U3O8, and ♥= UO3·H2O). | ||
The structures of UO2 and U4O9 are quite similar. The UO2 and U4O9 space groups (SG) are Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (SG no. 225) and I
m (SG no. 225) and I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d (SG no. 220), respectively. The Bragg reflection of both UO2 and U4O9 products correspond to the standard face-centered cubic (FCC) structure. Distinguishing these two products in the XRD patterns is difficult (Fig. 1, the red and blue curves). However, the lattice parameter of UO2 (a = 5.46 Å) is slightly larger than that of U4O9 (a/4 = 5.43 Å). According to Bragg diffraction, the 2 theta degree of the corresponding peaks in UO2 was slightly smaller than that of U4O9 (Fig. S1a†). On the other hand, the average U(V + VI)4O9 valence state is higher than U(IV) O2, which results in a higher binding energy for U4f (Fig. S1b and c†). There are significant signals which allowed us to distinguish between UO2 and U4O9. Experiments without the addition of acetone yield pure UO3·H2O (Table S1†). The product morphologies (Fig. S2†) and experimental conditions (Tables S1 and S2.2†) are listed in the (ESI†).
3d (SG no. 220), respectively. The Bragg reflection of both UO2 and U4O9 products correspond to the standard face-centered cubic (FCC) structure. Distinguishing these two products in the XRD patterns is difficult (Fig. 1, the red and blue curves). However, the lattice parameter of UO2 (a = 5.46 Å) is slightly larger than that of U4O9 (a/4 = 5.43 Å). According to Bragg diffraction, the 2 theta degree of the corresponding peaks in UO2 was slightly smaller than that of U4O9 (Fig. S1a†). On the other hand, the average U(V + VI)4O9 valence state is higher than U(IV) O2, which results in a higher binding energy for U4f (Fig. S1b and c†). There are significant signals which allowed us to distinguish between UO2 and U4O9. Experiments without the addition of acetone yield pure UO3·H2O (Table S1†). The product morphologies (Fig. S2†) and experimental conditions (Tables S1 and S2.2†) are listed in the (ESI†).
The oxidation degree of the products decreased with increasing pH values. This implies that the system's reducing efficiency increased with increasing pH values. The presence of U4O9 at a higher pH is due to the rapid growth packaging in uranium at higher residual oxidation states. This mechanism is explained in the following discussion. The particle's volume decreased with increasing pH values (Fig. S1†), which implies that there is a decrease in the number of particles with the equivalent amount of raw materials.
To understand the mechanism that controls compositions, we performed a series of experiments (details are listed in ESI†). All of the experiments showed similar regularity as previously mentioned.
We have observed the presence of a mixed-valence uranium oxide, U3O8, when synthesizing UO2 nanocrystals with diethylene glycol (DEG). We suggested that there must be more than one type of monomer in the growth process of uranium oxides.18 The mixed products in our experiments presented another kind of complexity. Fortunately, we obtained pure products for each phase and achieved morphological control (Fig. 2) by simply altering the experimental conditions (Tables S2.1 and S2.2†), which can help us to explore the control mechanisms.
 | ||
| Fig. 2 SEM images of U3O8 (a–e) and UO2 (f–i) nanocrystals produced at various reaction conditions. Morphologies: (a) nail-like, (b) bone-like, (c) prism; (d) spear-like; (e) bone-like; (f) sunken octahedron; (g) octahedron; (h) smooth octahedron; and (i) spherical. The details of the experiments are listed in Tables S2.1 (U3O8) and S2.2 (UO2).† | ||
LaMer16 presented a theory to explain the formation of mono-dispersed hydrosols in 1950 (Fig. S6†). This is a classic mechanism to describe nanomaterial nucleation and growth processes in the liquid phase. However, the LaMer theory has limitations in explaining a mixed-valence metal oxide and the formation of mixed composition products.
