
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual targeting of folate receptor-expressing glioma tumor-associated macrophages and epithelial cells in the brain using a carbon nanosphere–cationic folate nanoconjugate†
Chandra Kumar
Elechalawar‡
ae,
Dwaipayan
Bhattacharya‡
ad,
Mohammed Tanveer
Ahmed
ae,
Halley
Gora§
a,
Kathyayani
Sridharan
ae,
Piyush
Chaturbedy
b,
Sarmistha Halder
Sinha
c,
Madhan Mohan
Chandra Sekhar Jaggarapu
ae,
Kumar Pranav
Narayan
d,
Sumana
Chakravarty
a,
Muthusamy
Eswaramoorthy
 b,
Tapas Kumar
Kundu
c and
Rajkumar
Banerjee
*ae
b,
Tapas Kumar
Kundu
c and
Rajkumar
Banerjee
*ae
aApplied Biology Division, CSIR-Indian Institute of Chemical Technology, Hyderabad, 500 007, India. E-mail: banerjee@iict.res.in
bChemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O, Bangalore, 560 064, India
cTranscription and Disease Laboratory, Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O, Bangalore, 560 064, India
dDepartment of Biological Sciences, BITS Pilani, Hyderabad Campus, Jawahar Nagar, Shameerpet Mandal, Hyderabad 500078, India
eAcademy of Scientific & Innovative Research (AcSIR), Taramani, Chennai 600113, India
First published on 24th July 2019
Abstract
Glioblastoma multiforme (GBM), the highly invasive form of glioma, exhibits the highest mortality in patients with brain malignancies. Increasing glioma patients' survivability is challenging, as targeting only tumor-associated malignant cells would not reduce the overall aggressiveness of the tumor mass. This is due to the inadequacy in countering pro-proliferative, invasive and metastatic factors released by tumor-mass associated macrophages (TAMs). Hence, strategically, dual targeting both tumor cells and TAMs is necessary for effective glioma treatment and increased survivability. Conventional FR-targeting systems can easily target cancer cells that overtly express folate receptors (FRs). However, FRs are expressed only moderately in both glioma cells and in TAMs. Hence, it is more challenging to coordinate dual targeting of glioma cells and TAMs with lower levels of FR expression. A recently developed carbon nanosphere (CSP) with effective blood–brain barrier (BBB) penetrability was modified with a new folic acid–cationic lipid conjugate (F8) as a targeting ligand. The uniqueness of the cationic lipid–folate conjugate is that it stably associates with the negatively charged CSP surface at about >22 mol% surface concentration, a concentration at least 5-fold higher than what is achieved for conventional FR-targeting delivery systems. This enabled dual uptake of the CSP on TAMs and tumor cells via FRs. A doxorubicin-associated FR-targeting formulation (CFD), in an orthotopic glioma model and in a glioma subcutaneous model, induced the maximum anticancer effect with enhanced average mice survivability twice that of untreated mice and without any systemic liver toxicity. Additionally, we observed a significant decrease of TAM-released pro-aggressive factors, TGF-β, STAT3, invasion and migration related sICAM-1, and other cytokines indicating anti-TAM activity of the CFD. Taken together, we principally devised, to the best of our knowledge, the first FR-targeting nano-delivery system for targeting brain-associated TAMs and tumor cells as an efficient glioma therapeutic.
1. Introduction
Advanced nanomaterials are needed for improved drug delivery. To utilise the substantial therapeutic potential of nanomaterials, surface functionalization is of paramount importance. Functionalization includes antibody conjugation, small molecule labelling, functionalization with biologically active targeting molecules etc., thus facilitating efficient and promising imaging and therapy. Hence, the general challenge in surface modification of nanocarriers lies in designing multifunctional small molecules or fabricating bioactive large molecules.Among brain cancers, glioblastoma multiforme (GBM), the invasive form of glioma, induces maximum mortality1 in patients. The overall aggressiveness of GBM is not only due to the tumor-associated malignant cells but also due to a special tumor-associated micro-environment (TME) created within the tumor mass. The TME consists of malignant cancer cells, various heterogeneous cell components including cancer stem cells, tumor-associated vascular endothelial cells, fibroblasts, lymphatic cells and also macrophages. These tumor-associated macrophages (TAMs), the most abundant immune cells in tumor-masses, play predominant roles in tumor pro-proliferation, cell invasion, metastasis, angiogenesis, suppressing tumor-associated immune responses etc.2–4 GBM-associated glioma stem cells secrete periostin which recruits tumor-supportive TAMs.5 TAMs are differentiated monocytes that form in response to tumor-derived chemo-attractants such as CSF-1 (macrophage colony stimulating factor-1), various chemokines like CCL2/3/4/5/9 and VEGF. Levels of these proteins in tumor masses are proportionately related to the overall TAM count in the tumor environment,6 although no correlation was established between TAM count and patient prognosis. However, the majority of studies showed that high TAM numbers are linked with reduced patient survival. Many TME-expressed factors and corresponding receptors expressed on TAMs work in tandem to maintain the overall aggressiveness. One such incidence is the involvement of the expressed CSF-1 receptor (CSF-1R) which helps in the survival of TAMs. On depletion of the CSF-1R, the intracranial GBM propagation halts.7 But the TAM population does not deplete, as expression of other survival factors gains importance. Incidentally, most treated animals eventually acquire resistance to CSF-1R inhibition.8 The invasiveness of a glioma tumor mass is due to functional expression of many factors including TAM-expressed TGF-β, which in turn up-regulates STAT3 via IL-6 in the cells of tumor masses.9,10 Clearly, the direct target should be TAMs for effective and long-term therapeutic benefits. Hence, TAM-targeting is increasingly gaining importance. Recently, TAMs have been found to act as slow-release reservoirs for nanoformulations delivering drugs to tumor masses.11 However, delivery of nanoformulations through the blood–brain barrier (BBB) to in situ glioma masses and their TAMs is an additional challenge.
More recently, carbon nanospheres (CSPs) have been gaining attention due to their applications in various fields including drug delivery.12,13 Among them, glucose-derived CSPs are emerging as a new class of intracellular drug carriers in drug delivery. The non-toxic CSPs are readily dispersible in water, can target cell nuclei and have the unique property of crossing the blood–brain barrier (BBB) in vivo.14 Folate receptors (FRs), the glycosylphosphatidylinositol (GPI)-anchored protein, are expressed on the surface of the cell membrane and are often overtly expressed on many cancer cells including glioblastomas and also on the TAM membrane surface.15,16 As a FR binds and internalises folate or folic acid into cells, folate receptor targeting delivery systems especially for targeting FR-overexpressing cancer cells are well known. With folate as a targeting ligand, FR-targeting drug/gene nanodelivery systems are tested wherein in situ glioma tumors are targeted.17–19 Other than FR-targeting, other nanoformulations targeting gliomas are also reported.20–22 However, the importance of targeting TAMs for glioma treatment is not emphasized in those studies. There is no authentic report describing a delivery system directly targeting TAMs in tumor masses, especially those of glioma tumors through FRs.
Recently, we have developed a cationic lipid-conjugated folic acid as an FR-targeting ligand [F8, Scheme 1], which showed effective targeting ability against a melanoma model where melanoma cells express moderate to higher levels of FRs.23 Using F8 as a targeting ligand, here we describe a new BBB penetrable folate ligand-associated CSP for targeting FR-expressing, tumor associated malignant cells and TAMs. This cationic lipid-conjugated folate ligand (F8) electrostatically binds to negatively charged CSPs. With a high surface concentration of F8, the targeting CSPs showed enhanced glioma cell and TAM targeting simultaneously. Armed with a model anticancer drug, doxorubicin (DOX), the targeting nanoformulation exhibited significant glial cell killing, glioma tumor regression, and enhanced patient survivability. Here, TAM cells could also be targeted directly. This dual strategy of targeting TAMs and malignant cells exhibited a better outcome in glioma treatment, thereby emphasizing the importance of targeting TAMs.
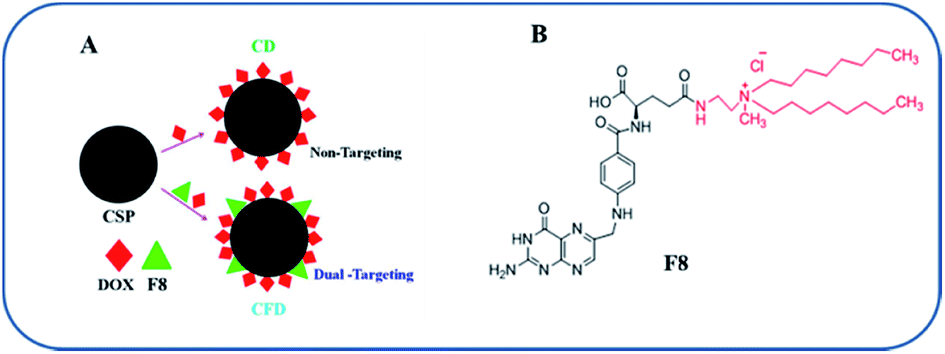 | ||
| Scheme 1 Nanomaterial fabrication and the folate receptor-targeting molecule: (A) the fabrication strategy of CSPs for the production of DOX-associated, non-targeting (CD) and targeting (CFD) nanoformulations. (B) Structure of the F8 molecule. | ||
2. Experimental
2.1 Chemicals and general procedures
Carbon nanospheres (CSPs) were synthesized according to a previously published protocol.15 Doxorubicin (DOX) was purchased from Alfa Aesar, India, and TAM isolation was performed using a Midi MACS separating kit containing a Midi MACS separator (130-042-302), LS columns (130-042-401) and CD11b micro beads (130-097-142) [Miltenyi Biotech Asia Pacific Pte Ltd, Singapore]. Folate-free RPMI-1640 medium (Cat no. 27016021) and NIR dye DIR (D12731) were purchased from Thermo Fisher Scientific, USA. A Proteome Profiler array (Mouse Cytokine Array Panel A) (ARY006) was purchased from R&D Systems, USA. Antibiotics were purchased from HiMedia, India. All the other reagents were purchased from local suppliers and used without further purification.2.2 Cell culture and antibodies
The mycoplasma-free GL261 cell line was procured from the National Cancer Institute, USA, and NIH3T3 was procured from the National Centre for Cell Sciences (Pune, India). Cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 in air in folate free RPMI (Gibco) containing 10% fetal bovine serum (South American Origin, Gibco, USA) and 1% penicillin–streptomycin–kanamycin. 85–90% confluent cultures of cells were used in both in vitro and in vivo experiments. The antibody against FRs (sc-28997) was purchased from Santa Cruz Biotechnology. Antibodies against FR4-PE (130-102-455), F4/80-FITC (130-102-327), CD-11b FITC (130-081-201), and CD-68 (130-102-534) were purchased from Miltenyi Biotec Asia Pacific Pte Ltd, Singapore.2.3 Preparation of CSP conjugates
5 mL of 1.5 mM solution of FA8 (M. wt 757 g mol−1) was made using HPLC grade methanol. 10 mg of CSPs was added to the FA8 solution and kept stirring for 12 h at room temperature. CSPs were separated by centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The amount of FA8 adsorbed to CSPs was calculated from the amount present in the supernatant methanol solution of FA8 obtained by measuring the absorbance at 280 nm using a spectrometer at 270 °C. A prior calibration plot was obtained by measuring the FA8 solution at different concentrations. About 40% of FA8 was adsorbed on 10 mg of CSPs. CSP-F8-DOX or CSP-DOX was prepared by adding 10 mg of CSP-FA8 or CSP to 5 mL of 2 mM methanol solution of DOX and it was stirred for 12 h at room temperature. The CSP conjugates were separated using the centrifugation method mentioned above. The amount of DOX adsorbed to CSP conjugates (CSP-FA8, CSP, and CSP-C8) was calculated by measuring the absorbance of DOX at 495 nm in the supernatant methanol solution. 20% DOX was loaded on the CSP surface.
