
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom a precursor to an etchant: spontaneous inversion of the role of Au(III) chloride for one-pot synthesis of smooth and spherical gold nanoparticles†
Seokyoung
Yoon
 a,
Chansong
Kim
b,
Byoungsang
Lee
b and
Jung Heon
Lee
*abc
a,
Chansong
Kim
b,
Byoungsang
Lee
b and
Jung Heon
Lee
*abc
aSKKU Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea. E-mail: jhlee7@skku.edu; Fax: +82-502-302-1918; Tel: +82-31-290-7404
bSchool of Advanced Materials Science and Engineering, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
cBiomedical Institute for Convergence at SKKU (BICS), Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
First published on 15th April 2019
Abstract
We report the inversion of the role of Au(III) chloride, from a gold precursor to an etchant, for the synthesis of smooth and spherical AuNPs with nanoscale size tunability in a one-pot-system. Inversion of the role of Au(III) chloride was achieved by regulating the ratio between the reducing agent and Au(III) chloride.
Introduction
Among metal nanoparticles, gold nanoparticles (AuNPs) have been actively studied because of their high biocompatibility, stability, and excellent catalytic, electrical, and optical properties.1–6 In particular, the localized surface plasmon resonance (LSPR) of AuNPs allows them to be used for diverse optical applications, including sensing,7–9 surface-enhanced Raman scattering,10 surface-enhanced fluorescence,11 and plasmon resonance energy transfer.12 It is widely known that the LSPR of AuNPs is greatly affected by their size, shape, and morphology. Thus, numerous studies have been conducted to control these factors.13–15 However, AuNPs synthesized via bottom-up strategies based on wet chemistry usually have faceted structures and are poly-crystalline and polydisperse in size and shape.16Recently, single-crystalline, smooth and round AuNPs have been developed with uniform optical properties. For instance, Xia et al. reported the synthesis of single-crystalline spherical AuNPs via a seed-mediated method.17,18 Yi et al. reported the preparation of highly spherical and monocrystalline AuNPs using a cyclic process.19 In addition, Mirkin and co-workers synthesized universal AuNP seeds through iterative reduction and oxidation processes.20 Notably, even nanoscale changes in the gap size can considerably change the interparticle coupling, leading to dramatic variation in the electromagnetic coupling of clustered metamolecules and superlattice metamaterials.21,22 Thus, fine size-tunability of AuNPs over the nanometer scale is very important.
We herein present a novel strategy to synthesize smooth and highly spherical AuNPs with nanometer scale tunability and monodispersity through a facile and reproducible etching method. It should be pointed out that this process is induced by spontaneous inversion of the role of Au(III) chloride (AuCl4−) from a precursor into an etchant without the introduction of an additional etchant.