In this study, we present a multi-monomer growth model to control the uranium oxide composition based on hundreds of experiments. We suggest that this model can be thought of as an extension of the LaMer theory and can be potentially extended to other multi-valence metal oxides.
In this model, each compound's valence (in solution) is considered to be an independent monomer that abides by the LaMer theory. As the concentration increases, the monomers tend to aggregate and nucleate to reduce system energy when they overstep their minimum nucleation concentration. Impurities always appear when the reaction time is insufficient or due to temperature. We can obtain a pure product by prolonging the reaction time.18 However, sometimes prolonging the reaction time did result in pure product B being obtained and the products appeared as a mixture throughout the experiment, from beginning to end [Tables S3.1 and S3.2†].
We assume that two types of monomers, known as A and B, are in solution. The monomers include the formation of atoms, molecules or clusters. For example, we aimed to obtain the product B. Monomer A, which has a higher solubility, transferred to monomer B via a chemical reaction in solution. Due to its high solubility, monomer A remained below its minimum nucleation concentration (solid orange curve in Fig. 3). The concentration of monomer B increased with time, which led to its nucleation and growth (Fig. 3, blue solid curves). There are two potential pathways to produce mixture products or impurities. We hypothesize that the solubility of monomer A is higher than that of monomer B and also rapidly increases with increasing temperature. In the first pathway, the reaction ends and the temperature decreases rapidly before monomer A is completely depleted. The minimum nucleation concentration decreases with decreasing temperature. This is due to the fact that the critical solubility decreases when the temperature decreases. This leads to the concentration of residual monomer A (Fig. 3a, orange curve) exceeding the new minimum nucleation concentration at T2 (Fig. 3a, dashed orange line). In the second pathway, the generation rate of monomer A is much faster than the depletion rate, which leads to a continuous increase in the concentration of monomer A (Fig. 3b, dashed orange line). After sufficiently long reaction times, monomer A can approach the minimum nucleation concentration. Both pathways promote the nucleation and growth of monomers A (Fig. 3a and b, orange curves), which further results in the production of mixed products or impurities.
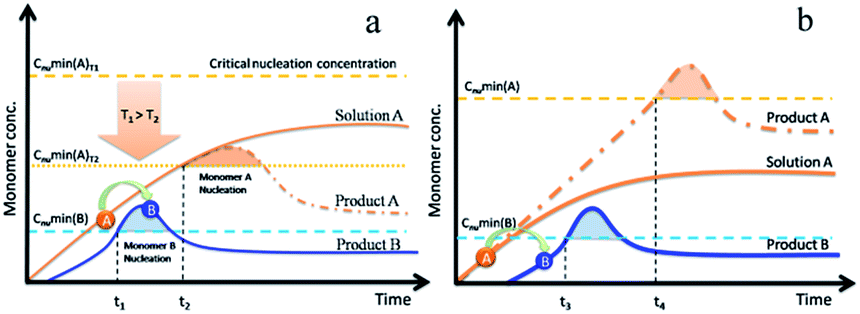 | ||
| Fig. 3 Illustration of the bi-monomer growth mechanism for producing impurities or mixtures. (a) Ending the reaction by rapidly decreasing reaction temperature from T1 to T2. Minimum nucleation concentration (dashed orange lines) of monomer A at T2 is under the concentration of residual monomer A (orange curves). (b) The reaction rate from A to B is too slow, which leads to an increase in the concentration of monomer A above its minimum nucleation concentration with a sufficient reaction time (t3 < t4). | ||
We have verified these two hypotheses by experiments in this study [ESI, Section 2†]. For the first situation, prolonging the reaction time contributes towards the yield of pure product B. However, this is useless for the second situation and we must alter the kinetic conditions.
Kvashnina28 and Gregory29 verified the uranium chemical state in mixed-valence uranium oxides by using high energy resolution fluorescence detection X-ray absorption near edge spectroscopy (HERFD-XANES) at the uranium M4 edge. Research results indicated U(VI)–U(V) in U3O8 and U(V)–U(VI) in U3O7 and U4O9. They did not observe a significant U(VI) contribution in U4O9 or a U(IV) contribution in U3O8.
