
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of amine functional groups on silica nanoparticles: a multi-method approach†
Ying
Sun
,
Filip
Kunc
,
Vinod
Balhara
,
Brian
Coleman
,
Oltion
Kodra
,
Mohammad
Raza
,
Maohui
Chen
,
Andreas
Brinkmann
 ,
Gregory P.
Lopinski
and
Linda J.
Johnston
*
,
Gregory P.
Lopinski
and
Linda J.
Johnston
*
National Research Council Canada, Ottawa, ON, Canada K1A 0R6. E-mail: Linda.Johnston@nrc-cnrc.gc.ca
First published on 22nd February 2019
Abstract
Surface chemistry is an important factor for quality control during production of nanomaterials and for controlling their behavior in applications and when released into the environment. Here we report a comparison of four methods for quantifying amine functional groups on silica nanoparticles (NPs). Two colorimetric assays are examined, ninhydrin and 4-nitrobenzaldehyde, which are convenient for routine analysis and report on reagent accessible amines. Results from the study of a range of commercial NPs with different sizes and surface loadings show that the assays account for 50–100% of the total amine content, as determined by dissolution of NPs under basic conditions and quantification by solution-state 1H NMR. To validate the surface quantification by the colorimetric assays, the NPs are modified with a trifluoromethylated benzaldehyde probe to enhance sensitivity for quantitative 19F solid state NMR and X-ray photoelectron spectroscopy (XPS). Good agreement between the assays and the determination from solid-state NMR is reinforced by elemental ratios from XPS, which indicate that in most cases the difference between total and accessible amine content reflects amines that are outside the depth probed by XPS. Overall the combined results serve to validate the relatively simple colorimetric assays and indicate that the reactions are efficient at quantifying surface amines, by contrast to some other covalent modifications that have been employed for functional group quantification.
Introduction
Surface functional groups play a key role in determining the properties of nanomaterials and controlling their interactions with other materials. The surface of nanomaterials is frequently functionalized, either to improve the stability or enhance the compatibility with other materials for nano-enabled products or to facilitate targeting and modulate protein corona formation for biomedical applications. The ability to adequately control surface chemistry is important to ensure quality control during production of the material and to determine the fate of the material when incorporated in devices, when used for biological applications or when released to the environment. A wide range of methods is available for the identification and quantification of surface functional groups on nanomaterials.1–17 They vary in their sensitivity, applicability to specific classes of nanomaterials, and time and resource requirements. Methods such as NMR, thermogravimetric analysis (TGA), and elemental analysis measure total functional group content. X-ray photoelectron spectroscopy (XPS) identifies atomic composition and has a depth penetration on the order of ∼10 nm. Titration methods are selective for surface accessible acidic or basic groups. Labelling strategies coupled with colorimetric and fluorescence assays, solid state NMR or XPS have also been widely employed to provide an assessment of reagent-accessible groups which is more relevant for some applications. Despite the wide range of methods, surface group quantification is still challenging; in some cases only qualitative identification of the surface groups is carried out and it is still uncommon to use multiple methods and validated protocols.2,18,19 Depending on the ultimate application of the nanomaterial, it may be important to be able to detect and identify the surface group and to understand the number of groups on the surface and their overall spatial distribution.We and others have recently reported a quantitative 1H NMR (qNMR) method to measure total amine content after dissolution of the silica matrix for a range of commercial and in-house synthesized aminated silica nanoparticles (NPs).3,20 Our initial study focused on silica due to its ease of synthesis and widespread use for a variety of applications, and amine functionalization because it is one of the most common types of silica modification. The qNMR results were compared to a simple colorimetric assay using ninhydrin for selected samples, thus providing a comparison of the total and surface accessible amine content.20 In addition to providing a detailed protocol that illustrates the factors that require optimization for the dissolution/qNMR assay, this work demonstrated that the amine content from commercially sourced silicas was variable, ranging from a small fraction of the estimated monolayer coverage to well in excess of a monolayer. This variability is probably due in part to the use of 3-aminopropyltriethoxysilane (APTES) for surface functionalization. Silica functionalization by reaction of aminoalkylalkoxy silanes with silanols is widely used but known to suffer from challenges associated with polymerization of the initial silane in solution, possible formation of heterogeneous multilayers on the surface in the presence of excess silane and the lack of stability of the aminated surfaces.21–23
In the present study we compare the results from two colorimetric assays for quantification of accessible surface amines to results obtained by labeling amines with trifluoromethylated probes that provide high sensitivity for XPS and quantitative solid state 19F NMR.2,5,19,24,25 The two colorimetric assays are based on the reaction of ninhydrin with primary amines to generate a dye that is released from the surface and quantified in the supernatant and the reaction of an aldehyde with surface amines to generate an imine product that can be cleaved for quantification after isolation of the NPs. These two assays may differ in their ability to detect surface-accessible amines, particularly at high surface coverage where steric effects are expected to become important. The use of fluorinated probes takes advantage of the high gyromagnetic ratio and natural abundance of 19F to allow quantitative ssNMR of intact silica NPs and also provides a sensitive probe for XPS measurements. The results from these four methods are compared to those obtained by 1H qNMR on dissolved silica for selected samples. This comprehensive comparison provides useful information on the advantages and limitations of the various methods and guidance as to the appropriate choice of technique(s) depending on the level of accuracy and uncertainty required.
Materials and methods
Materials
Multiple batches of 20, 50, 80, 100, and 120 nm amine-modified silica NPs were purchased from NanoComposix USA as ethanol suspensions (≈10 mg mL−1) or dry powders; the NPs were prepared by the Stöber process and functionalized with the 3-aminopropyl group. Ethanol suspensions of these aminated NPs have been shown previously to be stable towards loss of surface groups.20 The mass fraction of silica in NP suspensions was determined gravimetrically by drying at 125 °C to constant mass. Dry NPs were dispersed by preparing a 10 mg mL−1 ethanol or methanol suspension and sonicating for 20–25 min. Characterization data (as supplied by the manufacturer) for representative samples are summarized in Table S1.† 4-Nitrobenzaldehyde (4-NBA), 3,5-bistrifluoromethylbenzaldehyde (BTFBA), deuterium oxide 99.9%, sodium deuteroxide (10 M in D2O), trifluoroacetic anhydride and TraceCERT® maleic acid were purchased from Sigma Aldrich (Canada). 3,5-Bis-(trifluoromethyl)-benzoic acid (BTFMBA), a certified reference material, was purchased from the National Metrology Institute of Japan. Deionized water (18.2 MΩ cm, MilliQ) was used throughout. Centrifugation of NP suspensions, where necessary, was carried out at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm.
000 rpm.