In general, researchers have used various types of etchants to control the shape and size of AuNPs.23–25 In this study, we used a AuCl4−–cetyltrimethylammonium chloride (CTAC) complex for the oxidative etching of AuNPs, which was previously reported by Halas et al.26 and studied in detail by Liz-Marzán et al.27 In our study, gold sources can act as a gold precursor during the initial AuNP synthesis process and as an etchant during the subsequent etching process. The exchange of the role of AuCl4− was achieved by controlling the molar ratio between the reducing agent and AuCl4−. In addition, we found that a balance between the surface diffusion rate and etching rate of Au atoms on the AuNP surface is very important for the subsequent etching process to induce a size decrease without any morphological transformation. We could fine-tune the size of AuNPs in a facile manner by extracting the samples in a timely sequence, similar to that reported for quantum dots.28
Results and discussion
We synthesized AuNPs through a seed-mediated and stepwise growth process (Fig. 1).18 By rapidly reducing the gold precursor (AuCl4−) with sodium borohydride (NaBH4) in the presence of cetyltrimethylammonium bromide (CTAB), we synthesized single-crystalline gold clusters (Au-clusters). Subsequently, an ascorbic acid (AA) solution was injected, as a reductant, into the mixture containing CTAC, Au-clusters, and AuCl4− to synthesize primary AuNPs. Thus, Au-clusters functioned as seeds for the synthesis of primary AuNPs with CTAC ligands. For larger AuNPs, denoted as secondary AuNPs, primary AuNPs were used as seeds instead of Au-clusters. Accordingly, the gold source solution was slowly injected into the mixture of primary AuNPs, CTAC, and AA to synthesize secondary AuNPs. Final sizes of both primary and secondary AuNPs were found to be inversely proportional to the amount of seeds used. To obtain smooth and spherical AuNPs, the growth must be controlled by thermodynamics rather than kinetics.29,30 Hence, we used CTAC instead of CTAB in which bromide ions have strong binding affinity to Au atoms and ions as a capping agent, because its weak binding affinity to the AuNP surface and the gold ion enables the balance between surface diffusion of gold atoms and gold deposition on the AuNP surface.31 Furthermore, the capping of AuNPs with CTAC is highly beneficial: CTAC can be easily replaced with other ligands for further applications.31 CTAC capped AuNPs can also be an ideal building block for self-assembly and potential drug carriers.31,32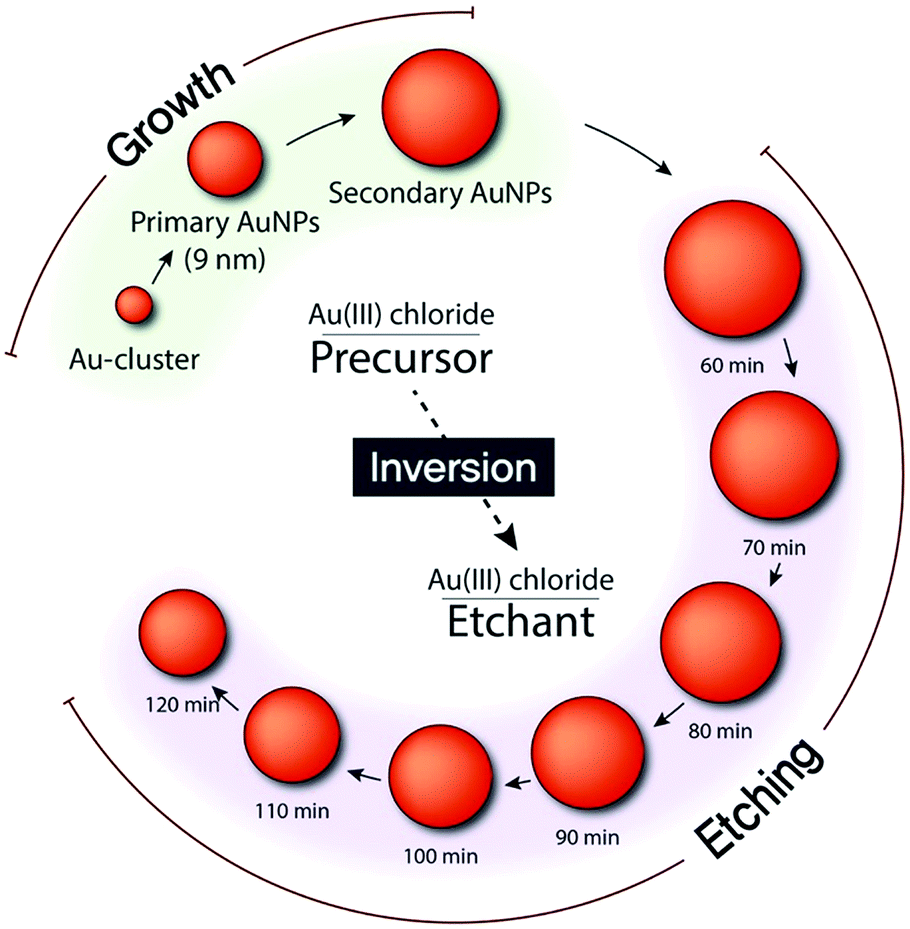 | ||
| Fig. 1 Schematic of stepwise growth and etching of AuNPs with Au(III) chloride functioning as a precursor and etchant. | ||
For the feasibility of the synthesis, we conducted the whole process at room temperature (25 °C). The surface diffusion rate is mainly dependent on the temperature, and for gold atoms, it is sufficiently high at room temperature.17,33–35 Thus, we tried to control the rate of gold deposition by adjusting the amount of AA to attain an optimum value, at which smooth spherical AuNPs would be formed. Fig. 2a shows the transmission electron microscopy (TEM) image of the primary AuNPs. As observed, the primary AuNPs are highly monodisperse with a size of 9.2 nm. UV-vis spectroscopy data are presented in Fig. S2a† magenta line. Notably, when primary AuNPs were further reduced with different amounts of AA, secondary AuNPs with different morphologies were produced (see Fig. 2b–d).