In this study, the final products include UO3·H2O, U3O8, UO2, and U4O9, simply synthesized from raw uranyl(IV) acetate hydrate (UAH). Thus, the following equation represents the chemical reduction route:
| Precursors → U(VI) → U(V) → U(IV) | (1) |
The precursor is thought to be liquid uranyl (UO22+), which is characterized by high solubility, or the solid U2(NH3)O3H2O when significant amounts of ammonia were added. Monomers may exist in the form of atoms, monomers, hydrate compounds or clusters. The solubility of various uranium compounds is different and the solubility of UO3·xH2O is much higher than UO2·xH2O.30–32 Despite that, no other information on the solubility of U(V) exists. We assume that the critical solubility of U2O5 is between that of UO3 and UO2. This is insignificant to understand the mechanisms that induce multi-monomer growth.
The composition is usually controlled by thermodynamics. When the precursor decomposed into the U(VI) monomer, the redox reaction did not occur. There was only one type of monomer, occurring as U(VI), that resulted in the production of UO3·H2O (Fig. 4a). We can use the LaMer theory to understand this situation. With an appropriate reducing condition, the monomer U(VI) was reduced to a U(V) monomer but the continuous reaction was limited by thermodynamics because there were two types of monomers, i.e. U(VI) and U(V), in the system. The U(V) monomer increased with time while the U(VI) monomer remained below its minimum nucleation concentration due to depletion during the redox reaction. Theoretically, this scenario may yield mono-valence U2O5 (orthorhombic) nanocrystals, while the final observed product was U3O8, which has an orthorhombic structure similar to that of U2O5. U2O5 is a metastable phase and is only observed under pressure. The monomer U(VI) aggregated, nucleated and grew together with U(V), which resulted in the formation of a stable U3O8 phase (Fig. 4b).
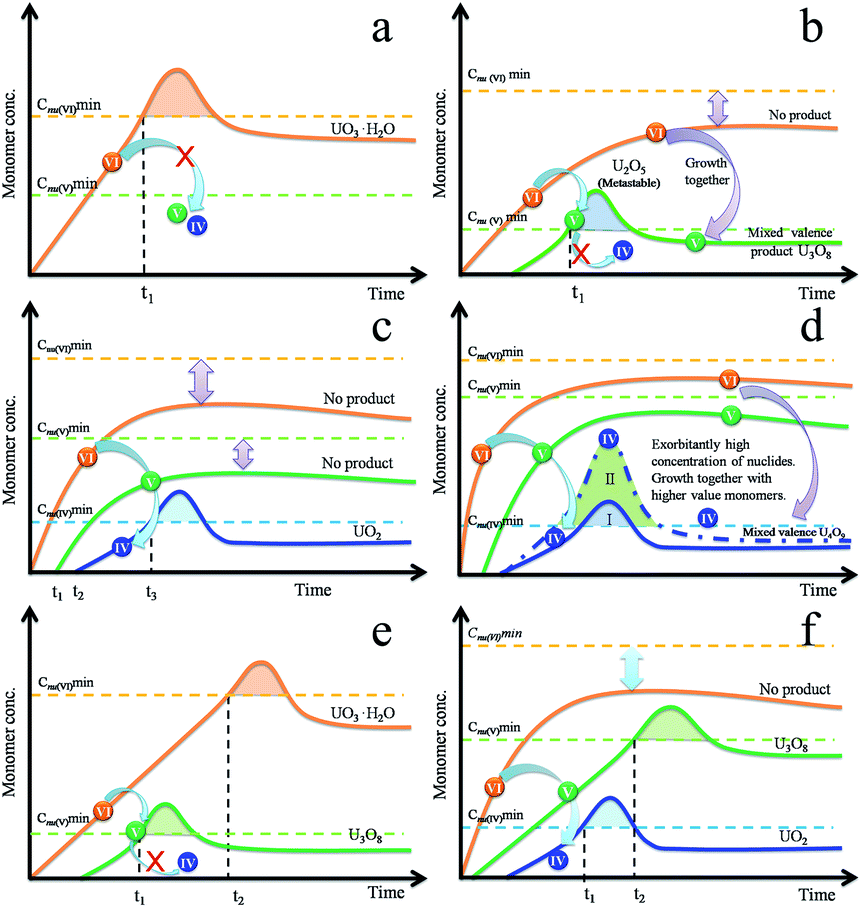 | ||
| Fig. 4 Illustration of the multi-monomer growth mechanism that produces pure (a–d) and mixed (e and f) uranium oxide nanocrystals. | ||
UO2 was obtained when the reducing efficiency was enhanced and the U(IV) monomer was produced. U(IV) and U(V) also remained below their minimum nucleation concentration as in Fig. 4c. However, the formation of U4O9 was slightly different. The nucleation stage still depends on the concentration of U(IV) monomer as UO2. With a higher rate of reduction, the amount of nuclides increases, which results in the minimization of the particle volume with an equivalent amount of raw materials. The amount of nuclides increases significantly (Fig. 4d-II) with increasing supersaturation degree, which results in shortening of the diffusion distance of the monomer in solution. Due to the high concentration of particles, the residual U(V)/U(VI) monomers were covered into nanocrystals, then formed a stable phase U4O9. As a result of rapid growth, U4O9 formed as a sphere, not a polyhedron (Fig. S2f†).