000 rpm for 10 min. The amount of FA8 adsorbed to CSPs was calculated from the amount present in the supernatant methanol solution of FA8 obtained by measuring the absorbance at 280 nm using a spectrometer at 270 °C. A prior calibration plot was obtained by measuring the FA8 solution at different concentrations. About 40% of FA8 was adsorbed on 10 mg of CSPs. CSP-F8-DOX or CSP-DOX was prepared by adding 10 mg of CSP-FA8 or CSP to 5 mL of 2 mM methanol solution of DOX and it was stirred for 12 h at room temperature. The CSP conjugates were separated using the centrifugation method mentioned above. The amount of DOX adsorbed to CSP conjugates (CSP-FA8, CSP, and CSP-C8) was calculated by measuring the absorbance of DOX at 495 nm in the supernatant methanol solution. 20% DOX was loaded on the CSP surface.
2.4 Characterization of CSP conjugates
The hydrodynamic diameter size and surface charge zeta potentials of CSP-F8-DOX (CFD), CSP-DOX (CD), CSP-F8 (CF8), CSP-FA-D (CFA-D), CSP-C8-D (CC8-D) and CSP-C8 (CC8) were measured by electrophoretic mobility using either a Zeta sizer 3000 HSA (Malvern, UK) or Lite Sizer™ 500 particle analyzer (Anton Paar GMBH, Germany). Briefly, 20 μL of each CSP conjugate was suspended in 1 mL of deionised water. A brief probe sonication of the suspension of CSP conjugates for 10–15 min minimised batch-to-batch size variations of respective CSP conjugates. Hydrodynamic diameters and surface charges of the suspension of respective CSP conjugates were then measured.2.5 Cellular uptake studies
Cellular uptake studies of FR targeting CSP-FA8-DOX and non-targeting control DOX containing CSPs were conducted by fluorescence microscopy and FACS analysis in GL261 cells. Cells were seeded at a density of 5 × 105 cells per well on square coverslips in a 6-well plate before 24 h of treatment. After 3 h of treatment cells were washed with phosphate buffered saline and pictures were taken with an inverted confocal Olympus FV10i microscope.2.6 In vitro cell viability assay/toxicity assay
Cells at a density of 5000 per well were seeded in 96-well plates and allowed to grow for 18–24 h before the treatment. Cells were treated with various conjugates carrying the drug and free non-conjugated drug and allowed to grow for 48 h. Cell viability was determined using an MTT based assay. MTT (5 mg mL−1 in PBS, 10 μL) was added to the cells and incubated for 4 h. The resulting formazan crystals were dissolved with 50 μL of 1:1 (v/v) DMSO/methanol. Absorbance was determined with a microplate reader (ELISA) at 550 nm wavelength.
2.7 Animals
C57BL/6J male mice (6–8 weeks old, each weighing ∼20–22 g) were purchased from the National Institute of Nutrition, Hyderabad, India. All in vivo experiments were performed strictly according to the guidelines provided by the statutory Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) [Chapter 4, Section 15(1) of the Prevention of Cruelty to Animals Act 1960, India]. The animal protocol [no. IICT/BMG/RKB/28/09/2015/03] was approved by the ‘Institutional Animal Ethical Committee’ (IAEC) of CSIR-Indian Institute of Chemical Technology, Hyderabad, India [Registration no. 97/GO/RBi/S/1999/CPCSEA, dated 1-Oct-2015].2.8 Orthotopic brain tumor implantation and a subcutaneous model for treatment
An orthotopic model was established using an earlier reported method.24 In brief, 4–6 week old male C57BL/6J mice were anaesthetized by intra-peritoneal injection of a ketamine–xylazine mixture. Using a 5 μL Hamilton syringe, 1.5 × 104 cells suspended in 3 μL of phosphate buffered saline (PBS) were stereotactically injected through an entry site at the bregma (2 mm to the right of the sagittal suture and 3 mm below the surface of the skull) using a stereotactic frame (Harvard Apparatus, USA). Animals were observed every day after tumor cell challenge, and mice that developed symptoms indicative of brain tumors such as paralysis, lethargy, or wasting symptoms were euthanized. In C57BL6/J mice, a subcutaneous model was developed by inoculating 5 × 105 GL261 cells in the left, lower abdomen of each mouse. When the tumor size reached 100 mm3 the mice were divided into 4 different groups and treatment with CFD, CD, free DOX and 5% glucose (untreated) started on alternate days with injection of overall 5 doses of DOX at an amount of 5 mg kg−1.2.9 In vivo imaging
The glioma bearing mice were established as described above. After tumor inoculation, on the 10th day 2 mg kg−1 DiR-loaded CSPs or CF8 was intraperitoneally injected into glioma tumor bearing (n = 3) mice. The distribution of fluorescence was observed using an IVIS Spectrum (PerkinElmer) in vivo imaging system after anaesthetising the animals. At 6 h and 24 h post injection, the in vivo images of intact animals were taken. Mice were then euthanized via CO2 overdose and organs (brain, heart, lungs, liver, spleen, and kidneys) were excised and imaged using the IVIS Spectrum.2.10 Immunohistochemical studies
For immunochemistry studies, mouse brains obtained from doxorubicin conjugated CSPs and CSP-FA8 treated glioma-bearing mice (n = 3) were fixed with 4% formaldehyde overnight, washed with PBS and stored in 20% glycerol solution (PBS). 10 micron frozen tissue sections were prepared with a Leica tissue sectioning biosystem. For folate receptor (FR) staining studies, the sections were rinsed with PBS for 10 min followed by incubation with 0.5% Triton X-100 in PBS for 60 min at room temperature. Slides were kept in a blocking solution of 5% BSA and 5% NHS with 0.5% Triton X-100 in PBS for 1.5 h. Then the sections were incubated with rabbit anti-FR (1:200) in the blocking solution overnight at 40 °C. After bringing the sections to RT and resting for 30 min, the sections were rinsed with PBST (0.1% Tween-20) 4 times each for 10 min. Then the sections were incubated with FITC labelled goat anti-rabbit antibody (1:500) for 2 h at RT followed by rinsing with PBST-20 four times. DAPI, 2 μg mL−1, was added to the sections and they were mounted with cover slips. The distribution of fluorescence was observed using an FV10i confocal microscope (Olympus).
2.11 Tumor associated macrophage (TAM) isolation and ex vivo uptake studies
Tumor associated macrophage isolation was performed following a procedure reported earlier.25,26 In brief, C57BL/6J mice were subcutaneously implanted with 5 × 105 GL261 cells. After 20 days, when the tumor size reached 1500 mm3, the mice were separated into three groups and treated with CFD, CD and DOX. After 8 h, tumors were isolated and tumor associated macrophages collected according to a standard protocol. The tumors were cut into small pieces and 10 mL of digestion medium (containing collagenase) was added. The erythrocytes were removed by treating the cell mass with erythrocyte lysis buffer, centrifuging and removing the supernatant. For isolating tumor associated macrophages, the above pellet was resuspended in MACS buffer and magnetic bead-conjugated antibodies (Miltenyi Biotec) against the CD11b surface marker were added at a concentration of 10 μL/107 cells. The cell suspension was added to a MACS LS column and placed under a magnetic field. The unbound cells were washed off, and the bound CD11b+ macrophages were isolated by removing the magnetic field. These unbound cells are the heterogeneous population of many kinds of tumor mass-associated cells, such as malignant cancer cells, tumor associated endothelial cells, fibroblasts, DCs etc. These TAM-free tumor cells were also preserved for estimating FR expression levels and for uptake studies. For in vivo uptake studies, GL261 subcutaneous tumor bearing mice (n = 3) were given single intraperitoneal injections of either DOX, DOX conjugated CSPs or CSP-F8. After 18 h tumors were isolated, and TAMs from tumor were analysed through flow cytometry (BD FACS Canto II).2.12 Anti-glioma effect
An orthotopic model was established using the above method. After tumor implantation (day 0), on day 4, tumor bearing mice were randomly sorted into four groups. Each group (n = 6) was administered DOX, DOX conjugated CSPs and CSP-FA8 through intra-peritoneal injections. For the subcutaneous model, 5 × 105 GL261 cells in 200 μL of PBS were injected in the right flank of mice on day 0. After 12 days, the mice were divided into 4 groups (n = 5). Each group was administered DOX, DOX conjugated CSPs and CSP-F8 separately via intra-peritoneal injections. The untreated group was administered 5% glucose only. Each mouse received five doses on every alternate day. Tumor volumes were measured with slide calipers and calculated using the 1/2 × a × b2 formula, where ‘a’ is the longest dimension and ‘b’ is the shortest. Mice were observed thereafter for survivability.2.13 Western blot study and chemokine profile analysis
All the tumors from the mice belonging to the respective treatment or untreated groups (i.e., DOX, CD, CFD and untreated) were isolated and pooled, and lysates were prepared. After homogenization of tumor tissues in 1× PBS, Triton X-100 was added to 1% final concentration and the tissues were kept on ice for 10 min. Centrifugation was performed at 10000 rpm for 10 min at 4 °C. Quantification of protein in lysates was carried out using the BCA method. Proteins and chemokines were analysed using Western blot and a mouse cytokine array, respectively.