 | ||
| Fig. 2 TEM images of (a) primary AuNPs and secondary AuNPs prepared with (b) 60, (c) 110, and (d) 160 μL of AA. Scale bar: 50 nm; inset scale bar: 20 nm. | ||
When 60 μL of AA was used, the size distribution was broad because of the insufficient amount of the reducing potential, as shown in Fig. 2b and S1a† (scanning electron microscopy (SEM) image). On the other hand, when 160 μL of AA was used, branched AuNPs were formed (Fig. 2d and S1c†). Because the presence of excessive AA led to over-deposition of gold atoms – on the AuNPs – with specific crystal-growth directions, it was hard to achieve a balance between gold deposition and gold surface diffusion on the AuNP surface. Consequently, AuNPs did not grow spherically under this condition. In contrast, 110 μL of AA was found to be appropriate to achieve a balanced rate between gold deposition and surface diffusion, and in turn, spherical secondary AuNPs were obtained (Fig. 2c and S1b†). Thus, if a reductant that has either stronger or weaker reducing power than AA was used, the shape of the AuNPs will become different and the amount of reducing agent needed to synthesize spherical AuNPs will be significantly varied. To confirm the uniformity of the synthesized AuNPs, we analysed them with UV-vis spectroscopy and Dynamic Light Scattering (DLS) (Fig. S2†). Only a single DLS peak was observed for the spherical secondary AuNPs. On the other hand, the DLS spectra of the branched AuNPs showed double peaks attributed to the rotational diffusion mode arising from their anisotropic shape (Fig. S2b†).36,37
When 110 μL of AA was used, we found that the size of the secondary AuNPs slowly decreased, while the spherical shape was retained as the reaction progressed. Thus, by extracting samples in a timely sequence from the same batch, we were able to obtain smooth AuNPs with 1 nm-scale size tunability (Fig. 3). We identified that the crystalline structure of AuNPs, which went through the etching process, retained the inherent spherical single crystal structure, as approved by high-resolution TEM (Fig. 3b). This etching process is proved by the decrease of extinction at 400 nm which presents the concentration of Au(0) (Fig. 3c).27 The intensity of the absorbance peak is affected by the concentration and size of the AuNPs.17 In this experiment, the number of AuNPs is kept identical due to the restriction of nucleation at the growth stage. Thus, the variation in the intensity of extinction and size of the AuNPs is related. The intensity of the UV-vis spectra decreased as the etching proceeded, indicating a decrease in the size of the AuNPs (Fig. 3c and d). In addition, the AuNPs showed absorbance values comparable to those calculated on the basis of Mie theory (Fig. S3†). This confirmed the formation of smooth and spherical AuNPs with high monodispersity.