If the decomposition rate is much faster than the reduction rate, the U(VI) and U(V) monomer concentration continued to increase even after being depleted during the simultaneous redox reaction. As time progressed, each monomer approaches their critical nucleation concentration. As a result, this reduced a fresh round of nucleation and growth for the new phases (Fig. 4e and f). In this situation, prolonging the reaction time is useless when attempting to obtain pure target products.
Fortunately, we have verified all of the situations described above with experiments presented in this study (ESI, Section 2†). The morphologies can be tuned by adjusting the experimental conditions in a minor range after composition was under control; for example, by changing the amount of ammonia, concentration of UAH and the ratio of acetone to DI water (ESI, Section 1 and 2†). On the one hand, the monomer generation rate, which might affect the amount of nuclides, may be affected by the acetone concentration, the initial uranyl amount and the pH value which influences the nucleation and growth kinetics. On the other hand, the ammonia might control the shape via surface adsorption. The shape of UO3·H2O clearly changed with the addition of ammonia (Fig. S2†). In addition, the organic impurities, which also potentially affected the uranium oxide nanocrystals morphology, increased with pH and reaction time.
Controlling the nanocrystal shape during synthesis is both simple chemistry and complex physics.14 As previously mentioned, controlling both the composition and morphology via a multi-monomer growth mechanism is quite complex. The question, therefore, is how to achieve this kind of control. In this study, we found a significant reaction system to attempt to answer this question. Due to the fact that acetone has no ability to act as a reducing agent, it is difficult to explain the synthesis of nanomaterials solely based on acetone as the reducing agent. The question, therefore, becomes how did acetone contribute to reduction at different pH values.
The gas chromatography mass spectrum (GC-MS) results showed that the products in solution phase include diacetone alcohol (DAA), mesityl oxide (MO), isomesityl oxide (IMO), 2,4-dimethyl furan, phorone, trimethyl benzene (TMB) etc.
We summarized the main reaction pathways for acetone (Fig. S6†). The aldol addition reaction has been observed on various metal oxide surfaces including U3O8, UO2,33 TiO2,34,35 MgO,36 ZrO2, La2O3, SrO2, and CaO.37 Only route 5 and 6 are redox reactions,38 which indicates that DAA and MO potentially acted as the reducing agent. Other experiments verified the reducing efficiency of MO but the morphologies of the solid products were irregular. The GC-MS results revealed that the oxidation product of MO is TMB (Table S6.2†). The composition of the products changed when the MO concentration was altered.