2.14 H & E staining for histology analysis
The acute toxicity of the formulation in various organs such as the heart, liver, kidney etc. was histologically examined following a single injection of CFD to mice. 24 h post treatment, the collected organs were frozen using the Leica tissue freezing medium (Leica, Germany). The frozen tissues were sectioned with a thickness of 5 microns using a Leica CM1850 Instrument (Leica Microsystem, Germany). The sections were imaged in bright field using a Nikon Eclipse TE2000 microscope (Nikon, Japan) following tissue staining with hematoxylin & eosin (H & E).2.15 Statistical analysis
Error bars represent mean values ± SD. The statistical significances of the samples were determined using the two-tailed Student's t-test, Graph Pad Prism software. p values less than 0.05 were considered to be statistically significant.3. Results
3.1 Synthesis and characterization of CSP conjugates
The F8 structure [Scheme 1] was designed to accommodate a dioctyl methyl ammonium moiety in the distal end of the carboxylic group, which is often used for folate ligand conjugation. Synthesis of F8 was accomplished as per a synthetic protocol described earlier.14 The C8 control molecule was prepared (ESI Scheme S1†) and characterized through 1H NMR and ESI mass spectrometry. CSPs were then simultaneously associated with F8 and DOX, or only DOX. These were then reconstituted in aqueous solution [Scheme 1]. The hydrodynamic diameters (size) and surface charges (zeta potential) of different CSP conjugates were measured using a Zetasizer. The sizes of CSP-F8-DOX (CFD) and CSP-DOX (CD), respectively, are in the range of 360 nm (Fig. 1A). The polydispersity index (PDI) of such CSP conjugates is less than 0.3, thereby clearly indicating the fairly homogeneous size distribution of CSP conjugates (Fig. 1C). IR spectra of CSPs and their conjugated products indicate the association of ligands and/or drugs on CSPs (Fig. S1†). The size of DOX and F8-conjugated CSP [i.e., CFD] is stable up to 48 h in 50% serum in PBS (Fig. S2†). Additionally, SEM images show a homogeneously spherical structure of CFD and CD conjugates (Fig. 1B). The surface charges of different conjugates [CSP, CSP-F8 (CF8), CFD, CD, CFA-D, and CC8-D] are in the range of −22 to −37 mV (Fig. 1C). The mol% of F8 associated with the surface of CSPs was measured by calculating residual F8 un-associated (measured from F8 absorbance max at 284.5 nm) with CSPs. The associated F8 is ∼22% (w/w) with respect to CSPs. The zeta potential measurement data indicate that the association of the cationic folate ligand F8 significantly reduced the negative surface charge of CSPs. DOX entrapment on CSP-F8 and CSP surfaces was in between 50 and 60% with respect to the weight of CSPs. The DOX and F8 entrapment or association efficiencies with CSPs are calculated based on standard curves [Fig. S3–S5†].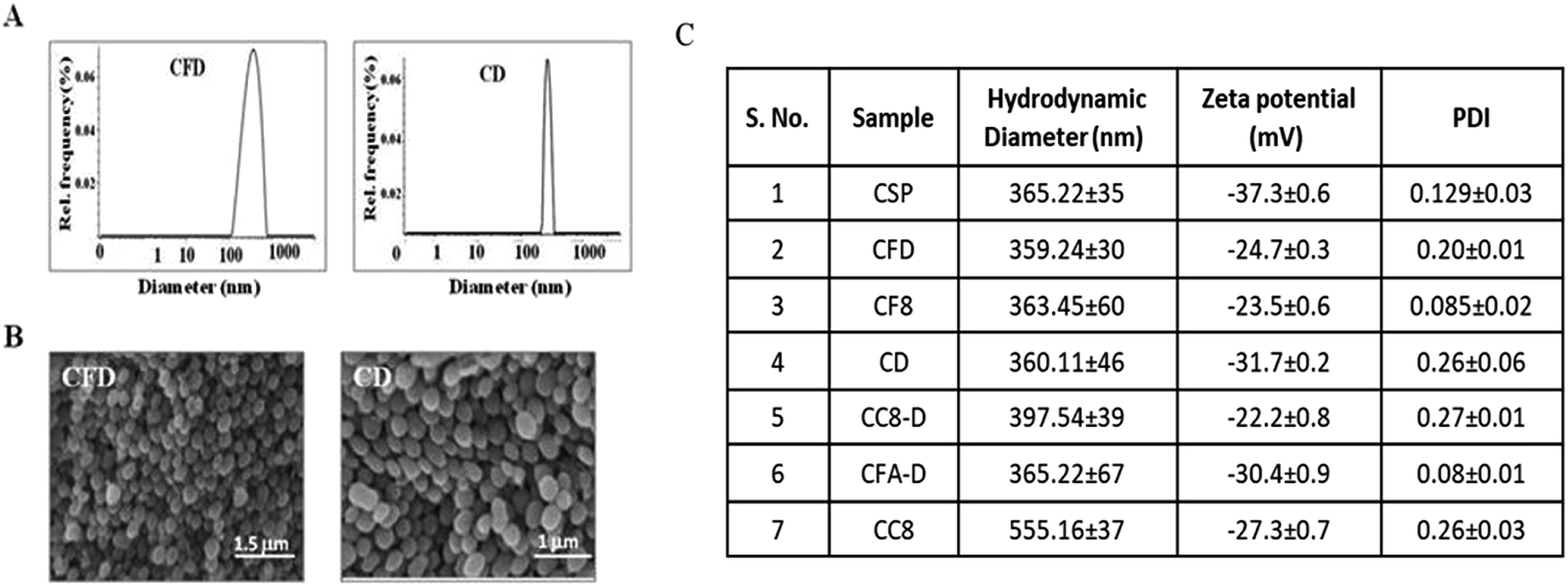 | ||
| Fig. 1 (A) Size of CFD and CD measured with DLS. (B) SEM images of CFD and CD. CD represents CSP-DOX and CFD represents CSP-F8-DOX. (C) Table shows the size and charge of various carbon nanospheres. CSP represents pristine, non-functional carbon nanospheres; CF8 represents CSP conjugated with F8, CD represents CSP conjugated with DOX, CFD represents CSP simultaneously conjugated with F8 and DOX, CC8-D represents CSP conjugated with C8 lipid and DOX, CFA-D represents CSP conjugated with folic acid (FA) and DOX and CC8 represents CSP conjugated with C8 lipid. | ||
3.2 F8 enables FR-mediated cellular uptake of CSPs in GL261 cells
Cellular uptake studies of either free DOX, DOX associated CSP-F8 [i.e., CFD] or CSP [i.e., CD] were conducted in mouse glioma GL261 cells following 3 h of treatment using fluorescence microscopy and flow cytometry. Results visually show that the DOX uptake is higher in CFD-treated GL261 cells than in cells treated with CD (Fig. 2A). Flow cytometric uptake data also reveal the highest red fluorescence shift in CFD treated GL261 cells (indicated with green in Fig. 2B) compared to the shifts obtained from cells treated with CD and DOX. Hence, the presence of the F8-ligand helped CFD to gain better access in GL261 cells. However, to test whether F8 or folic acid (FA) is better in enabling efficient cellular uptake, we compared the cellular uptake of FA-associated CD (i.e., CFA-D) with that of CFD using flow cytometry analysis. Fig. 2B (2nd panel) shows that the cellular uptake of CFD is more than that of CFA-D. This clearly indicates that F8-association rather than FA-association helps to achieve better cellular uptake of nanospheres. This also corroborates our logic of designing the cationic lipid–folic acid conjugate, which seemingly shows stronger affinity for FR-expressing GL261 cells. Among all, the uptake of CFD in GL261 cells is significantly inhibited in the presence of folic acid, indicating that its uptake is truly FR-mediated (Fig. 2C). | ||
| Fig. 2 Cellular uptake of doxorubicin (DOX) and viability studies in cancer and non-cancer cells. (A) Microscopy images of mouse glioma GL261 cells treated with CFD, CD and pristine DOX. (B and C) Flow cytometric analysis of glioma cells treated with CFD (green), CD (red), DOX (black), CC8-D (pink) and CFA-D (blue, FA denotes folic acid). (D and E) Cytotoxicity profiles of different DOX-conjugated CSPs in GL261 cancer cells and NIH-3T3 non-cancer cells. | ||
3.3 CFD shows efficient and selective cancer cell killing
The cellular cytotoxicities of DOX-associated, FR-targeting carbon nanospheres (CFD) and non-targeting carbon nanospheres (CD) and free DOX were evaluated and compared in GL261 cells and in non-cancerous fibroblast NIH 3T3 cells using the standard MTT assay. CFD shows the lowest IC50 at 400 nM in FR-positive GL261 cells. The IC50 values of CD (≥2.5 μM) and free DOX (≥2 μM) were significantly higher than that of CFD in GL261 cells indicating that CFD is a most potent cytotoxic material, more potent than even free DOX in vitro (Fig. 2D). Moreover, the cytotoxic effects of these materials were also checked in FR-negative NIH 3T3 cells. No cellular toxicities were noticed following the treatment with CFD, CD or DOX (Fig. 2E). Overall, the data imply that CFD is the most potent and selective cytotoxic material among other nanospheres.3.4 CF8 accumulates with higher efficiency than CSPs in orthotopic glioma tumors developed in mice
Next, we wanted to examine the uptake of CF8 in orthotopic glioma tumors in mice. For this, an NIR dye (DiR) was conjugated with CSPs and CF8. Tissue distributions of CF8-DiR and CSP-DiR were examined in orthotopic GL261 tumor bearing C57BL/6J mice by quantifying the amount of visible dye. The nanomaterials were injected intra-peritoneally in mice at two different time points (6 h and 24 h) and the distribution patterns of nanomaterials in the brain and other organs at these two time points were obtained. Fig. 3A shows that the accumulation of the nanomaterial CF8-DiR is significantly higher than that of CSP-DiR in the brain at both time points. After ex vivo harvesting of different organs followed by an external washing of organs (e.g., brain) with PBS, we noticed that the uptake of nanoparticles in the brain was reduced relative to what was observed during live conditions. Even then, the brain uptake of CF8-DiR was significantly higher than that of CSP-DiR in all time points tested (Fig. 3B).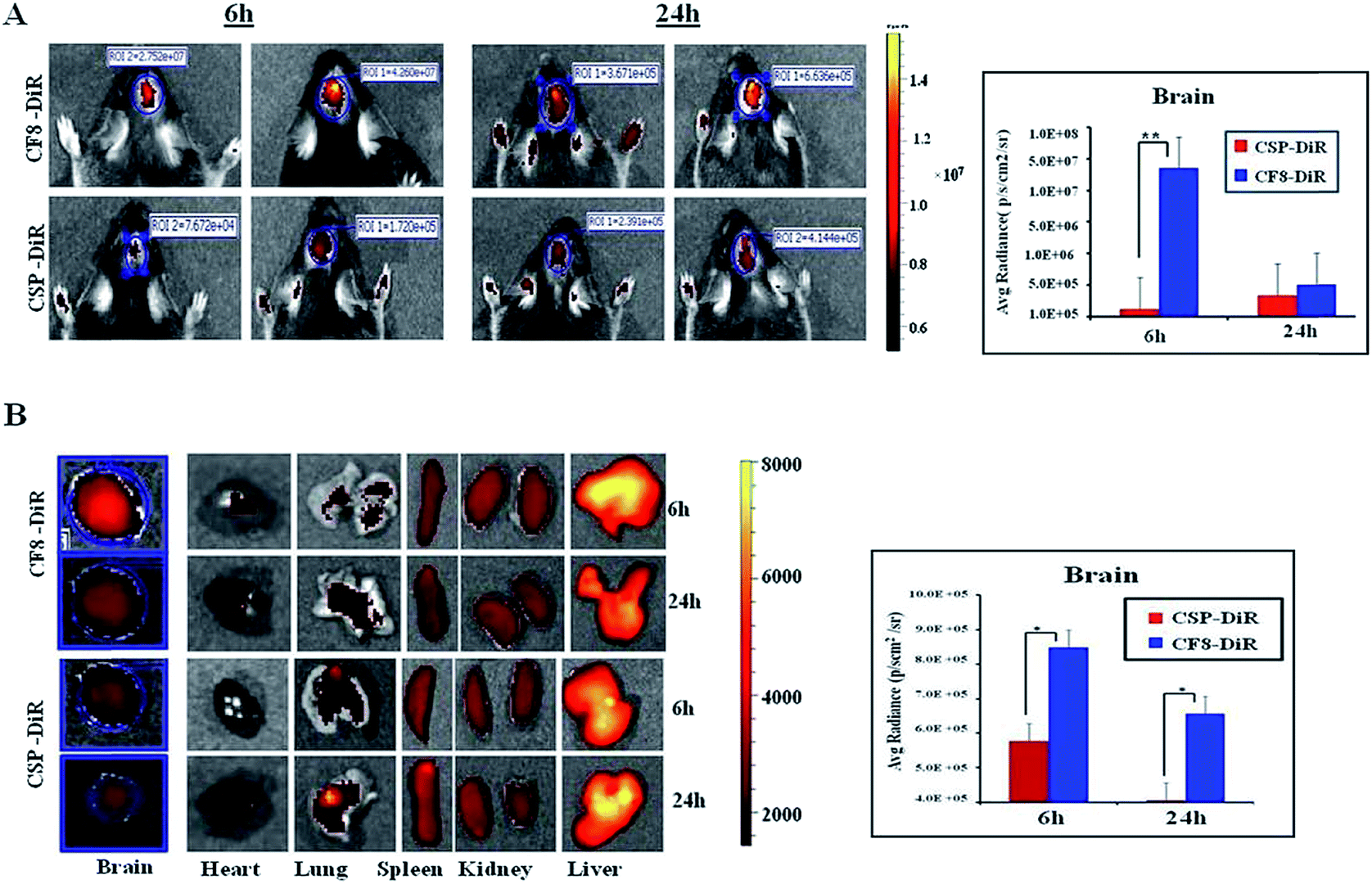 | ||