Ninhydrin assay
A 500 μL aliquot of silica NP dispersion (containing ∼5 mg of NPs) was pipetted into a microcentrifuge tube and centrifuged for 20 min. Ethanol (200 μL) was removed and replaced with 200 μL of water and the sample was sonicated in a bath sonicator to redisperse the NPs. In some cases the redispersed suspensions were measured by DLS to ensure that centrifugation had not caused agglomeration of the NPs; an example is shown in Table S2.† The sample was transferred to a glass test tube and 100 μL KCN and 75 μL of phenol (Kaiser Kit, Anaspec) were added sequentially. The tube was vortexed and an excess (75 μL) of ninhydrin reagent was added. The tube was stoppered with a glass marble and placed in a preheated water bath at 97 °C for 10 min. The tube was removed, cooled in ice water and poured into a 5 mL volumetric flask. The tube was rinsed with 60% EtOH which was added to the volumetric flask to give a final volume of 5 mL. A 200 μL aliquot was placed in a centrifuge tube, diluted to a final volume of 1 mL in 60% EtOH and centrifuged for 20 min. An aliquot of supernatant was removed and its absorbance was measured immediately at 570 nm in a 1 cm path length cell using a UV-vis spectrometer with dilutions as needed to ensure that the optical density was below 0.8. The assay was calibrated using the same procedure for a minimum of 5 concentrations of octylamine. The optical density of the supernatant did not change over a period of 80 min, indicating that dye was stable over the time needed for this experiment.4-Nitrobenzaldehyde assay
A 500 μL aliquot of silica NP dispersion in ethanol was pipetted into a microcentrifuge tube and centrifuged for 20 min. The supernatant was removed and the NPs were redispersed by bath sonication (for at least 5 min) in 1 mL methanol containing an excess of 4-NBA (∼100 fold, based on the estimated amine content corresponding to full monolayer coverage) and then reacted by heating overnight at 45 °C at 1100 rpm in a Ther-Mix heated mixer (Vitl Life Science Solutions). The particles were purified by centrifugation for 20 min, after which the supernatant was discarded and the particles were redispersed with fresh methanol to remove unreacted 4-NBA. The centrifuge/redisperse steps were repeated a total of 4 times. The NPs were isolated by centrifugation/removal of the supernatant, re-dispersed in hydrolysis solution, (1:1 methanol/H2O) and incubated at 45 °C overnight. Two additional rounds of hydrolysis were performed for 1 h each. The three hydrolytic washes were saved and diluted for measurement of optical density at 275 nm. Monitoring of individual hydrolysis solutions indicated that 4-NBA was removed quantitatively with three hydrolysis steps. Calibration curves were prepared using 4-NBA in hydrolysis solution.
19F solid state NMR
BTFBA-conjugated silica NPs were prepared by adding 5 mL of 10 mg mL−1 ethanol dispersion to a scintillation vial with a magnetic stir bar and adding an excess of BTFBA (∼100 fold). The solution was stirred overnight at room temperature. NPs were purified by four centrifugation/supernatant removal, ethanol addition and redispersion cycles. To prepare dry particles for solid-state NMR measurement, the dispersions were centrifuged for 20 min, the supernatants were carefully removed, and residual solvent was removed by freeze-drying overnight. Trifluoromethylaminated silica NPs were prepared by mixing 200 mg aminated silica with 5 mL trifluoroacetic anhydride and 2 mL toluene and stirring at room temperature overnight in a closed round bottom flask. Excess solvent was removed in a stream of nitrogen gas and the silica was dried at 100 °C overnight. The NPs were further purified by washes with cold methanol and acetone, followed by drying overnight at 100 °C.A Bruker 200 MHz Avance solid state NMR spectrometer equipped with a 3.2 mm Bruker double resonance probehead and magic-angle spinning at a frequency of 18 kHz were used for all experiments. Silica NPs (∼8 mg) were placed in the sample rotor. The probe was tuned to the 19F Larmor frequency given by 188.7 MHz prior to every measurement. The published protocol for 19F quantitative solid-state NMR26 was employed, specifically metrological traceability was achieved by using 3,5-bis(trifluoromethyl)benzoic acid (BTFMBA) as external calibrant. Furthermore, we employed the ERETIC (Electronic REference To access In vivo Concentrations) and EASY (Elimination of Artifacts in NMR Spectroscopy) methods.27,28 A relaxation delay of 20.89 s and a spectral width of 100 kHz were used for all experiments. We co-added 3328 individual transients with 4096 complex data points and identical receiver gain. The 90° pulse was optimized prior to each experiment, typically found between 2.3 and 2.5 μs.
qNMR of dissolved silica NPs
Silica NPs were dissolved by shaking (1200 rpm) in a heated mixer at 45 °C in 0.4 M NaOD for 3 h. Internal calibrant (maleic acid) was added and qNMR was performed at 20 °C (±1 °C) with a Bruker Avance III 400 MHz spectrometer equipped with a 5 mm BBFO probe. Full details on the optimized silica dissolution and qNMR procedure have been reported.20XPS
The samples for XPS were prepared by spin coating dispersions containing NPs onto cleaned Au substrates (Arrandee, Germany). Substrates (12 × 12 mm) were rinsed with dichloromethane, isopropyl alcohol and water, and then cleaned with Piranha (3:1, H2SO4:H2O2) for 2 min and rinsed extensively with water. The substrates were dried with N2 and ozone treated for 30 min. 5 mg of silica NPs were dispersed in 1 mL of methanol via bath sonication for 30 min and then briefly bath sonicated immediately prior to spin-coating onto a clean Au substrate. Each substrate was spin-coated (12 s at 500 rpm and 2 min at 2000 rpm) 3 times with 50 μL volumes. Spectra were obtained using a Kratos Analytical Axis Ultra DLD spectrometer with monochromatized Al Kα X-rays. Each sample was measured at three distinct points. At each point survey scans were obtained in order to determine the relative atomic composition of the sample and detect any impurities that may be present. High resolution spectra in regions corresponding to strong core level transitions for the major elements present on these samples (Au 4f, Si 2p, O 1s, C 1s, N 1s and F 1s) were also acquired. These high resolution spectra were used for more accurate quantification of the sample composition and also provided additional information on the chemical state of the elements. The intensity of the Au signal was compared for substrates before and after spin coating the NPs. An attenuation in Au intensity of 80% or greater was observed after NP deposition, consistent with at least monolayer coverage. Data analysis was carried out with the CasaXPS analysis software using relative sensitivity factors for the Kratos instrument. For quantification the integrated area under each peak was determined, after subtracting the background. To subtract the background, the Shirley method was used for Au 4f, Si 2p and O 1s regions, whereas a linear background was chosen for the N 1s and F 1s peaks.