Extended experiments indicate that acetone could only be transferred to DAA at 200 °C without the addition of other agents (Table S6.1†). MO was detected after the addition of UAH. The MO fraction increased with the addition of ammonia. The amounts of organic by-products increase at higher pH values (Table S7†). Idiss et al.24 reported that aldolization of acetone occurs on the surface of uranium oxide and other metal oxides. This indicates that a self-catalysis process occurs at some point in this reaction system.
The whole process can be summarized as follows. In the first stage, the addition of sufficient ammonia generated the precursor [U2(NH3)O6·H2O]. In the second stage, the precursor decomposed into a U(VI) monomer at high temperatures. The concentration of MO, which acted as an actual reducing agent, increased with time via acetone aldolization in basic conditions. This reaction rate was significantly affected by the pH value. The concentration of MO could have affected the reducing efficiency of the entire reaction system. The increased amount of ammonia promoted MO production, which acts as the actual reducing agent. Then, the U(VI) monomer was reduced to U(V) with a lower MO concentration and U(V) was reduced to U(IV) with a relatively high MO concentration. The MO concentration, which was affected by pH, acetone and uranyl concentration, could affect the redox rate. Thus, the complex physics for controlling both composition and morphology, was achieved easily with such a simple method.
In conclusion, we have developed a new oxidation–reduction reaction system with acetone, which is usually used as a purge agent or dissolvent. This reaction system is capable of controlling both the composition and morphology of uranium oxides micro/nanomaterials in the solution phase. We have presented two important mechanisms that control the composition of multi-value metal oxides. We developed an extended multi-monomer growth model based on LaMer's theory, which was used to understand the formation of mixed-value metal oxides and mixture products. More importantly, the self-catalysis mechanism allows us to regulate the complex processes that control different monomer transformations. The results of this study have great potential to be used in the construction of other multi-value elemental compounds or mixed-valence metal oxides.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support of Millions of Leading Engineering Talent from the first batch of the Ten Thousand Talent Program and the Science and Technology Foundation of China Academy of Engineering Physics (No. 2015B0302047).Notes and references
- Z. L. Wang, Adv. Mater., 2007, 19, 889–892 CrossRef CAS.
- G. J. Hutchings, C. S. Heneghan, I. D. Hudson and S. H. Taylor, Nature, 1996, 384, 341 CrossRef CAS.
- Z. T. Zhang, M. Konduru, S. Dai and S. H. Overbury, Chem. Commun., 2002, 2406–2407, 10.1039/b204987b.
- X. Peng, Adv. Mater., 2003, 15, 459–463 CrossRef CAS.
- V. Biju, T. Itoh, A. Anas, A. Sujith and M. Ishikawa, Anal. Bioanal. Chem., 2008, 391, 2469–2495 CrossRef CAS PubMed.
- M. Zhu, J. Zhou, Z. Hu, H. Qin and X. Peng, ACS Photonics, 2018, 5, 4139–4146 CrossRef CAS.
- H. Qin, R. Meng, N. Wang and X. Peng, Adv. Mater., 2017, 29, 1606923 CrossRef PubMed.
- J. Zhou, M. Zhu, R. Meng, H. Qin and X. Peng, J. Am. Chem. Soc., 2017, 139, 16556–16567 CrossRef CAS PubMed.
- Q. A. Pankhurst, N. T. K. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- L. Xu, Y. Liu, Z. Chen, W. Li, Y. Liu, L. Wang, Y. Liu, X. Wu, Y. Ji, Y. Zhao, L. Ma, Y. Shao and C. Chen, Nano Lett., 2012, 12, 2003–2012 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- Z. Wu, S. Yang and W. Wu, Nanoscale, 2016, 8, 1237–1259 RSC.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2008, 48, 60–103 CrossRef PubMed.