| Fig. 3 Comparison of in vivo accumulation of FR-targeting and non-targeting carbon nanospheres in orthotopic GL261 tumor bearing C57BL/6 mice following IP injections. Here CSPs are further conjugated with either an NIR dye (DiR) or DOX. (A) NIR-imaging of the brain region of mice treated with DiR-labeled CSP-F8 (CSP-F8-DiR) and CSP (CSP-DiR) after 6 h and 24 h of treatment. The graph represents the uptake and comparison of quantified DiR dye in the brain of mice treated with CSP-F8-DiR and CSP-DiR, respectively (n = 3). **p < 0.01. (B) Bio-distribution profiles of DiR-conjugated CSPs and CSP-F8 in different organs including the brain excised after sacrificing the mice from respective treatment groups after 6 h and 24 h. The graph represents the comparison of brain uptake in different treatment groups at different time points (n = 3). *p < 0.05. | ||
The accumulated amounts of nanomaterials in the brain (Fig. 3A and B) were decreased for both CF8-DiR and CSP-DiR 24 h later. Notably, even though the amounts of nanoparticle uptake in the brain are different, the uptake amounts of CF8-DiR and CSP-DiR in other organs were similar (Fig. 3B). To substantiate the notion that the observed bio-distribution pattern is because of nanoparticle-associated DiR and not because of free DiR in vivo, we injected free DiR dye intraperitoneally and found a completely different picture. Free DiR dye remains largely localized to its point of injection and even after 24 h one can observe its negligible presence in the brain (Fig. S6†), thus assuring that the bio-distribution in Fig. 3 is due to nanoparticle-associated DiR. The overall data clearly indicate that F8-conjugated nanomaterial has the propensity to have higher brain accumulation than non-targeting nanomaterials. As F8 exhibits affinity for FRs, and glioma cells implanted in the brain naturally express FRs, there is a possibility that this condition has led to enhanced brain accumulation of CF8-DiR compared to CSP-DiR.
3.5 Enhanced accumulation of CFD in FR-expressing glioma lesions in the brain
Next, we wanted to confirm if DOX-associated nanomaterials (CFD and CD) behaved similarly to NIR dye associated nanomaterials. On the 10th day, after orthotopic GL261 tumor implantation in the brain, mice were divided into three groups (n = 3) and separately injected with a single dose of DOX, CFD or CD. After 18 h, mice were sacrificed to collect brains and they were cryo-sectioned for immunohistochemical analysis. It was noticed that the CFD-accumulation (red) in the brain was significantly more than CD-accumulation, especially in FR-expressing regions (green) in the brain (Fig. 4A). Free DOX-accumulation in the brain was visually negligible in FR-expressing brain tumor regions. As a matter of fact, the FR-expression level was checked in non-tumor regions of the brain. It was found that the FR-expression was too low to detect properly, indicating that only cells in the brain tumor regions express FRs (Fig. 4B). Notably, in the non-tumor region, an almost negligible amount of CFD accumulation was observed (Fig. 4B). The overall data show that CFD accumulation is significantly higher than CD accumulation in the FR-expressing tumor region. The data are important as specific delivery of drug molecules to the tumor region and not to the non-cancer region is the most sought after observation in chemotherapy.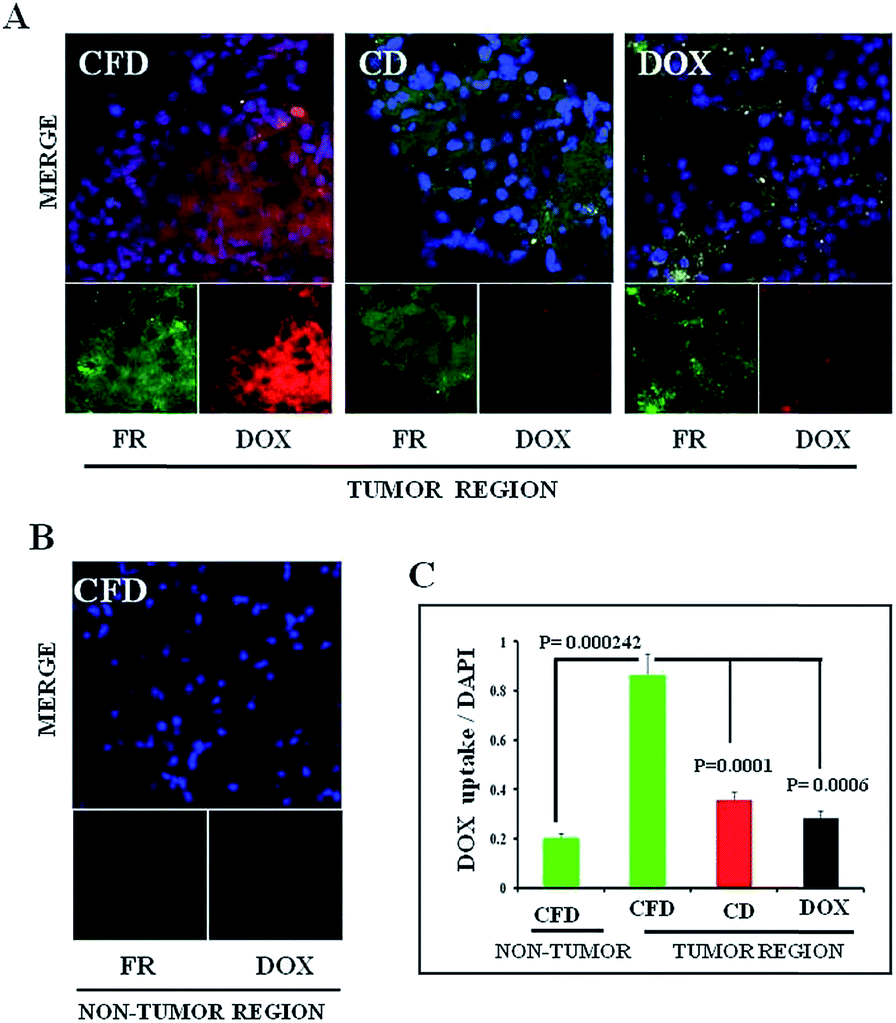 | ||
| Fig. 4 Comparison of in vivo accumulation of DOX containing FR-targeting (CFD) and non-targeting carbon nanospheres (CD), and simple DOX in brain sections of orthotopic GL261 tumor bearing mice following IP injections. (A) Confocal fluorescent images of tumor-bearing brain sections obtained from mice treated, respectively, with CFD, CD and DOX. The top panel shows the merged image of blue (DAPI, nuclear staining), green (FR expression) and red (DOX accumulation) fields and the bottom panels are images, respectively, obtained in green and red fields. (B) FR expression level and accumulation of CFD in CFD treated mice in the tumor-free region of the brain. Lower panels are green (FR expression, left panel) and blue (DAPI/nuclear staining, right panel) fields, and the upper panel is a merged image of green and red fields. (C) Quantification of red fluorescence in non-tumor and tumor regions of mice separately treated with CFD, CD, and DOX. | ||
3.6 Isolation of TAMs and estimation of FR expression in TAM population obtained from the tumor microenvironment
Tumor associated macrophages (TAMs), which express FRs, play a predominant role in cancer progression, invasion and metastasis.16We hence intended to find out if an FR-targeting nanomaterial (CFD) has any selective uptake in TAMs. For this study, isolation of TAMs from the GL261 tumor was essential. Since the tumor size in the orthotopic mouse brain is very small, the isolation of TAMs from the in situ brain tumor is difficult. So, TAMs were isolated from a subcutaneous GL261 tumor in mice using an antibody against the TAM marker CD11b. The TAM isolation (Fig. 5A) was performed using CD11b antibody-labelled magnetic beads. First, surface markers of TAMs in the magnetically separated CD11b+ TAM cell fraction were checked. This was followed by the examination of expression markers such as F4/80 and CD68 (Fig. 5B), which are established macrophage markers.27 Most importantly, no expression of LY6C and low expression of MHC-II (here, no initial shift was observed) were found, which characteristically represents alternatively activated (M2) macrophages (or TAMs) that are immunosuppressive and tumor promoting in nature.28 In contrast, cell populations exhibiting combinations of high LY6C, low MHC-II inflammatory monocytes, or high LY6C, high MHC-II immature macrophages, or intermediate LY6C, high MHC-II represent classically activated M1 macrophages; all these cells possess the ability to eradicate tumors.28 Hence, CD11b+ TAMs isolated from the GL261 tumor were cross-checked for the extent of expression of FRs and F4/80 in these cells by flow cytometry, and the status of CD11b+/FR+ and F4/80+/FR+ populations was analysed, respectively (Fig. 5C). It was observed that 41–44% of total TAMs have these two populations, indicating that the FR-positive population among CD11b+ TAMs is indeed real. We also find that the glioma lesions in brain sections do express F4/80, indicating the presence of TAMs in the brain (Fig. S7†). As TAMs also express FRs, it is logically expected that TAMs in the brain could be targeted through their expressed FRs using CFD.