 | ||
| Fig. 3 Variation in size and UV-vis spectra of secondary AuNPs as a function of reaction time. (a) TEM images of secondary AuNPs with reaction time varying from 60 min to 120 min. Scale bar: 50 nm; inset scale-bar: 20 nm. (b) Selected area electron diffraction pattern for individual secondary AuNPs (reaction time: 60 min). (c) UV-vis spectra of secondary AuNPs with different reaction times. (d) Variation in the extinction and diameter of secondary AuNPs as a function of reaction time. | ||
We hypothesized that unreduced AuCl4− ions induced etching of the AuNPs, because of the lack of AA and the variation of the molar ratio between AA and AuCl4− ([AA]/[AuCl4−]) occurring across the reaction (Fig. S4,† schematic of the AuNP-etching model). According to the stoichiometric reduction reaction of gold, an [AA]/[AuCl4−] ratio of 1.5 was required to fully reduce AuCl4− to Au(0). For 110 μL of AA, [AA]/[AuCl4−] corresponded to 1.1. Thus, it was not sufficient to fully reduce the whole AuCl4− in the growth solution, resulting in the eventual etching of the AuNPs. In addition, [AA]/[AuCl4−] continuously changed because the AuCl4− solution was slowly injected into the growth solution, instead of a one-shot injection. In this sense, [AA]/[AuCl4−] in the latter part of the reaction is relatively smaller than the ratio in the former part of the reaction, which leads to the retardation of the reduction reaction.
The reduction of AuCl4− in the presence of AA occurs in two steps. In the intermediate stage of the reduction of AuCl4− to Au(0), AuCl2− is formed because of the low reducing power of AA.16 To prove our hypothesis, we monitored the concentrations of AuCl4− and AuCl2− over time using UV-vis spectroscopy (Fig. 4). We separately measured the extinctions of AuCl4−–CTAC and AuCl2−–CTAC complexes, to monitor the amounts of AuCl4− and AuCl2−, respectively. Notably, the inherent absorbances of AuCl4− and AuCl2− without CTAC were different from those with CTAC (see the UV-vis spectra of CTAC and AuCl4− in Fig. S5a†). For the yellowish AuCl4−–CTAC complex, two peaks were observed at 228 nm and 321 nm. The extinction at 321 nm contributed to the yellowish color, as both the color and extinction at 321 nm disappeared after the addition of AA (Fig. 4a). On the other hand, for the AuCl2−–CTAC complex, which was transparent under ambient light, a distinct peak at 243 nm was observed. These results indicated that the amounts of AuCl4−–CTAC and AuCl2−–CTAC complexes in the AuNP solution mixture could be separately measured. To monitor the exact concentrations of AuCl4−–CTAC and AuCl2−–CTAC complexes, we separated the as-synthesized solution (AS) into supernatant (SP) and nanoparticle solutions (NP) and measured the extinctions of AuCl4−–CTAC and AuCl2−–CTAC complexes individually (Fig. S5b†).
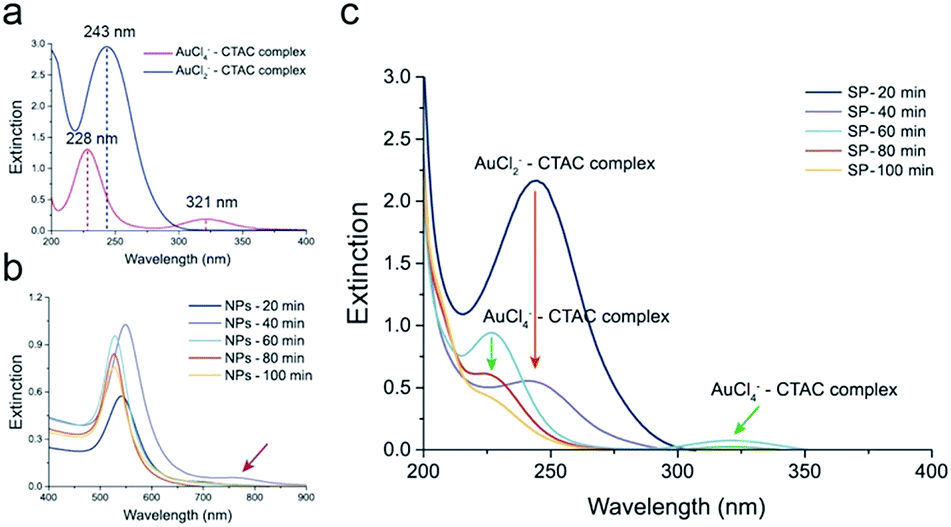 | ||
| Fig. 4 Mechanism of AuCl4−–CTAC-derived AuNP etching. (a) UV-vis spectra of AuCl4−–CTAC and AuCl2−–CTAC complexes. (b) UV-vis spectra of NP obtained from AS with different reaction times. (c) UV-vis spectra of SP obtained from AS with different reaction times. | ||
In the initial stage of the growth, two extinction peaks were observed at 541 nm and 695 nm for AuNPs reduced for 20 min (NPs-20 min) and at 549 nm and 760 nm for AuNPs reduced for 40 min (NPs-40 min) (see Fig. 4b). This indicates the formation of anisotropic AuNPs with a branched morphology, similar to the case of gold nanorods previously reported.38 However, once the growth proceeded due to the surface diffusion of gold atoms, smooth and spherical AuNPs were formed, and a single extinction peak was observed at 528, 526, and 526 nm for AuNPs reduced for 60, 80, and 100 min, respectively (Fig. 4b and S6†). After 20 min of the growth, the characteristic extinction peak of the AuCl2−–CTAC complex appeared at 243 nm. The peak intensity decreased within 40 min of growth because of the reduction of the injected AuCl4−–CTAC complex by AA in the solution, and its consumption, resulting in the deposition on AuNPs (Fig. 4c, red arrow). After 60 min of the reaction, the AuCl4−–CTAC complex extinction peaks emerged at 228 nm and 321 nm, indicating the inversion of the role of AuCl4− from a gold precursor into an etchant.