- C. Pu, H. Qin, Y. Gao, J. Zhou, P. Wang and X. Peng, J. Am. Chem. Soc., 2017, 139, 3302–3311 CrossRef CAS PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- J. Ji, Y. Huang, J. Yin, X. Zhao, X. Cheng, S. He, X. Li, J. He and J. Liu, ACS Appl. Nano Mater., 2018, 1, 3935–3944 CrossRef CAS.
- Q. Yan, Y. Mao, X. Zhou, J. Liang, M. Ye and S. Peng, J. Nucl. Mater., 2018, 512, 417–422 CrossRef CAS.
- K. Chen and D. Xue, CrystEngComm, 2012, 14, 8068–8075 RSC.
- M. J. Monreal and P. L. Diaconescu, Nat. Chem., 2010, 2, 424 CrossRef CAS PubMed.
- C. Young, J. Petrosky, J. M. Mann, E. M. Hunt, D. Turner and T. Kelly, Phys. Status Solidi RRL, 2016, 10, 687–690 CrossRef CAS.
- S. H. Taylor and S. R. O'Leary, Appl. Catal., B, 2000, 25, 137–149 CrossRef CAS.
- S. H. Taylor, C. S. Heneghan, G. J. Hutchings and I. D. Hudson, Catal. Today, 2000, 59, 249–259 CrossRef CAS.
- H. Idriss, Surf. Sci. Rep., 2010, 65, 67–109 CrossRef CAS.
- S. Krivovichev, P. Burns and I. Tananaev, Structural chemistry of inorganic actinide compounds, Elsevier, 2006 Search PubMed.
- R. Zhao, L. Wang, Z.-J. Gu, L.-Y. Yuan, C.-L. Xiao, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, CrystEngComm, 2014, 16, 2645–2651 RSC.
- S. Hu, H. Li, H. Liu, P. He and X. Wang, Small, 2015, 11, 2624–2630 CrossRef CAS PubMed.
- K. O. Kvashnina, S. M. Butorin, P. Martin and P. Glatzel, Phys. Rev. Lett., 2013, 111, 253002 CrossRef CAS PubMed.
- G. Leinders, R. Bes, J. Pakarinen, K. Kvashnina and M. Verwerft, Inorg. Chem., 2017, 56, 6784–6787 CrossRef CAS PubMed.
- V. Neck and J. Kim, Radiochim. Acta, 2001, 89, 1–16 CAS.
- T. Fanghänel and V. Neck, Pure Appl. Chem., 2002, 74, 1895–1907 Search PubMed.
- R. Jovani-Abril, M. Gibilaro, A. Janßen, R. Eloirdi, J. Somers, J. Spino and R. Malmbeck, J. Nucl. Mater., 2016, 477, 298–304 CrossRef CAS.
- H. Madhavaram, P. Buchanan and H. Idriss, J. Vac. Sci. Technol., A, 1997, 15, 1685–1691 CrossRef CAS.
- W. Choi, J. Y. Ko, H. Park and J. S. Chung, Appl. Catal., B, 2001, 31, 209–220 CrossRef CAS.
- W. Xu and D. Raftery, J. Catal., 2001, 204, 110–117 CrossRef CAS.
- V. K. Díez, C. R. Apesteguía and J. I. Di Cosimo, J. Catal., 2006, 240, 235–244 CrossRef.
- G. Zhang, H. Hattori and K. Tanabe, Appl. Catal., 1988, 36, 189–197 CrossRef CAS.
- C. O. Veloso, J. L. F. Monteiro and E. F. Sousa-Aguiar, in Studies in Surface Science and Catalysis, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, vol. 84, pp. 1913–1920 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: More details about experimental conditions, characterization and analysis are listed in ESI. See DOI: 10.1039/c8na00392k |
| This journal is © The Royal Society of Chemistry 2019 |