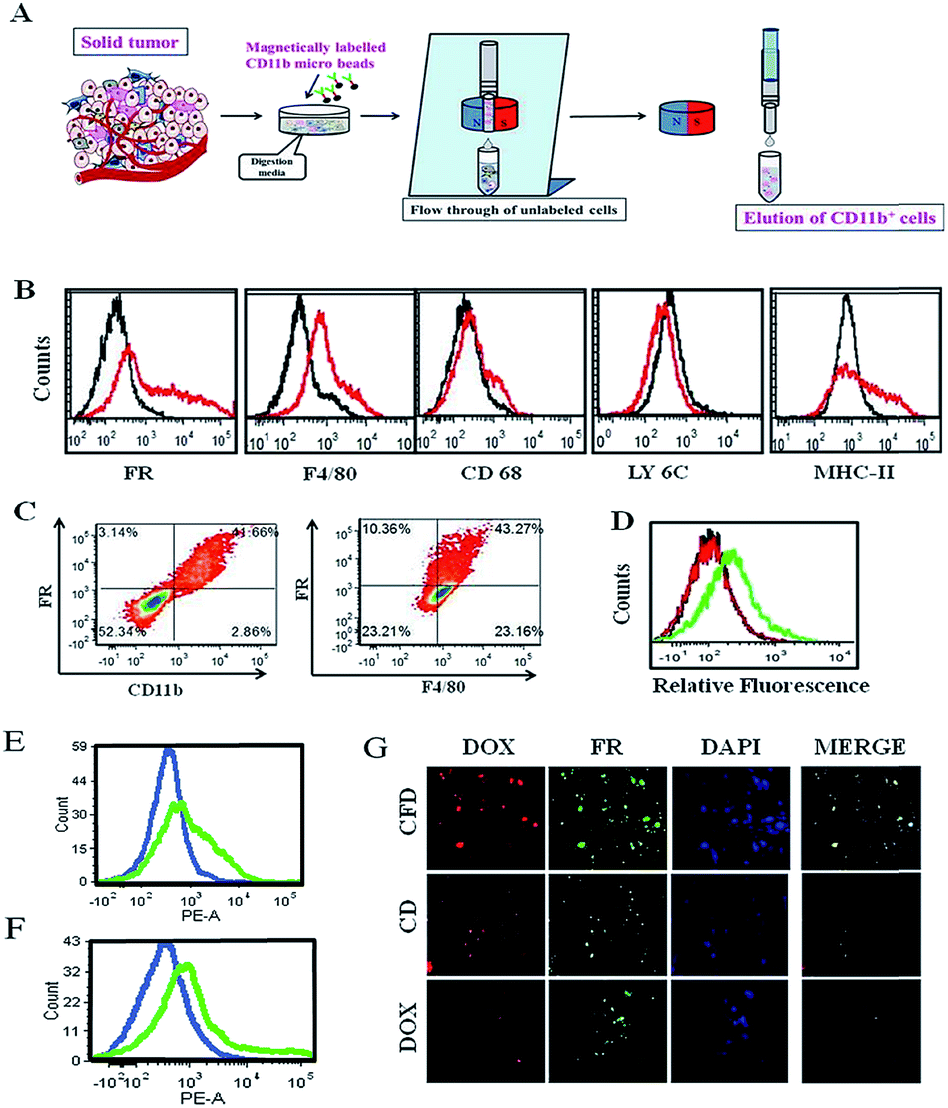 | ||
| Fig. 5 FR-targeting carbon nanospheres locate in tumor associated macrophages (TAMs) in the tumor environment obtained from the subcutaneous GL261 tumor model. The TAMs are isolated using magnetically labeled CD11b-associated micro-beads. (A) Scheme showing the separation method for TAM isolation from a solid tumor. (B) Flow cytometric analyses of tumor-associated surface marker expressions on TAMs isolated from a subcutaneous GL261 tumor in mice. Expression levels of different surface markers (red) on TAMs and their corresponding subtype IgG controls (black) are represented accordingly. (C) The percentages of CD11b+ and F4/80+ macrophages simultaneously expressing FRs. (D) Flow cytometric analysis of TAMs isolated from tumor-bearing mice treated with CFD (green), CD (red) and DOX (black). (E) Flow cytometric analysis of differences in CFD uptake in TAMs (green) and TAM-free, isolated tumour cells (blue) from the tumor-bearing mice. (F) Flow cytometric analysis to exhibit the differences in FR-expression levels in TAMs (green) and TAM-free, isolated tumour cells (blue) from the tumor-bearing mice. (G) Confocal microscopy images of TAMs isolated from CFD-, CD- or DOX-treated tumor-bearing mice. | ||
3.7 CFD accumulates in TAMs in vivo
For this study, after subcutaneous GL261 tumor inoculation, when the tumor size reached 1500 mm3 volume, mice were divided into three groups and separately treated with CFD, CD or free DOX. After 12 h, CD11b+ TAMs were isolated from tumors of sacrificed mice in different treatment groups. The extent of uptake of DOX in TAMs from different treatment groups was analysed by flow cytometry. Higher uptake of DOX was obtained in TAMs from CFD-treated mice. Uptake of CD and free DOX in TAMs was much lower compared to CFD-uptake (Fig. 5D). Incidentally, the amounts of uptake of CD and free DOX in TAMs are similar. We further compared the uptake of CFD in TAMs and in isolated tumor cells. For this, CD11b-void cells were separately isolated. These cells are a heterogeneous mixture of all tumor-associated cells except TAMs. Clearly the uptake of CFD is more in TAMs than in tumour cells (Fig. 5E). This is corroborated by the observation that the FR expression is also evidently higher in TAMs than in tumour cells (Fig. 5F). Thus, the uptake of CFD is more or less directly proportional to the FR expression level. Additionally, the uptake pattern of CFD, CD and DOX (shown in Fig. 5D) was also confirmed by fluorescence microscopy images of isolated TAMs from mice of respective treatment groups (Fig. 5G). Clearly, these FR-positive TAMs treated with CFD have higher propensity to take up DOX. Virtually, the uptake of CD and DOX in these TAMs was significantly low. From Fig. 5B, C and F it is clear that TAMs express FRs, possibly even more than the gross tumor cells, and CFD by virtue of the FR-targeting ligand F8 is selectively and efficiently taken up by these TAMs.3.8 Tumor growth inhibition study
Further, with a view to examine the efficacy of the orthotopic tumor growth inhibitory effect of the TAM and tumor cell (dual) targeting CFD system orthotopic GL261 glioma bearing mice were developed. On day 4, post orthotopic tumor implantation in the brain, mice were randomly divided into four groups (n = 6) and separately administered CFD, CD, free DOX or 5% glucose (untreated control group) intraperitoneally on every alternate day with a total of five doses. The DOX amount so injected in all treated groups was maintained at 5 mg kg−1 body weight of mice in all doses. Importantly, mice treated with CFD had significantly improved survivability compared to mice in untreated and other treated groups (Fig. 6A). We believe that this enhanced survivability of CFD-treated orthotopic tumor bearing mice was presumably due to the dual targeting effect of DOX associated CFD on FR-expressing tumor cells and TAMs. For visibly assessing the effect of CFD on the tumor mass, we performed an in vivo tumor regression study in subcutaneous GL261 tumor-bearing mice. After 12 days of subcutaneous tumor inoculation, mice were divided into 4 groups (n = 5) and treated separately similar to the above-mentioned treatment groups, and the mouse survivability was observed. It is clear that CFD treated mice have the highest overall survivability compared to other treatment groups (Fig. 6B). Fig. 6C shows the trend of subcutaneous tumor regression over 22 days, after which mice were sacrificed and tumors from different groups were harvested and their sizes were compared (shown on the top of Fig. 6C). The visual comparison of tumor sizes conforms with the respective tumor volume (Fig. 6D), which proves the efficient in vivo effect of CFD on tumor targeting and tumor-regression ability.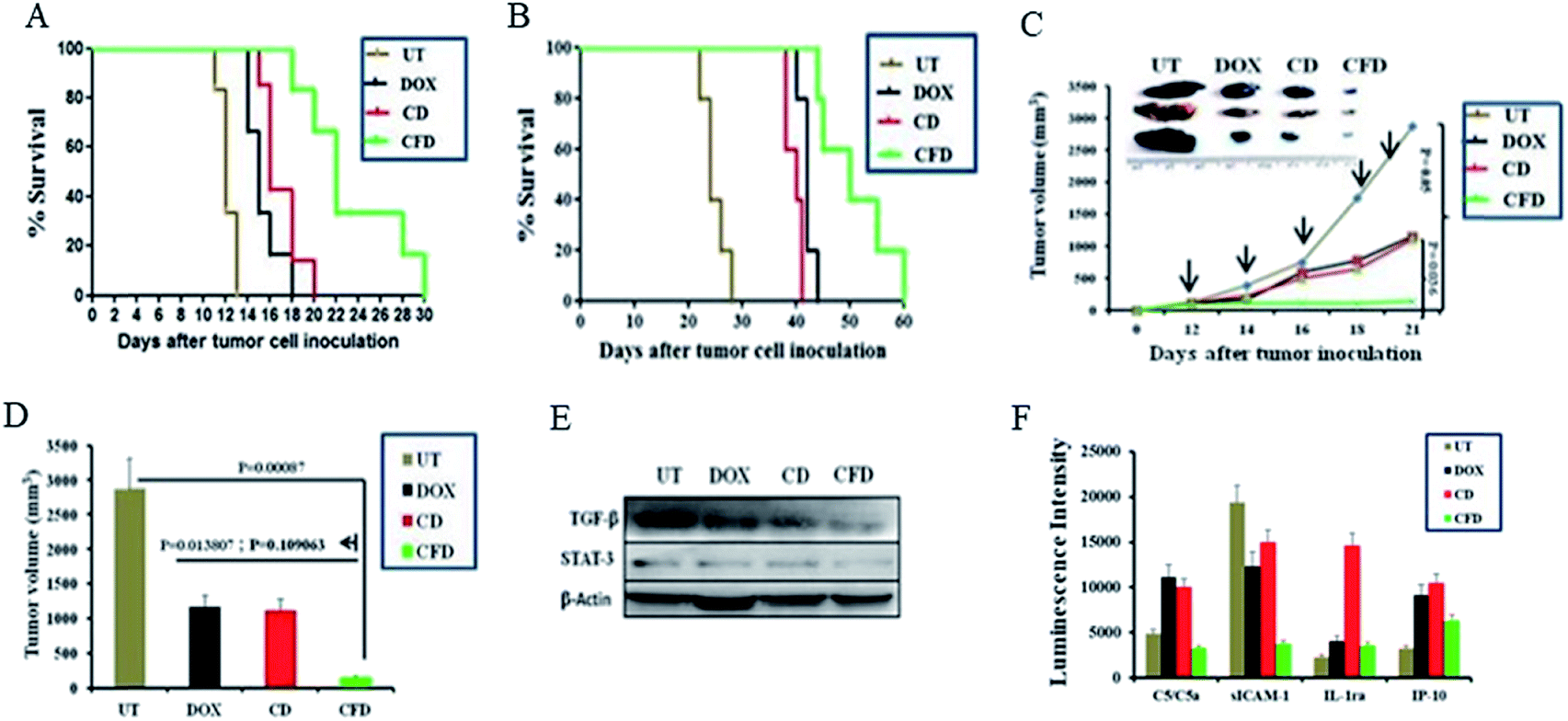 | ||
| Fig. 6 FR-targeting nanospheres inhibit tumor growth in mice. (A) Cumulative survival study of orthotopic GL261 brain tumor bearing mice after treatment with CFD, CD or pristine DOX on days 4, 6, 8, 10 and 12 after tumor cell inoculation. UT represents the group treated with 5% glucose. In all DOX-treated groups the amount of DOX was 5 mg kg−1 body weight of mice. (B) Cumulative survival study of subcutaneous GL261 tumor bearing mice after CFD, CD, DOX treatments. UT represents 5% glucose treatment. (C) Graph indicates the tumor regression curve after subcutaneous GL261 tumor inoculation followed by respective treatments for the represented groups on days 12, 14, 16, 18 & 20. The image represents tumors obtained from mice treated with 5% glucose, DOX, CD and CFD 22 days after inoculation of cells subcutaneously. (D) Subcutaneous tumor volume of indicated treatment groups after five treatments followed by sacrificing of mice on day 22 from the day of subcutaneous inoculation of GL261 cells in C57BL/6J mice. (E) Expression of TGF-β and STAT-3 with respect to β-actin in brain tumor-lysates from mice treated with 5% glucose (UT), DOX, CD and CFD, respectively. (F) Levels of various chemokines in brain tumor (n = 3) lysates obtained from mice treated with 50% glucose (UT), DOX, CD and CFD, respectively. | ||