As AA was consumed during the reduction reaction, AA became insufficient in the latter part of the reaction. Thus, retardation of reduction of the AuCl4−–CTAC complex and etching of AuNPs could have happened. Although the amount of AA left was small, the intensity of the AuCl4−–CTAC complex extinction peaks continuously decreased because slow reduction reaction continued to progress (60–100 min in Fig. 4c). In consequence, the injected AuCl4− was found to play time-dependent multiple roles: a gold precursor in the beginning of the reaction and an etchant in the end.
The role of AuCl4− as an etchant depended on the retardation of the reduction reaction caused by the lack of AA. We varied the amount of AA to check its role in detail during the synthesis of secondary AuNPs. The extinction peak showed that the retardation of reduction depended on the amount of AA, as expected. When a small amount of AA was used (70 μL), relatively high-intensity extinction peaks of the AuCl4−–CTAC complex emerged early in the reaction indicating early inversion of AuCl4− into an etchant (40 min in Fig. S7a†). As the etching rate was very high under this condition, there was no balance between etching and diffusion, which resulted in the formation of non-spherical AuNPs (Fig. S7c†). However, the extinction peaks of the AuCl4−–CTAC complex did not appear when a large amount of AA was used (130 μL), indicating the absence of inversion (Fig. S7b and c†). Through these data, we confirmed that an optimum amount of AA is a prerequisite to change the role of the AuCl4−–CTAC complex: from a gold precursor to an etchant in the one-pot system. In addition, we prepared large AuNPs in the range 70–80 nm using the same method (Fig. S8†), as these AuNPs with considerably large scattering cross-section can be used for diverse optical applications.
Conclusions
In summary, we successfully demonstrated that the size of smooth and spherical AuNPs could be fine-tuned across various size ranges through etching in a one-pot system. Notably, we found that the role of AuCl4− could be changed: from a gold precursor to an etchant, depending on the amount of the reducing agent. A balance between the chemical etching rate and surface diffusion rate was found to be critical to maintain the spherical shape of AuNPs. As demonstrated, this strategy is a simple and powerful method to control the size of highly uniform AuNPs, and it can be used for various applications, such as plasmonic coupling and optical interference between AuNPs and fluorescent dye molecules, depending on the size of the AuNPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grants funded by the Ministry of Science and ICT for the Bio-inspired Innovation Technology Development Project (NRF-2018M3C1B7021997) and a grant funded by the Ministry of Education for Basic Science Research Program (NRF-2018R1D1A1B07043213).Notes and references
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- Y. Mikami, A. Dhakshinamoorthy, M. Alvaro and H. García, Catal. Sci. Technol., 2013, 3, 58–69 RSC.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- J. H. Joo, B. H. Kim and J. S. Lee, Small, 2017, 13, 1701751 CrossRef PubMed.