Results
Colorimetric assays
The ninhydrin assay has been widely used for both qualitative and quantitative determination of amines in peptides and proteins and has been applied in a few cases for amines on nanomaterials and thin films, including silica NPs and planar silica surfaces.29–35 The reaction of primary amines with ninhydrin occurs by an initial nucleophilic displacement of a ninhydrin hydroxyl group by amine, followed by a complex mechanism that generates a colored product known as Ruhemann's purple (Scheme 1).29 The generation of free dye in solution for colorimetric analysis has the advantage of being less likely to suffer from steric effects when quantifying high surface coverages of amine. Initial studies used a literature procedure in ethanol solvent and the reported extinction coefficient for the product dye to assess suitability of the assay.32 The results for multiple replicates of the assay for 50 and 120 nm aminated silica NPs showed good reproducibility, with measured amine contents that were significantly in excess of an estimated monolayer coverage, based on the mean TEM diameter and 4 aminopropyl siloxanes/nm2 (Table S3†).36 However, quantification of several different batches for three NP sizes showed a large batch-to-batch variability in amine content, with most samples having only fractional monolayer coverage (Table S4†).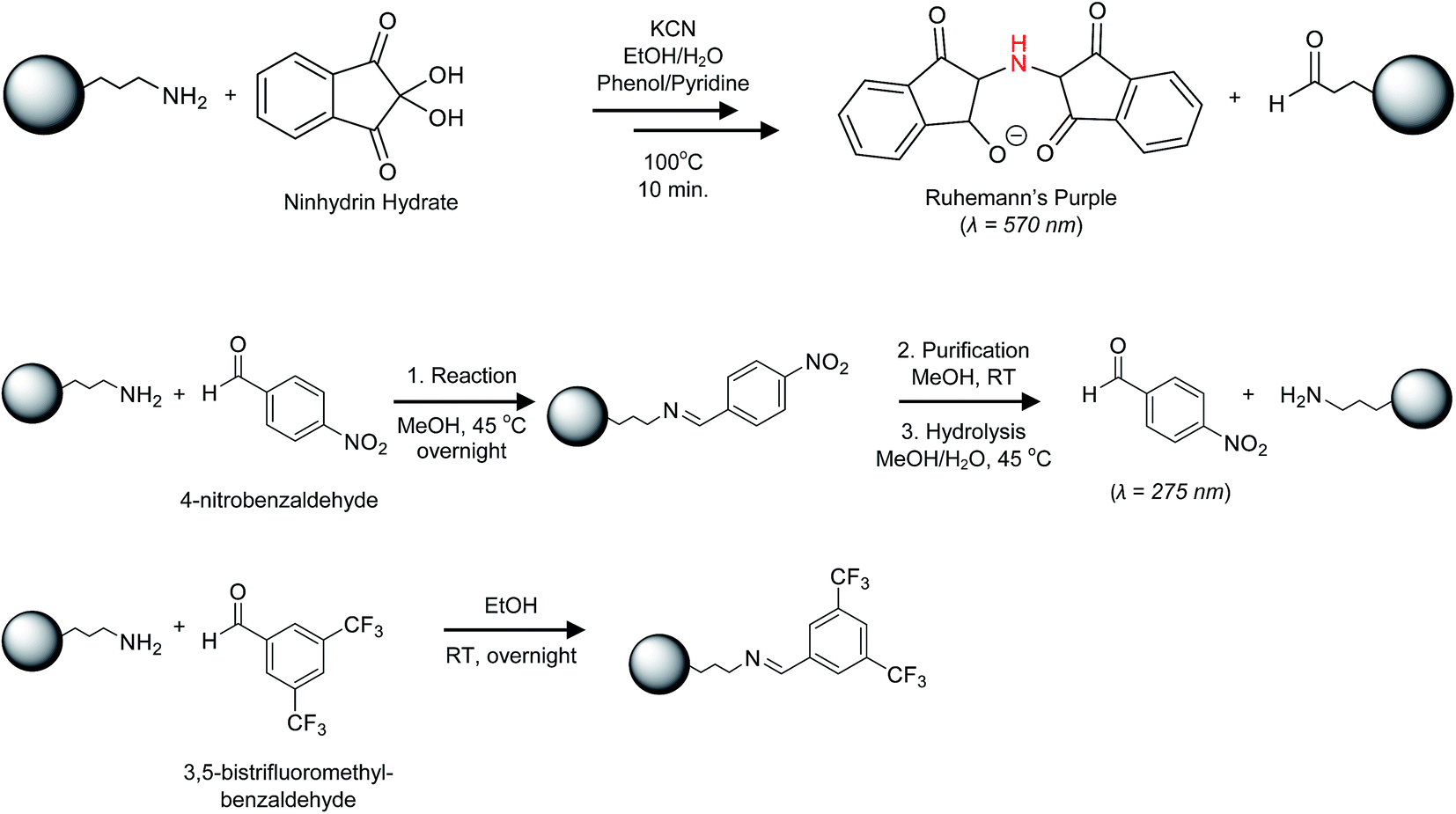 | ||
| Scheme 1 Reactions of ninhydrin, 4-NBA and BTFBA with aminated silica. | ||
Literature examples have reported a number of variations on the procedure for the ninhydrin reaction;29–35 furthermore, the product dye is unstable under some conditions and its absorption maximum and extinction coefficient vary with solvent. Therefore, reaction times and solvent (ethanol vs. aqueous ethanol mixtures) were varied for reactions of an excess of ninhydrin with both octylamine and 50 nm aminated silica NPs in order to select the optimum assay conditions (60% aqueous ethanol and 10 min reaction time at 100 °C; Fig. S1 and S2†). The stability of the dye under these reaction conditions was verified by absorbance measurements (see Materials and Methods). The ninhydrin reaction with octylamine and leucine under the optimized reaction conditions was then used to calibrate the product yield (Fig. S3 and S4†). APTES was also tested but gave a lower and more variable product yield, possibly due to its tendency to polymerize. The data for octylamine was used for calibration purposes since this stable and non-volatile primary amine is a better model for the propylamine groups on the silica surface. The estimated amine contents for multiple batches of aminated silica NPs with nominal sizes of 20, 50, 80, 100 and 120 nm using the optimized procedure are summarized in Table 1.
| Sample | Ninhydrin assaya μmol g−1 | 4-Nitrobenzaldehyde assaya μmol g−1 | qNMRb μmol g−1 | Accessible/total aminec | Fractional monolayer coveraged |
|---|---|---|---|---|---|
| a The assay results represent an average of at least 3 and typically 6 or 9 replicate experiments. b Data for B2 and B3 from ref. 20. c Surface-accessible amine from the 4-NBA assay and total amine from qNMR. d Fractional monolayer coverage = total amine from qNMR/estimated monolayer coverage. Estimated monolayer coverages in μmol g−1 assuming 4 aminopropyl siloxanes/nm2 and using the surface area calculated from mean TEM diameter (see Table S1 for details) are 920 (20 nm), 360 (50 nm), 230 (80 nm), 180 (100 nm) and 150 (120 nm). | |||||
| 20 nm, B2 | 320 ± 20 | 459 ± 5 | 655 ± 3 | 0.70 | 0.71 |
| 50 nm, B1 | 121 ± 3 | 215 ± 2 | |||
| 50 nm, B2 | 47 ± 1 | 66 ± 12 | 64 ± 1 | 1.03 | 0.18 |
| 50 nm, B3 | 48 ± 8 | 53 ± 3 | 57 ± 1 | 0.93 | 0.16 |
| 80 nm, B1 | 18 ± 2 | ||||
| 80 nm, B2 | 103 ± 4 | 132 ± 15 | 145 ± 3 | 0.91 | 0.63 |
| 80 nm, B3 | 76 ± 2 | 113 ± 4 | 130 ± 2 | 0.87 | 0.56 |
| 100 nm, B1 | 91 ± 5 | ||||
| 100 nm, B2 | 152 ± 13 | 169 ± 15 | 186 ± 1 | 0.91 | 1.03 |
| 100 nm, B3 | 98 ± 2 | 117 ± 1 | 180 ± 2 | 0.64 | 1.02 |
| 120 nm, B2 | 117 ± 4 | 122 ± 2 | 179 ± 3 | 0.68 | 1.19 |
Reaction with 4-nitrobenzaldehyde to generate an imine, followed by isolation of the NPs and hydrolysis of the imine to regenerate the aldehyde for UV-vis quantification (Scheme 1) was examined as an alternate to the ninhydrin assay.13,37–39 This assay has been used previously to assess amine content of mesoporous silicas.13Table 1 and Fig. S5 and S6† provide the results for amine quantification using the 4-NBA assay for most of the same sizes and batches of silica that were examined by the ninhydrin assay. The measured mean amine content was higher using the 4-NBA assay for all samples, although in several cases the values were within the range defined by the standard deviations for the two assays. The similar or higher accessible amine content suggests that steric crowding on the silica surface is not a factor for the 4-NBA reaction. Quantification limits of 0.8 and 1.2 μmol g−1 were estimated for the ninhydrin and 4-NBA assays (see ESI† for details) using the optimized assay conditions.