3.9 Western blot and cytokine profile analysis
Next, in order to investigate the dual targeting effects of CFD in the tumor microenvironment, Western blot and cytokine profile analyses were performed on tumor lysates obtained from tumors of orthotopic GL261 glioma mice (n = 3) treated with CFD, CD, free DOX or 5% glucose (UT). Western blot analysis (Fig. 6E) shows that the expression of TGF-β and STAT-3 was prominently reduced by CFD. These proteins, typically expressed by TAMs, help tumor-associated cells’ proliferation and invasion.10,29 Cytokine profile analysis of tumor lysates shows the changes in the expression of certain common cytokines. In comparison to the untreated sample, CFD treatment led to a decrease in the expression of macrophage-released cytokine C5a and another cytokine released by tumor epithelial cells called sICAM-1, which are involved in tumor progression.30–34 In contrast, CFD also mediated the increase of certain tumor-inhibiting cytokines such as IL-1ra and IP-10 (Fig. 6F). As IL-1 is involved in tumor invasiveness and angiogenesis,34 the antagonist to the IL-1 receptor, i.e., IL-1ra which is expressed by immune cells, epithelial cells etc., antagonizes the IL-1-mediated tumor aggressiveness and pro-inflammation.35,36 Further, the IFN-γ-induced protein (IP10; also referred to as CXCL10), which is produced mostly from immune cells besides epithelial cells due to innate and adaptive immune responses, is associated with anti-tumorigenic angiostatic properties, involved in the recruitment of NK cells in tumor masses etc.37,38 Clearly, CFD treatment leads to the suppression (e.g., C5/C5a and sICAM-1) or up-regulation (e.g., IL-1ra and IP-10) of cytokines in tumor masses pointing toward specific anti-cancer activity of the FR-targeting CFD formulation in tumor masses and tumor associated macrophages. Importantly, these data are corroborated in tumor-bearing brains also. The lysates of tumor-bearing brains from mice treated with the above-mentioned compounds also show that CFD indeed induced decreased expression of sICAM-1 and C5/C5a compared to other treatment groups [Fig. S8†].3.10 Histology analysis using H & E staining
Histology analyses of various organs of mice were performed. Organs such as the liver, heart and spleen were harvested from sacrificed mice IP injected with single injection of either CFD or PBS. Harvested organs were sectioned and stained with hematoxylin and eosin (H & E). The respective sections from two different groups were compared. The section images (Fig. 7) of CFD-treated mice showed no distinct or perceptible changes in physiological characteristics of organ-tissues as these were comparable to those of tissues obtained from PBS-treated mice. No tissue damage following CFD treatment indicates that CFD possibly has no short-term, non-specific toxicity to vital organs such as the liver during the course of treatment.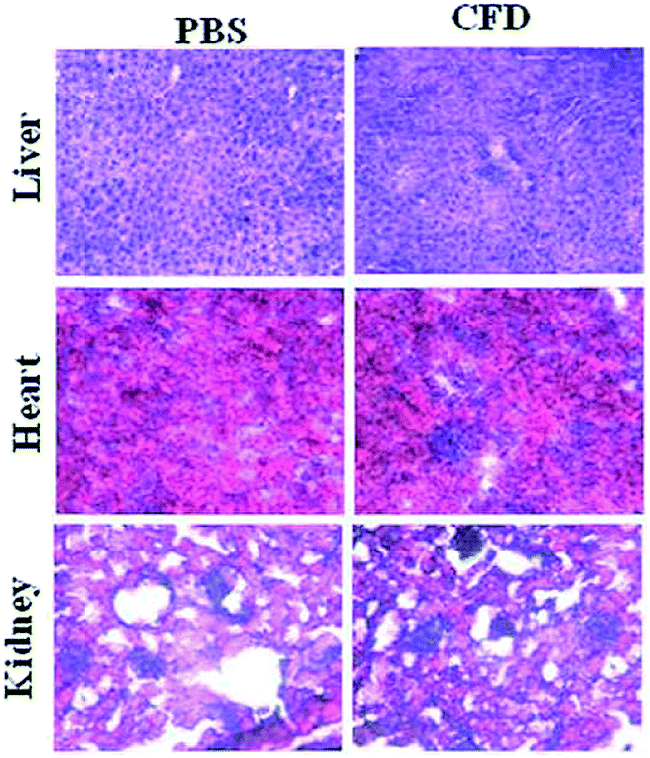 | ||
| Fig. 7 Representative H & E staining of sections of healthy organs (liver, heart and kidney) of mice. After 24 h of CFD administration, mice were sacrificed and organs were isolated. Sections were stained with H & E and compared with organ sections obtained from mice treated with only PBS. No perceptible tissue damage was demonstrated following CFD administration. | ||
4. Discussion
In spite of significant preclinical and clinical research toward developing efficient therapeutics against glioma the enhancement of overall survivability in glioma subjects and patients still remains challenging. Targeting the brain through the BBB was accomplished by targeting through various receptors. The nicotinic acetylcholine receptor is one such example, which is over-expressed on brain endothelial cells and neuromuscular junctions in the CNS.39–41 Using a D-peptide ligand to target these receptors for delivering doxorubicin Wei et al. reported a less than 1.5 times increase in median survivability of glioma nude mice compared to the median survivability of the untreated group.42 Cyclic RGD shows affinity for various integrin receptors over-expressed on glioblastoma and tumor-associated endothelial cells.43 Although controversial about how to target in situ glioblastoma through the BBB, Miura et al. reported cyclic RGD-based polymeric micelles with a platinum-based drug for glioblastoma regression in an orthotopic model in mice.44 These are representative examples to showcase how to target tumor-associated endothelial cells, but there is no evidence of targeting TAMs in brain tumor masses.TAMs are functionally linked with the receptors that increase malignancy. Cytokines such as TGF-β and IL-10 are frequently released in the TME and are implicated with polarization of M2 macrophages.45 Recently, Karlsson et al. reported targeting the MARCO receptor on TAMs using an anti-monoclonal MARCO antibody based targeted therapy in breast and colon tumor-associated TMEs.46 They reported antitumor activity resulting from reprogramming of TAM populations to become a pro-inflammatory phenotype, increasing immunogenicity in tumors. Herein, we demonstrate TAM targeting through the over-expressed folate receptor (FR). The FR is incidentally over-expressed on both TAMs and glioma cells. On this basis, we accomplished drug-delivery to tumor masses for effective tumor regression.
Carbon nanospheres (CSPs) are gaining more attention among carbon-based materials in biomedical applications. Selvi et al. reported these carbon nanospheres as a good candidate for crossing BBB and delivering drugs to the nucleus and intra-cellular compartments.14 The folate receptor is overexpressed on several epithelial cancer cells. Corbi et al. reported that overexpression of the FR is found on TAMs having an M2 like functional profile while exerting immunosuppressive functions in the TME.16 Hence, it could be logically hypothesized that upon association with the FR-targeting molecule (F8), the functional CSPs (i.e., CF8) would be able to deliver the drug cargo not only within the brain but also selectively to FR-expressing glioma cells and TAMs after crossing the BBB. The CSPs, being negatively charged, electrostatically accommodate F8 and hence it is possible for them to carry excess ligands. This is one of the limitations of other conventional FR-targeting delivery systems such as liposomes. Because of the enhanced amount of F8, CF8 would experience enhanced affinity for the FR. Additionally, as the BBB also over-expresses the FR,47 it is pertinent to think that CF8 would have relatively higher interaction with the BBB. In fact, it was found to be true as we observed 100-fold higher uptake of CF8 than CSPs in the brain area within 6 h. The uptake dissipated eventually by 24 h but by then the interaction with the BBB and eventual entry in the brain would ensure higher uptake of CF8 in the brain. As CF8 targets FRs, one can expect selective uptake of CF8 in FR-expressing glioma lesions. Further, CF8 upon conjugation with doxorubicin (i.e., CFD) showed higher in vitro toxicity with enhanced cellular uptake in GL261 than that of the non-targeting CSPs.
To confirm CFD-mediated DOX delivery to TAMs especially through FRs, it was necessary to check the level of FRs in TAMs and the selective uptake of FR-targeting CFD in TAMs. Following CFD-treatment, CD11b+ TAMs were isolated. The isolated TAMs showed maximum uptake for CFD compared to the non-targeting CSPs, which indicates that FRs on the TAM surface are functionally active for active drug transport. For the treatment of SLE disease, Philip S. Low and co-workers have shown that FR-positive macrophages adorned with folate-linked highly immunogenic ‘haptens’ are conveniently recognised and destroyed by body's innate immune response.48 This strategy was extended to cancer immunotherapy also.49 It was also shown that activated macrophages express FRs and are hence used as drug targets. However, FRs are not expressed in F4/80+ peritoneal macrophages obtained from non-tumor associated models.50 Our data show that TAMs obtained by using the marker CD11b+ express FRs and also carry the F4/80 marker. This clearly indicates that simultaneous expression of FRs in tumor specific TAMs that bear the F4/80 marker is evident and is potentially useful for active drug targeting. But the important question would be to know if glioma tumor lesions also contain TAMs. This is important as the original idea is to deliver drugs to both tumor cells and TAMs in glioma lesions in the brain. Our data (Fig. S7†) show that brain sections carrying tumor lesions certainly express F4/80 which is one of the markers for macrophages. And as TAMs express FRs, it is expected that the region of tumor in the brain contains FR-expressing tumor cells and F4/80+ TAMs. Clearly, an FR targeting drug delivery system (here CFD) will have access to both TAMs and tumor cells in tumor masses in situ. Although it has been shown by FACS data that CFD gains access to TAMs, this example of active drug transport to TAMs especially for the glioma model, to the best of our knowledge, was not reported before.
One can notice that there is up-regulated FR expression in tumor regions in the brain section. Together with higher DOX uptake in the tumor-associated brain in CFD-treated mice, we believe it offers an additional advantage for FR-targeting anti-glioma therapeutics. This also substantiates the FR as a reliable prognostic marker useful for glioma treatment. CFD could increase the average survivability more than twice that of the untreated control, indicating possible adverse effects on glioma cells as well as TAMs in tumor lesions. The adverse effect on TAMs was evident by the change of levels of cytokines which are expressed mostly by immune cells.
TAMs express TGF-β, which influences tumor-associated epithelial cells to proliferate via many pathways including the STAT-3 pathway. A decrease in TGF-β followed by the STAT-3 level in the tumor lysate advocates the possibility that both TAMs and tumor cells are adversely affected by CFD. TGF-β is known to induce EMT, which leads to metastasis. A decrease in TGF-β evidently induces EMT reversal and reduces metastasis. Similar to the use of DOX as a model anticancer agent, any other potent drug, in combination with DOX or otherwise, that synergizes the EMT reversal can be used for a better preclinical outcome. Clearly, findings from this study open the door for a new targeting strategy which includes targeting both FR-expressing tumor associated macrophages and tumor cells with a single drug delivery system thereby promising an effective strategy against aggressive tumors such as glioblastoma. The targeting system bears promise in future for delivering multitude of drugs in combination for a better anti-tumor efficacy. Although there was no noticeable toxicity in CFD-treated mice, a complete toxicological evaluation is certainly due before the system is projected further for clinical trials.
5. Conclusion
In summary, we have developed a new, dual-targeting system using functional glucose-based carbon nanospheres for selective delivery of a model anti-cancer drug, doxorubicin. The system does not require FR-over-expression for targeting but can target even TAMs that moderately express FRs and malignant cells in tumor stroma. The simultaneous targeting and killing of TAMs and malignant cells by CFD is schematically described (Scheme 2). Findings from in vitro and in vivo experiments revealed clear therapeutic benefits for using this dual targeting strategy for simultaneously targeting both FR-expressing tumor cells and TAMs in the tumor microenvironment. This viable strategy enables effective brain tumor regression and increased survivability.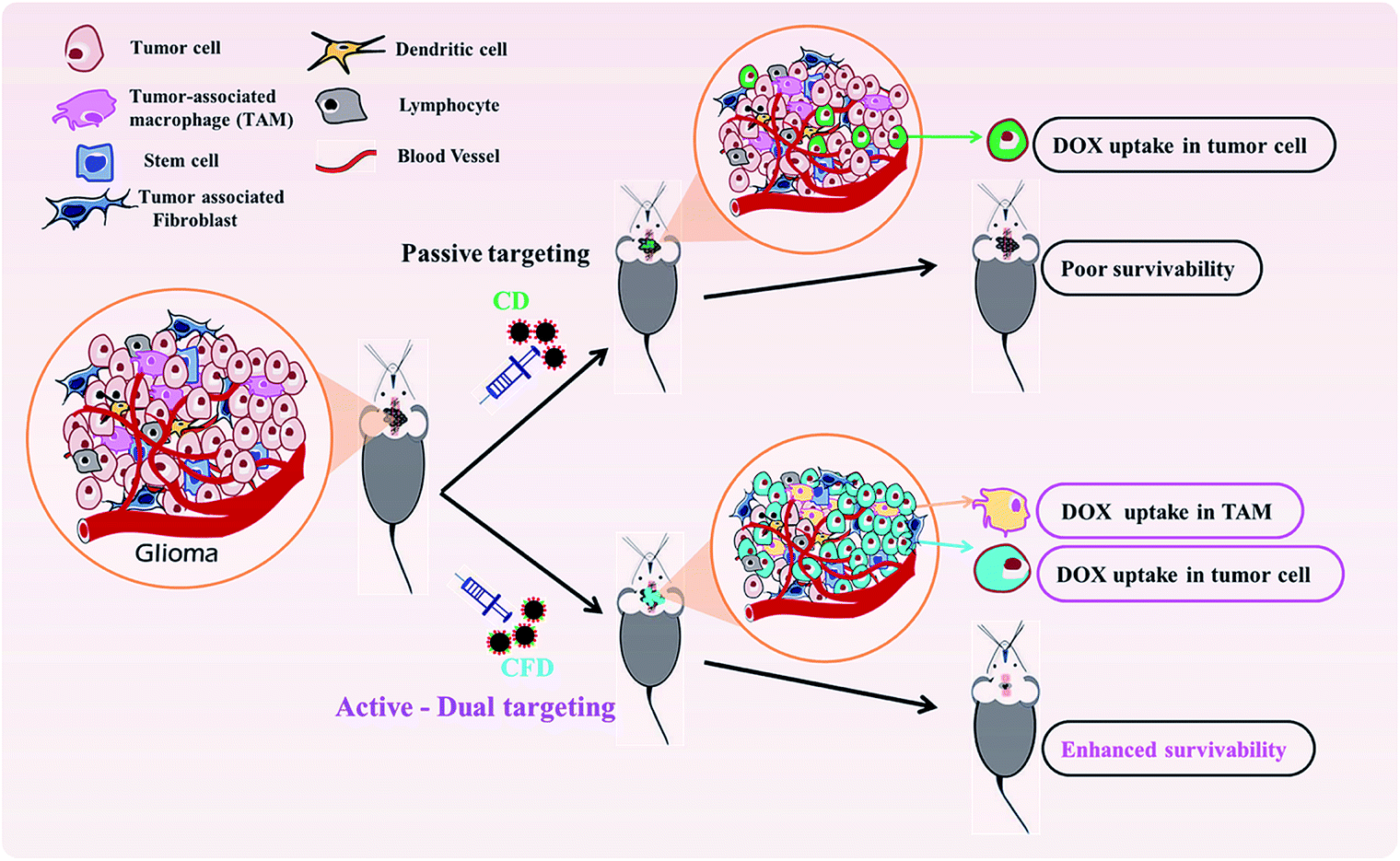 | ||
| Scheme 2 CFD-mediated dual targeting of TAMs and malignant tumor cells. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the CSIR-Mayo Clinic Partnership Grant (CMPP010), other CSIR network projects CSC0302, BSC0123, Govt. of India (to R. B.), and Department of Biotechnology sponsored project (BT/PR11756/NNT/28/764/2014) (to R. B. & T. K. K), Department of Science and Technology, Nanomission Project (SR/NM/NS-1058/2015-JNCASR), Govt. of India (to T. K. K. and M. E, JNCASR). C. K. E. and K. S. acknowledge CSIR, and M. T. A. acknowledges DBT doctoral research fellowships, respectively. This is IICT communication no. IICT/Pubs/2018/102.References
- R. Y. Bai, V. Staedtke and G. J. Riggins, Trends Mol. Med., 2011, 17, 301–312 CrossRef CAS PubMed.