For comparison to the colorimetric assays, the total amine content was measured by 1H qNMR after dissolution of silica NPs in strong base. A quantification limit of ∼10 μmol g−1 for 32 transients was estimated previously for this method, approximately 10 times higher than those for the colorimetric assays.20 The time required for the qNMR measurement (20 min) is similar to that for sample dilution and absorbance measurements for the assays. The sample preparation times for qNMR and the assays are comparable, although the silica dissolution and assay reaction times are different: 3 h, 10 min and overnight for qNMR, ninhydrin and 4-NBA, respectively. A representative qNMR spectrum and the data for total amine content are provided in Fig. S7† and Table 1, respectively.20 Note that residual ethanol is typically observed in the NMR spectra, even after vacuum drying, indicating that either surface adsorbed ethanol is not removed or that there is some ethanol trapped in interior pores.40 With one exception, the total amine content is higher than the amount of accessible amines reported by either ninhydrin or 4-NBA assays. The ratio of accessible (4-NBA) to total amines (qNMR) varies from 0.64 to ∼1 (Table 1). The two samples for which the accessible to total amine ratio is ∼1 are those which have the lowest amine coverage, expressed as the fraction of estimated full monolayer coverage. Although this observation is consistent with quantitative reaction of amines with 4-NBA at low surface coverage, there is no obvious trend for most of the other samples. For example, the 100 and 120 nm NPs all have monolayer or slightly higher surface coverages but the accessible to total amine ratios vary from 0.64 to 0.98; by contrast the 80 nm NPs have slightly more than half monolayer coverage, but the accessible/total amine ratios are approximately 0.9.
19F solid state NMR
Since the 1H qNMR method is destructive, requiring dissolution of the silica matrix, we explored the use of ssNMR for quantification of silica functional groups. Quantitative NMR in the solid state using heteronuclei such as 31P and 13C to achieve the necessary spectral resolution has limited sensitivity and is time-consuming. Therefore, we used a fluorine labelling strategy to take advantage of the high sensitivity and high natural abundance of 19F.2,24 The first approach involved trifluoracetylation of aminated silica NPs with trifluoroacetic anhydride (TFAA). The representative spectra of the reaction product after purification shown in Fig. 1 for 50 nm and 100 nm NPs have a resonance at −78 ppm that we assign to the CF3 group in the product amide. However, there is an additional shoulder at −75 ppm that appears in most samples and cannot be removed by additional purification of the silica NPs. Control experiments in which bare silica was subjected to the same reaction conditions using either TFAA or trifluoroacetic acid (a reaction product) did not produce signals at the same chemical shift as the shoulder detected for the aminated silica NPs. It is possible that the additional signal reflects a second amine-derived product such as the 2:1 imine adduct, which has been proposed elsewhere,41 that interferes with quantification based on the signal at −78 ppm. Nevertheless in the absence of confirmation of the source of the additional signal, we opted to test an alternate labeling approach.
 | ||
| Fig. 1 19F ssNMR of trifluoroacetylated silica NPs. | ||
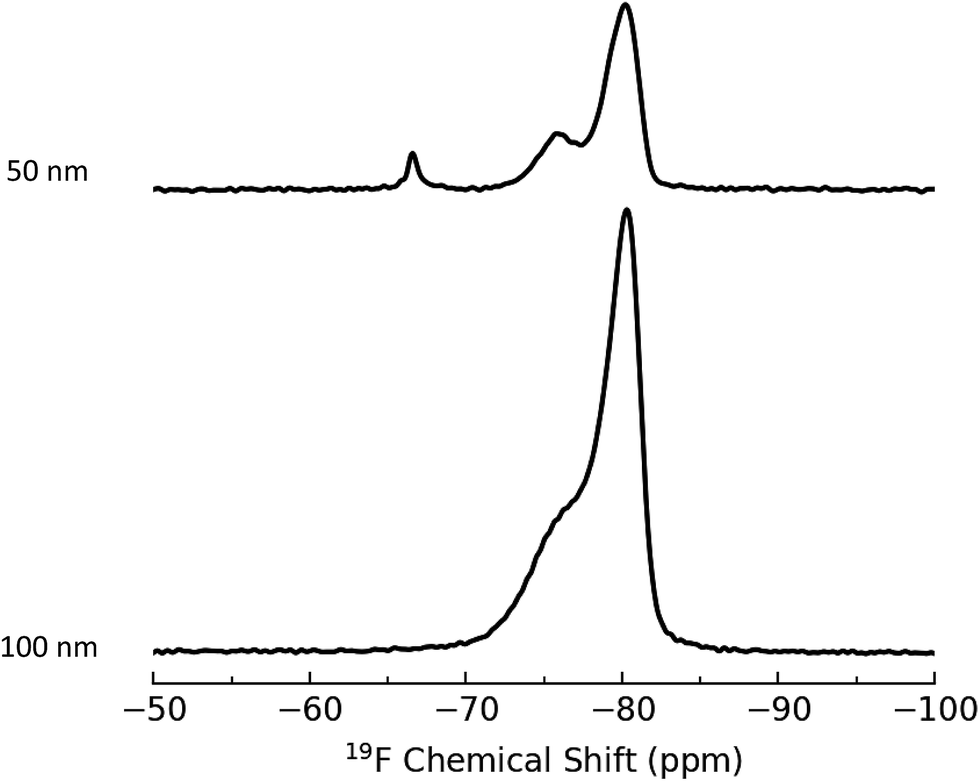
The second method employed for introduction of a fluorinated probe is based on imine formation, similar to the 4-NBA colorimetric assay described above and a 19F NMR assay developed to quantify primary amines in polymers.41 This approach used 3,5-bis(trifluoromethyl)benzaldehyde, BTFBA (Scheme 1), which has the advantage of introducing 2 trifluoromethyl groups for each amine moiety, increasing the sensitivity of the assay. Aminated silica NPs were reacted with BTFBA and purified. Representative ssNMR spectra for BTFBA-modified silica NPs are shown in Fig. 2. The results indicate conversion to a single trifluoromethylated product (−68 ppm) although there are additional signals between −76 and −82 ppm that vary in relative intensity for the various samples. A control experiment in which 80 nm bare silica was subjected to the BTFBA reaction/purification conditions resulted in the same signals between −76 and −82 ppm (Fig. 2) and the 19F solution NMR of BTFBA also showed signals due to an impurity at lower field. This confirms that the signal at −76 is due to an impurity in the starting material; the sharp signal is assigned to an impurity that is not removed during the freeze drying process. Results for two replicate experiments for BTFBA-labeling of each of four aminated silica NPs for quantification by 19F qNMR are summarized in Table 2. For the 80 and 100 nm NPs, the amount of amine detected is similar to the value obtained using the 4-NBA colorimetric assay, whereas 19F NMR gives an amine content that is ∼20% lower than that measured using 4-NBA for the 20 and 50 nm NPs. A quantification limit of 32 μmol g−1 was estimated for 32 transients in order to obtain a signal to noise of 10. For the 3328 transients acquired in this study the limit of quantification reduces to 3 μmol g−1.
 | ||
| Fig. 2 19F ssNMR of 80 and 100 nm BTFBA-labelled silica NPs. The control shows the spectrum of non-functionalized 80 nm NPs subjected to the BTFBA reaction conditions. | ||
 | ||
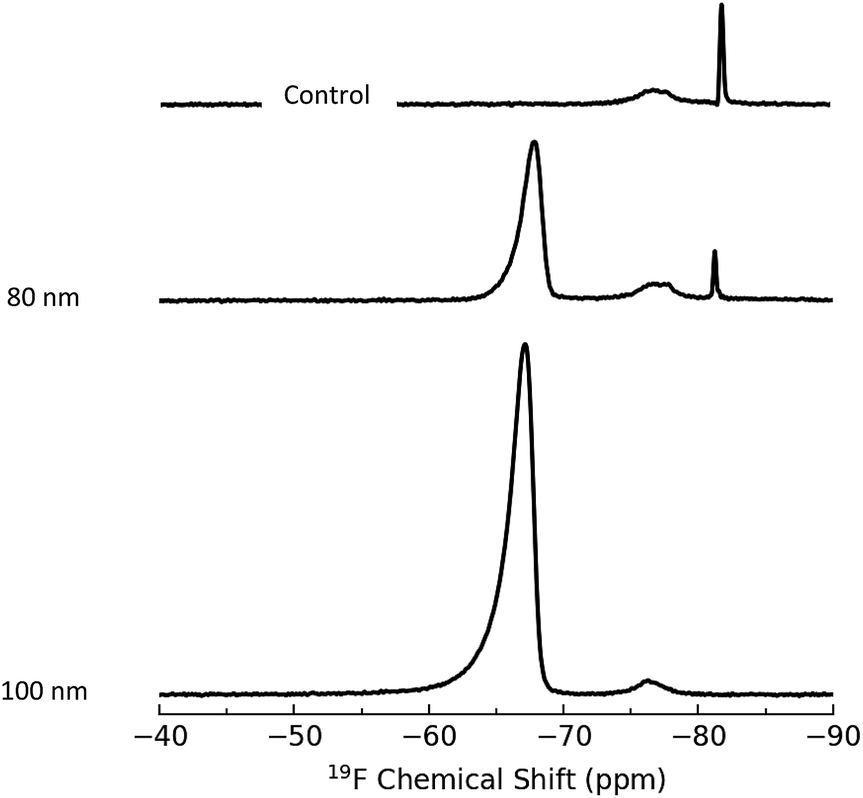
| Fig. 3 High resolution XPS scans of the C 1s, N 1s and F 1s regions for aminated silica NPs before (100 nm B3) and after modification with BTFBA (100 T1, 80 T1, 50 T1 and 20 T1); see Table 3 for further details on sample identification. | ||
| Sample | Averagea (μmol g−1) | Fractional monolayer coverageb |
|---|---|---|
| a Averages are based on replicate experiments for two independently prepared BTFBA-labelled samples for each NP size. b μmol amine measured by 19F ssNMR/estimated monolayer coverage. | ||
| 20 nm, B2 | 380 ± 1 | 0.41 |
| 50 nm, B3 | 40.1 ± 0.2 | 0.11 |
| 80 nm, B3 | 109 ± 3 | 0.47 |
| 100 nm, B3 | 125 ± 6 | 0.69 |
X-ray photoelectron spectroscopy
XPS measures the kinetic energy distribution of photoelectrons ejected from a sample upon irradiation with a monochromatic X-ray source. The observed peaks can be assigned to electrons ejected from core levels associated with particular elements, allowing for identification and quantification of the atomic composition. The probing depth of this method is determined by the inelastic mean free path of the photoelectrons which depends on kinetic energy and is in the range of 1 to 4 nm for the core levels of interest here. This sensitivity to the near surface region makes XPS well-suited for quantification of surface functional groups.XPS was measured for aminated silica NPs functionalized with BTBFA and deposited on Au substrates (Fig. S8†), as well as for several aminated NPs as controls. The only elements detected in survey scans (see Fig. S9†) were Au from the substrate, Si and O from the silica NPs, and C, N and F from the functionalization of the particles. High resolution spectra in regions corresponding to strong core level transitions for these elements were used for quantification of the sample composition and to provide information on the chemical state of the elements. Representative high resolution spectra for the C 1s, N 1s and F 1s regions are shown in Fig. 3. Table 3 shows the ratios of atomic compositions which are derived from the ratio of integrated intensities in the high resolution spectra and corrected for the relative sensitivity factors for each element. As seen in the table, the observed Si/O ratios are close to 0.5 as expected for SiO2 NPs. The N/Si ratio is a measure of the relative coverage of amine groups and thus can be compared with the amine content determined by qNMR and the colorimetric assays in Table 1. The N/Si ratio is seen to exhibit good reproducibility for the two different runs with the same size NPs. Furthermore, this ratio is seen to closely track the fraction of a monolayer coverage estimated from the qNMR data (Fig. 4). The 100 nm particles have the highest amine content (expressed as the fractional surface coverage) followed by the 20 nm and 80 nm NPs. The 50 nm particles appear to have a rather low amine content, in agreement with the qNMR observations. Similar results for amine content are observed in the case of the control samples which did not undergo the coupling reaction with BTFBA. Although the N/Si ratio tracks the amine content determined by qNMR, we note that the uncertainties in this quantity are rather high (up to 10–20%). This can be attributed to the somewhat weak N 1s signals, evidenced by comparing the noise levels for these peaks with that of the C 1s and F 1s in Fig. 3. This suggests that using the N 1s peak is not ideal for quantification and that uncertainties can be lowered by introducing F-labelled functional groups, such as in BTFBA. We note that the use of XPS for quantitative analysis requires that the core level signals remain stable during measurement. In fact, it has been observed that particularly in the case of the N 1s, changes in the peak position and integrated intensity during measurement can occur.42 In the present case, measurement of the N 1s signal as a function of time indicated that loss of the N 1s intensity during data acquisition would result in <5% underestimate of the amount of nitrogen on the particles (see Fig. S10†).
| Si/O | N/Si | F/Si | N/C | F/N | CF3/Ctotal | |
|---|---|---|---|---|---|---|
| a T1 and T2 correspond to XPS measurements for the two independently-prepared BTFBA-labelled samples used for ssNMR (Table 2); control samples are shown in the last 3 rows. | ||||||
| 20 nm, B2 T1 | 0.53 ± 0.01 | 0.075 ± 0.003 | 0.42 ± 0.01 | 0.077 ± 0.002 | 5.7 ± 0.3 | 0.086 ± 0.004 |
| 20 nm, B2 T2 | 0.54 ± 0.01 | 0.074 ± 0.001 | 0.43 ± 0.01 | 0.077 ± 0.001 | 5.9 ± 0.1 | 0.091 ± 0.004 |
| 50 nm, B3 T1 | 0.45 ± 0.01 | 0.027 ± 0.005 | 0.052 ± 0.003 | 0.035 ± 0.005 | 2.0 ± 0.3 | |
| 50 nm, B3 T2 | 0.44 ± 0.01 | 0.028 ± 0.003 | 0.065 ± 0.004 | 0.027 ± 0.001 | 2.3 ± 0.2 | |
| 80 nm, B3 T1 | 0.45 ± 0.02 | 0.085 ± 0.017 | 0.47 ± 0.02 | 0.065 ± 0.002 | 5.7 ± 0.9 | 0.082 ± 0.013 |
| 80 nm, B3 T2 | 0.48 ± 0.01 | 0.070 ± 0.001 | 0.41 ± 0.01 | 0.069 ± 0.002 | 5.8 ± 0.3 | 0.086 ± 0.006 |
| 100 nm, B3 T1 | 0.49 ± 0.01 | 0.095 ± 0.001 | 0.63 ± 0.01 | 0.077 ± 0.001 | 6.6 ± 0.2 | 0.127 ± 0.002 |
| 100 nm, B3 T2 | 0.49 ± 0.01 | 0.098 ± 0.009 | 0.47 ± 0.01 | 0.075 ± 0.003 | 4.9 ± 0.6 | 0.080 ± 0.008 |
| 50 nm, B2 | 0.46 ± 0.01 | 0.019 ± 0.001 | 0.004 ± 0.001 | 0.031 ± 0.001 | 0.21 ± 0.03 | |
| 80 nm, B3 | 0.46 ± 0.01 | 0.063 ± 0.005 | 0.011 ± 0.001 | 0.098 ± 0.002 | 0.18 ± 0.03 | |
| 100 nm, B3 | 0.45 ± 0.01 | 0.101 ± 0.006 | 0.017 ± 0.002 | 0.116 ± 0.003 | 0.16 ± 0.04 | |
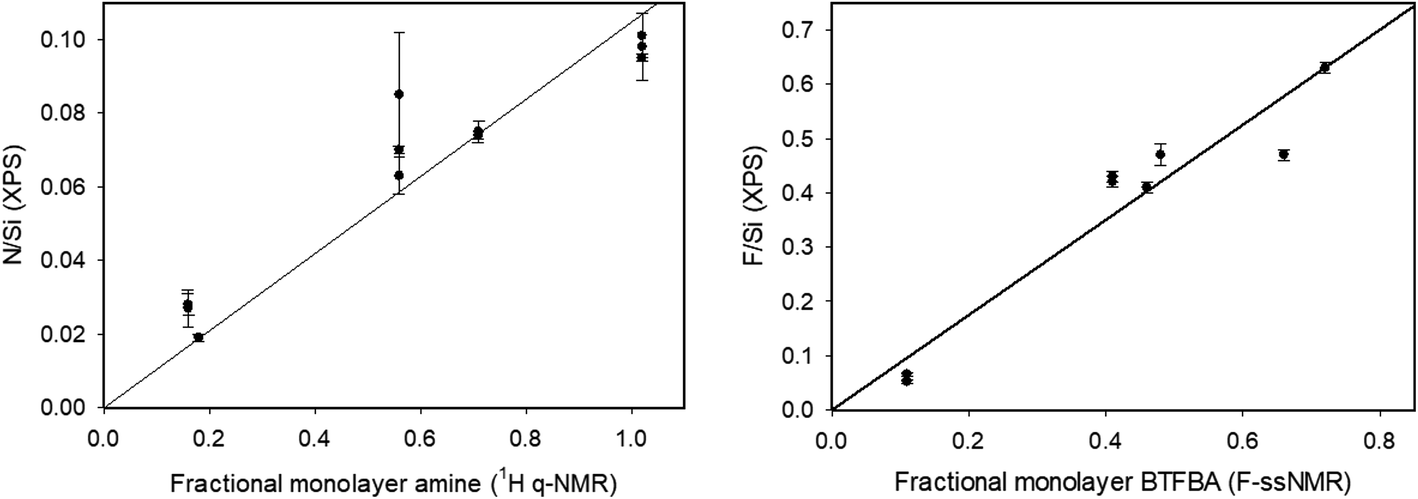 | ||
| Fig. 4 Correlation of fractional monolayer coverage (based on 1H qNMR and 19F ssNMR BTFBA-amine content) with XPS N/Si and F/Si ratios. | ||