- C. E. Lewis and J. W. Pollard, Cancer Res., 2006, 66, 605–612 CrossRef CAS.
- K. D. Elgert, D. G. Alleva and D. W. Mullins, J. Leukocyte Biol., 1998, 64, 275–290 CrossRef CAS PubMed.
- C. Sunderkötter, M. Goebeler, K. Schulze-Osthoff, R. Bhardwaj and C. Sorg, Pharmacol. Ther., 1991, 51, 195–216 CrossRef.
- W. Zhou, S. Q. Ke, Z. Huang, W. Flavahan, X. Fang, J. Paul, L. Wu, A. E. Sloan, R. E. McLendon, X. Li, J. N. Rich and S. Bao, Nat. Cell Biol., 2015, 17, 170 CrossRef CAS PubMed.
- C. Murdoch, A. Giannoudis and C. E. Lewis, Blood, 2004, 104, 2224–2234 CrossRef CAS PubMed.
- S. M. Pyonteck, L. Akkari, A. J. Schuhmacher, R. L. Bowman, L. Sevenich, D. F. Quail, O. C. Olson, M. L. Quick, J. T. Huse, V. Teijeiro and M. Setty, Nat. Med., 2013, 19, 1264–1272 CrossRef CAS PubMed.
- D. F. Quail, R. L. Bowman, L. Akkari, M. L. Quick, A. J. Schuhmacher, J. T. Huse, E. C. Holland, J. C. Sutton and J. A. Joyce, Science, 2016, 352 Search PubMed.
- H. Chong, Y. Vodovotz, G. W. Cox and M. H. Barcellos-Hoff, J. Cell. Physiol., 1999, 178, 275–283 CrossRef CAS.
- S. Wan, E. Zhao, I. Kryczek, L. Vatan, A. Sadovskaya, G. Ludema, D. M. Simeone, W. Zou and T. H. Welling, Gastroenterology, 2014, 147, 1393–1404 CrossRef CAS PubMed.
- M. A. Miller, Y. R. Zheng, S. Gadde, C. Pfirschke, H. Zope, C. Engblom, R. H. Kohler, Y. Iwamoto, K. S. Yang, B. Askevold and N. Kolishetti, Nat. Commun., 2015, 6, 8692 CrossRef CAS PubMed.
- P. Zhang, Z. A. Qiao and S. Dai, Chem. Commun., 2015, 51, 9246–9256 RSC.
- A. Nieto-Márquez, R. Romero, A. Romero and J. L. Valverde, J. Mater. Chem., 2011, 21, 1664–1672 RSC.
- B. R. Selvi, D. Jagadeesan, B. S. Suma, G. Nagashankar, M. Arif, K. Balasubramanyam, M. Eswaramoorthy and T. K. Kundu, Nano Lett., 2008, 8, 3182–3188 CrossRef CAS PubMed.
- J. M. Saul, A. Annapragada, J. V. Natarajan and R. V. Bellamkonda, J. Controlled Release, 2003, 92, 49–67 CrossRef CAS PubMed.
- A. Puig-Kröger, E. Sierra-Filardi, A. Dominguez-Soto, R. Samaniego, M. T. Corcuera, F. Gómez-Aguado, M. Ratnam, P. Sanchez-Mateos and A. L. Corbi, Cancer Res., 2009, 69, 9395–9403 CrossRef PubMed.
- D. Cheng, N. Cao, J. Chen, X. Yu and X. Shuai, Biomaterials, 2012, 33, 1170–1179 CrossRef CAS PubMed.
- N. Chen, C. Shao, Y. Qu, S. Li, W. Gu, T. Zheng, L. Ye and C. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 19850–19857 CrossRef CAS PubMed.
- Y. C. Chen, C. F. Chiang, S. K. Wu, L. F. Chen, W. Y. Hsieh and W. L. Lin, J. Controlled Release, 2015, 211, 53–62 CrossRef CAS PubMed.
- H. Gao, Z. Yang, S. Zhang, S. Cao, S. Shen, Z. Pang and X. Jiang, Sci. Rep., 2013, 3, 2534 CrossRef CAS PubMed.
- J. Wang, Y. Yang, Y. Zhang, M. Huang, Z. Zhou, W. Luo and J. Tang, Adv. Funct. Mater., 2016, 26, 7873 CrossRef CAS.
- L. Zeng, L. Zou, H. Yu, X. He, H. Cao, Z. Zhang, Q. Yin, P. Zhang, W. Gu, L. Chen and Y. Li, Adv. Funct. Mater., 2016, 26, 4201–4212 CrossRef CAS.
- C. K. Elechalawar, K. Sridharan, A. Pal, M. T. Ahmed, M. Yousuf, S. S. Adhikari and R. Banerjee, Biomater. Sci., 2017, 5, 1898–1909 RSC.
- H. Okada, H. Tahara, M. R. Shurin, J. Attanucci, K. M. Giezeman-Smits, W. K. Fellows, M. T. Lotze, H. Chambers and M. E. Bozik, Int. J. Cancer, 1998, 78, 196–201 CrossRef CAS PubMed.
- L. Cassetta, R. Noy, A. Swierczak, G. Sugano, H. Smith, L. Wiechmann and J. W. Pollard, Adv. Exp. Med. Biol., 2016, 211–229 CrossRef CAS PubMed.
- D. Laoui, E. Van Overmeire, J. Keirsse, K. Movahedi and J. A. Van, Bio-Protoc., 2014, 4, e1294 CrossRef.
- J. G. Quatromoni and E. Eruslanov, Am. J. Transl. Res., 2012, 4, 376 Search PubMed.
- R. Noy and J. W. Pollard, Immunity, 2014, 41, 49–61 CrossRef CAS.
- N. B. Hao, M. H. Lü, Y. H. Fan, Y. L. Cao, Z. R. Zhang and S. M. Yang, Clin. Dev. Immunol., 2012, 2012 Search PubMed.
- J. Panse, K. Friedrichs, A. Marx, Y. Hildebrandt, T. Luetkens, K. Bartels, C. Horn, T. Stahl, Y. Cao, K. Milde-Langosch and A. Niendorf, Br. J. Cancer, 2008, 99, 930 CrossRef CAS PubMed.
- H. Nitta, Y. Murakami, Y. Wada, M. Eto, H. Baba and T. Imamura, Oncol. Rep., 2014, 32, 1715–1719 CrossRef CAS PubMed.
- C. Rosette, R. B. Roth, P. Oeth, A. Braun, S. Krammerer, J. Ekblom and M. F. Denissenko, Carcinogenesis, 2005, 26, 943–950 CrossRef CAS PubMed.
- B. A. Teicher and S. P. Fricker, Clin. Cancer Res., 2010, 16, 2927–2931 CrossRef CAS PubMed.
- Y. Gong, E. Scott, R. Lu, Y. Xu, W. K. Oh and Q. Yu, PLoS One, 2013, 8, e77366 CrossRef CAS PubMed.
- E. Voronov, D. S. Shouval, Y. Krelin, E. Cagnano, D. Benharroch, Y. Iwakura, C. A. Dinarello and R. N. Apte, Proc. Natl. Acad. Sci., 2003, 100, 2645–2650 CrossRef CAS PubMed.
- S. Perrier, F. Darakhshan and E. Hajduch, FEBS Lett., 2006, 580, 6289–6294 CrossRef CAS PubMed.
- J. A. Belperio, M. P. Keane, D. A. Arenberg, C. L. Addison, J. E. Ehlert, M. D. Burdick and R. M. Strieter, J. Leukocyte Biol., 2000, 68, 1–8 CAS.
- K. Kajitani, Y. Tanaka, K. Arihiro, T. Kataoka and H. Ohdan, Breast Cancer Res. Treat., 2012, 134, 139–155 CrossRef PubMed.
- P. Kumar, H. Wu, J. L. McBride, K. E. Jung, M. H. Kim, B. L. Davidson, S. K. Lee, P. Shankar and N. Manjunath, Nature, 2007, 448, 39 CrossRef CAS PubMed.
- S. Son, W. Hwang do, K. Singha, J. H. Jeong, T. G. Park, D. S. Lee and W. J. Kim, J. Controlled Release, 2011, 155, 18–25 CrossRef CAS PubMed.
- C. Zhan, Z. Yan, C. Xie and W. Lu, Mol. Pharm., 2010, 7, 1940–1947 CrossRef CAS PubMed.
- X. Wei, C. Zhan, Q. Shen, W. Fu, C. Xie, J. Gao, C. Peng, P. Zhang and W. Lu, Angew. Chem., Int. Ed., 2015, 54, 3023–3027 CrossRef CAS PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef CAS.
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto, H. Koyama, H. Cabral, N. Nishiyama and K. Kataoka, ACS Nano, 2013, 7, 8583–8592 CrossRef CAS PubMed.
- F. O. Martinez, L. Helming and S. Gordon, Annu. Rev. Immunol., 2009, 27, 451–483 CrossRef CAS PubMed.
- A. M. Georgoudaki, K. E. Prokopec, V. F. Boura, E. Hellqvist, S. Sohn, J. Östling, R. Dahan, R. A. Harris, M. Rantalainen, D. Klevebring and M. Sund, Cell Rep., 2016, 15, 2000–2011 CrossRef CAS PubMed.
- D. Wu and W. M. Pardridge, Pharm. Res., 1999, 16, 415–419 CrossRef CAS.
- B. Varghese, N. Haase and P. S. Low, Mol. Pharm., 2007, 4, 679–685 CrossRef CAS PubMed.
- Y. Lu, E. Sega, C. P. Leamon and P. S. Low, Adv. Drug Delivery Rev., 2004, 56, 1161–1176 CrossRef CAS PubMed.
- W. Xia, A. R. Hilgenbrink, E. L. Matteson, M. B. Lockwood, J. X. Cheng and P. S. Low, Blood, 2009, 113, 438–446 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00056a |
| ‡ Equal contributing authors. |
| § Current Address: School of Veterinary Science, The University of Queensland, Gatton Campus, Queensland 4343, Australia. |
| This journal is © The Royal Society of Chemistry 2019 |