Similarly, the F/Si ratio is a measure of the relative amount of fluorine attached to the particles after reaction with BTFBA. This ratio is expected to follow the same trend as the 19F NMR results. The 20, 80 and 100 nm NPs exhibit F/Si ratios > 0.4, with 100 T1 exhibiting the highest F/Si ratio of 0.63. The 50 nm particles exhibit a much lower amount of F, consistent with the 19F NMR results. Fig. 4 shows the correlation of the F/Si and N/Si ratios from the XPS data with the fractional monolayer coverage estimated from the NMR data. The yield of the coupling reaction can be calculated from the F/N ratio as complete reaction of all amine groups would result in F/N = 6. For the 20 and 80 nm particles, the yield of the reaction is found to be >95%. For the 100 nm particles the two runs had yields of 110% and 82% respectively. The 50 nm particles showed a yield of 33–38%. So not only did these particles exhibit a low initial amine concentration, the yield of the coupling reaction was also found to be low, although it should be noted that this is not consistent with the 19F NMR results (Table 2). A further consistency check on the coupling reaction may possibly be provided by the N/C ratio which should be 0.33 for the control (aminated) NPs, decreasing to 0.083 after complete coupling with BTFBA. The fact that observed N/C ratios of ∼0.077 for the 20 nm and 100 nm particles are close to this expected value is consistent with the high yield of the coupling reaction. However, we note that for the 80 and 100 nm control NPs the observed N/C value is considerably lower than the expected value, suggesting the presence of carbon contamination (either of the Au substrate or the NPs) and indicating that it can be problematic to use the C 1s value for quantification.
In addition to detecting the F directly through observation of the F 1s core level, the CF3 groups of the BTFBA are also observed as a chemically shifted component in high resolution scans of the C 1s region. As seen in Fig. 3, the BTFBA modified particles exhibit a carbon peak at a binding energy of ∼292 eV, shifted from the main C 1s peak by ∼8 eV. Peaks in this region have been previously assigned to CF3 groups.19 Calculating the area under this feature as a fraction of the total C 1s signal yields the CF3/Ctot ratio which is expected to be 0.17. While the observed values of this ratio show the same trend as the F/N ratio, the lower than expected observed values of CF3/Ctot are not consistent with the high yield of the coupling reaction determined above. The fact that all the observed ratios are lower than the expected value suggests that carbon contamination is likely contributing to the Ctot value. We note that previous use of the CF3 feature for quantitative analysis was carried out on carbon based NPs where the C 1s signal from the particles is much greater than any contamination.19
Discussion and conclusions
A comparison of amine content for several silica NPs measured by the ninhydrin and 4-NBA colorimetric assays, 1H qNMR of solutions of dissolved silica and 19F ssNMR of BTBFA-labelled silica is shown in Fig. 5. As noted above, the two colorimetric assays give similar amine contents for some samples, with up to ∼40% higher amine content estimated for other samples using 4-NBA. This argues against a role for steric hindrance in reducing the amine coupling efficiency, since the ninhydrin reaction releases the colored product immediately into solution and is therefore less likely to be subject to steric effects at high monolayer coverage. The 1H qNMR method routinely measures a higher total amine content than either assay substantiating our earlier conclusions (from a limited data set) that silica modified with APTES has inaccessible amines;20 we had hypothesized that the higher total amine content was due to localized regions of amine multilayers that prevent full reaction with either ninhydrin or 4-NBA and/or amination of internal pores that are not accessible to reagents used in the colorimetric assays. The porosity of the silica used here is low compared to mesoporous silicas; however, recent reports have indicated microporosity for Stober silica.40 It is also possible that agglomeration of NPs may play a role, particularly for the smaller NPs, although we have attempted to ensure that assay conditions do not cause further agglomeration of NPs.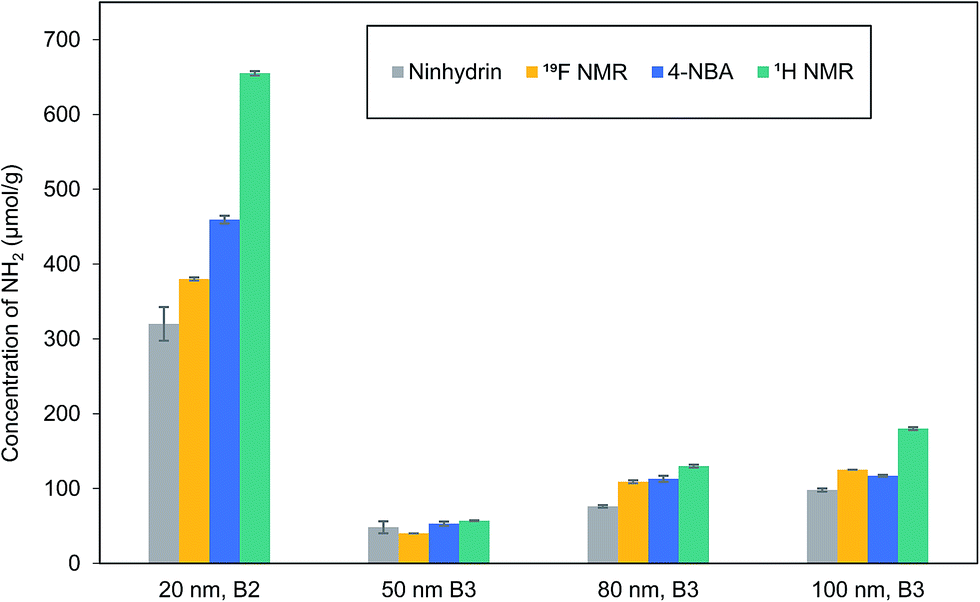 | ||
| Fig. 5 Comparison of results for colorimetric assays and NMR for selected silica NPs. | ||
Studies of amine and carboxylic acid functionalized polymer and clay NPS have determined that anywhere from a few percent to approximately all of the functional groups can be covalently modified.2,7,18,43,44 With the exception of an assay based on reaction of maleimide with cysteine,45 most studies have reported covalent modification of low fractions of total amine with larger probes or reagents generally having lower coupling efficiency. The results in Table 1 demonstrate that a minimum of 50% of total amines react with the reagents used for the two colorimetric assays for silica NPs, with over 70% modification achieved for most samples. Furthermore, the differences in coupling efficiencies for ninhydrin and 4-NBA are relatively modest, at least under the conditions of our assays which use a significant excess of reagent. These results can be compared to two previous studies of aminated mesoporous silicas. In one case covalent modification with fluorescein that was released from isolated particles and quantified by fluorescence gave a much lower amine content than an assay based on silica dissolution and quantification of released amines by reaction with fluorescamine to give a fluorescent product.12 In a second example, the accessible amine content measured using the 4-NBA assay was generally larger than the total content estimated from TGA.13 The larger difference between accessible and total amine content in these studies is consistent with the large internal surface area for mesoporous silicas.
The XPS results provide information that helps to distinguish between the various possible explanations for the higher total amine content. The F/N ratios are close to the expected value for full amination of surface amines (two CF3 groups/amine) which suggests that amines that are not detected in the two colorimetric assays are likely to be beneath the surface and therefore not within the probing depth of XPS. This argues against low coupling efficiency accounting for the lower accessible amine content. There is one exception in that the F/N ratio for 50 nm silica NPs (Table 3) is low, indicative of incomplete amination of the surface. However, this is inconsistent with results of the two assays and we hypothesize that the low total amine content (<20% of monolayer coverage) may introduce a larger error in the XPS determination, which may contribute to this discrepancy. It is noteworthy that recent reports using dynamic nuclear polarization enhanced 29Si NMR provide information on the heterogeneity of surface functional groups on silica NPs46,47 and a similar approach could in principle help to understand the differences between accessible and total amine content.
The XPS data in Table 3 provide relative atomic compositions that cannot be compared directly to the amine contents measured with the other methods. However, it is of interest to compare the N/Si and F/Si ratios with the fractional monolayer coverages calculated from the 19F ssNMR results for the BTFBA-labelled silica NPs. As shown in Fig. 4, there is good correlation between XPS and ssNMR data. Overall, this indicates that there is very good agreement between the amine contents estimated by the 4 different methods, all of which rely on initial reaction of accessible surface amines. However, the methods do differ significantly in terms of ease of use and general accessibility. Both ssNMR and XPS are useful methods for validating results of other assays. These methods are neither routine nor generally accessible methods for most laboratories, although XPS can be particularly useful for assessing sample contamination. By contrast, the colorimetric assays are compatible with routine measurements. Comparison to the silica dissolution 1H qNMR method will provide useful information on the ratio of accessible to total amine content and is a generally available and relatively easy-to-implement method. Note that elemental analysis, ICP-MS or ICP-OES have also been used for the identification of total functional groups; each of these methods provide total elemental content. The use of solution NMR method is an attractive alternative since the released functional group is quantified, allowing for analysis of NPs that have more complex surfaces or contain impurities.
The various methods tested also differ in their sensitivity. For 32 co-added transients, we had previously estimated a limit of quantification of 10 μmol g−1 for the qNMR assay20 and the estimate for 19F ssNMR is a factor of ∼3 higher. The ninhydrin and 4-NBA assays have a quantification limit that is approximately an order of magnitude lower than qNMR in solution for assay conditions that use a similar amount of silica NPs. The XPS results are more reliable for higher surface coverages of amines, although a more detailed data set would be required to estimate an approximate detection limit for this method. Overall this comparison indicates that the colorimetric assays can be used to measure lower amine content, but that qNMR has the advantage of providing information on the structure of released amines and/or impurities. Both are readily accessible methods; although the solution qNMR method can be readily generalized to a range of functional groups, it relies on dissolution of the silica and is only suitable for materials that can be dissolved without destroying the functional group.
In conclusion it is important to note that studies employing multiple methods will help to establish criteria and best practices for quantifying surface functional groups. This area of nanomaterial characterization is often neglected and the required standard protocols for inter-laboratory comparability are lacking. This is unfortunate since the surface is key to determining interactions of the material with its environment and ultimately its fate. For applications where further functionalization of the nanomaterial is required, it is typically the reagent accessible surface groups that are of primary interest. However, the ability to distinguish between surface accessible and total functional groups is important to understand the differences that may occur during solvent exchange and drying of particles during initial production and purification48–50 and their transformations when released to the environment or used in biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Council Canada, Environment and Climate Change Canada and the Natural Sciences and Engineering Research Council, Canada.Notes and references
- D. R. Baer, M. H. Engelhard, G. E. Johnson, J. Laskin, J. Lai, K. Mueller, P. Munusamy, S. Thevuthasan, H. Wang, N. Washton, A. Elder, B. L. Baisch, A. Karakoti, S. V. N. T. Kuchibhatla and D. Moon, J. Vac. Sci. Technol., A, 2013, 31, 050820 CrossRef PubMed
.
- A. Hennig, P. M. Dietrich, F. Hemmann, T. Thiele, H. Borcherding, A. Hoffmann, U. Schedler, C. Jäger, U. Resch-Genger and W. E. S. Ungera, Analyst, 2015, 140, 1804–1808 RSC
- C. I. C. Crucho, C. Baleizao and J. P. S. Farinha, Anal. Chem., 2017, 89, 681–687 CrossRef CAS PubMed
- D. Das, Y. Yang, J. S. O'Brien, D. Breznan, S. Nimesh, S. Bernatchez, M. Hill, A. Sayari, R. Vincent and P. Kumararathasan, J. Nanomater., 2014, 2014, 176015 Search PubMed
- A. Huber, T. Behnke, C. Wurth, C. Jaeger and U. Resch-Genger, Anal. Chem., 2012, 84, 3654–3661 CrossRef CAS PubMed
- M. W. Ambrogio, M. Frasconi, M. D. Yilmaz and X. Chen, Langmuir, 2013, 29, 15386–15393 CrossRef CAS PubMed
- M. Moser, N. Nirmalananthan, T. Behnke, D. Geibler and U. Resch-Genger, Anal. Chem., 2018, 90, 5887–5895 CrossRef CAS PubMed
- S. E. Lehman, Y. Tataurova, P. S. Mueller, S. V. Mariappan and S. C. Larsen, J. Phys. Chem. C, 2014, 118, 29943–29951 CrossRef CAS
- S. K. Davidowski and G. P. Holland, Langmuir, 2016, 32, 3253–3261 CrossRef CAS PubMed
- A. M. Clemments, P. Botella and C. C. Landry, ACS Appl. Mater. Interfaces, 2015, 7, 21682–21689 CrossRef CAS PubMed
- Y. Chen and Y. Zhang, Anal. Bioanal. Chem., 2011, 399, 2503–2509 CrossRef CAS PubMed
- H. Ritter and D. Bruhwiler, J. Phys. Chem. C, 2009, 113, 10667–10674 CrossRef CAS
- J. M. Rosenholm and M. Linden, Chem. Mater., 2007, 19, 5023–5034 CrossRef CAS
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS
- A. J. Morris-Cohen, M. Malicki, M. D. Peterson, J. W. J. Slavin and E. A. Weiss, Chem. Mater., 2013, 25, 1155–1165 CrossRef CAS
- D. R. Baer, D. J. Gaspar, P. Nachimuthu, S. D. Techane and D. G. Castner, Anal. Bioanal. Chem., 2010, 396, 983–1002 CrossRef CAS PubMed
- A. Hennig, H. Borcherding, C. Jaeger, S. Hatami, C. Wurth, A. Hoffmann, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, J. Am. Chem. Soc., 2012, 134, 8268–8276 CrossRef CAS PubMed
- P. M. Dietrich, A. Hennig, M. Holzweber, T. Thiele, H. Borcherding, A. Lippitz, U. Schedler, U. Resch-Genger and W. E. S. Unger, J. Phys. Chem. C, 2014, 118, 20393–20404 CrossRef CAS
- F. Kunc, V. Balhara, A. Brinkmann, Y. Sun, D. M. Leek and L. J. Johnston, Anal. Chem., 2018, 90, 13322–13330 CrossRef CAS PubMed
- L. D. White and C. P. Tripp, J. Colloid Interface Sci., 2000, 232, 400–407 CrossRef CAS PubMed
- M. Zhu, M. Z. Lerum and W. Chen, Langmuir, 2012, 28, 416–423 CrossRef CAS PubMed
- D. V. Okhrimenko, A. Budi, M. Ceccato, M. Cardenas, D. B. Johansson, D. Lybye, K. Bechgaard, M. P. Andersson and S. L. S. Stipp, ACS Appl. Mater. Interfaces, 2017, 9, 8344–8353 CrossRef CAS PubMed
- T. Fischer, P. M. Dietrich, C. Streeck, S. Ray, A. Nutsch, A. Shard, B. Beckhoff, W. S. E. Unger and K. Rurack, Anal. Chem., 2015, 87, 2685–2692 CrossRef CAS PubMed
- N. Kong, J. Zhou, J. Park, S. Xie, O. Ramstrom and M. Yan, Anal. Chem., 2015, 87, 9451–9458 CrossRef CAS PubMed
- A. Brinkmann, M. Raza and J. Melanson, Metrologia, 2019, 56, 024002 CrossRef
- S. Akoka, L. Barantin and M. Trierweiler, Anal. Chem., 1999, 71, 2554–2557 CrossRef CAS PubMed
- C. Jaeger and F. Hemmann, Solid State Nucl. Magn. Reson., 2014, 57–58, 22–28 CrossRef CAS PubMed
- M. Friedman, J. Agric. Food Chem., 2004, 52, 385–406 CrossRef CAS PubMed
- V. K. Sarin, S. B. H. Kent, J. P. Tan and R. B. Merrifield, Anal. Biochem., 1981, 117, 147–157 CrossRef CAS PubMed
- E. Soto-Cantu, R. Cueto, J. Koch and P. S. Russo, Langmuir, 2012, 28, 5562–5569 CrossRef CAS PubMed
- D. Iannazzo, A. Piperno, A. Ferlazzo, A. Pistone, C. Milone, M. Lanza, F. Cimino, A. Speciale, D. Trombetta, A. Saija and S. Galvagno, Org. Biomol. Chem., 2012, 10, 1025–1031 RSC
- G. Jarre, S. Heyer, E. Memmel, T. Meinhardt and A. Krueger, Beilstein J. Org. Chem., 2014, 10, 2729–2737 CrossRef CAS PubMed
- H.-T. Lu, Colloid J., 2013, 75, 311–318 CrossRef CAS
- D. R. Hristov, L. Rocks, P. M. Kelly, S. S. Thomas, A. S. Pitek, P. Verderio, E. Mahon and K. A. Dawson, Sci. Rep., 2015, 5, 17040 CrossRef CAS PubMed
- L. T. Zhuravlev, Langmuir, 1987, 3, 316–318 CrossRef CAS
- L. Girard, R. Nadarajah, Y. M. Bazin, J. Sun, X. X. Zhu and S. Giasson, Appl. Surf. Sci., 2016, 370, 476–485 CrossRef
- J. H. Moon, J. W. Shin, S. Y. Kim and J. W. Park, Langmuir, 1996, 12, 4621–4624 CrossRef CAS
- M. van de Waterbeemd, T. Sen, S. Biagina and I. J. Bruce, Micro Nano Lett., 2010, 5, 282–285 CrossRef
- S. Li, Q. Wan, Z. Qin, Y. Fu and Y. Gu, Langmuir, 2015, 31, 824–832 CrossRef CAS PubMed
- S. Ji, T. R. Hoye and C. W. Macosko, Macromolecules, 2005, 38, 4679–4686 CrossRef CAS
- J. G. McEvoy and Y. Thibault, Appl. Surf. Sci., 2019, 466, 970–981 CrossRef
- T. Felbeck, K. Hoffmann, M. M. Lezhina, U. H. Kynast and U. Resch-Genger, J. Phys. Chem. C, 2015, 119, 12978–12987 CrossRef CAS
- P. M. Dietrich, C. Streek, S. Glamsch, C. Ehlert, A. Lippitz, A. Nutsch, N. Kulak, B. Beckhoff and W. E. S. Unger, Anal. Chem., 2015, 87, 10117–10124 CrossRef CAS PubMed
- M. Moser, T. Behnke, C. Hamers-Allin, K. Klein-Hartwig, J. Falkenhagen and U. Resch-Genger, Anal. Chem., 2015, 87, 9376–9383 CrossRef CAS PubMed
- T. Kobayashi, D. SIngappuli-Aachchige, Z. Wang, I. L. Slowing and M. Pruski, Phys. Chem. Chem. Phys., 2017, 19, 1781–1789 RSC
- D. Lee, G. Monin, N. T. Duong, I. Z. Lopez, M. Bardet, V. Mareau, L. Gonon and G. De Paepe, J. Am. Chem. Soc., 2014, 136, 13781–13788 CrossRef CAS PubMed
- R. Franca, T. D. Samani, G. Bayade, L. Yahia and E. Sacher, J. Colloid Interface Sci., 2014, 420, 182–188 CrossRef CAS PubMed
- L.-K. Mireles, E. Sacher, L. Yahia, S. Laurent and D. Stanicki, Data in Brief, 2016, 7, 1296–1301 CrossRef PubMed
- J. T. Nurmi, V. Sarathy, P. G. Tratnyek, D. R. Baer, J. E. Amonette and A. Karkamkar, J. Nanopart. Res., 2011, 13, 1937–1952 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Additional data for optimization and calibration of colorimetric assays, 1H qNMR spectrum and XPS survey spectra and controls. See DOI: 10.1039/c9na00016j |
| This journal is © The Royal Society of Chemistry 2019 |