- E. Y. Kim, D. Kumar, G. Khang and D. K. Lim, J. Mater. Chem. B, 2015, 3, 8433–8444 RSC.
- H. Jung, J. Jung, Y. H. Kim, D. Kwon, B. G. Kim, H. B. Na and H. H. Lee, BioChip J., 2018, 12, 249–256 CrossRef CAS.
- J. Y. Noh, J. I. Kim, Y. W. Chang, J. M. Park, H. W. Song, M. J. Kang and J. C. Pyun, BioChip J., 2017, 11, 246–254 CrossRef CAS.
- M. Rana, M. Balcioglu, N. M. Robertson, M. S. Hizir, S. Yumak and M. V. Yigit, Chem. Sci., 2017, 8, 1200–1208 RSC.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- P. Reineck, D. Gómez, S. H. Ng, M. Karg, T. Bell, P. Mulvaney and U. Bach, ACS Nano, 2013, 7, 6636–6648 CrossRef CAS PubMed.
- Y. Choi, T. Kang and L. P. Lee, Nano Lett., 2009, 9, 85–90 CrossRef CAS PubMed.
- M. Grzelczak, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- F. Kim, K. Sohn, J. Wu and J. Huang, J. Am. Chem. Soc., 2008, 130, 14442–14443 CrossRef CAS PubMed.
- N. D. Burrows, S. Harvey, F. A. Idesis and C. J. Murphy, Langmuir, 2017, 33, 1891–1907 CrossRef CAS PubMed.
- S. D. Perrault and W. C. W. Chan, J. Am. Chem. Soc., 2009, 131, 17042–17043 CrossRef CAS PubMed.
- Y. Zheng, Y. Ma, J. Zeng, X. Zhong, M. Jin, Z. Y. Li and Y. Xia, Chem.–Asian J., 2013, 8, 792–799 CrossRef CAS PubMed.
- Y. Zheng, X. Zhong, Z. Li and Y. Xia, Part. Part. Syst. Charact., 2013, 31, 266–273 CrossRef.
- Y. J. Lee, N. B. Schade, L. Sun, J. A. Fan, D. R. Bae, M. M. Mariscal, G. Lee, F. Capasso, S. Sacanna, V. N. Manoharan and G. R. Yi, ACS Nano, 2013, 7, 11064–11070 CrossRef CAS PubMed.
- M. N. O'Brien, M. R. Jones, K. A. Brown and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 7603–7606 CrossRef PubMed.
- F. Shafiei, F. Monticone, K. Q. Le, X. X. Liu, T. Hartsfield, A. Alù and X. Li, Nat. Nanotechnol., 2013, 8, 95–99 CrossRef CAS PubMed.
- K. J. Park, J. H. Huh, D. W. Jung, J. S. Park, G. H. Choi, G. Lee, P. J. Yoo, H. G. Park, G. R. Yi and S. Lee, Sci. Rep., 2017, 7, 1557 CrossRef PubMed.
- X. Ye, M. R. Jones, L. B. Frechette, Q. Chen, A. S. Powers, P. Ercius, G. Dunn, G. M. Rotskoff, S. C. Nguyen, V. P. Adiga, A. Zettl, E. Rabani, P. L. Geissler and A. P. Alivisatos, Science, 2016, 354, 874–877 CrossRef CAS PubMed.
- T. S. Deng, J. E. S. van der Hoeven, A. O. Yalcin, H. W. Zandbergen, M. A. van Huis and A. van Blaaderen, Chem. Mater., 2015, 27, 7196–7203 CrossRef CAS.
- A. Carattino, S. Khatua and M. Orrit, Phys. Chem. Chem. Phys., 2016, 18, 15619–15624 RSC.
- C. M. Aguirre, T. R. Kaspar, C. Radloff and N. J. Halas, Nano Lett., 2003, 3, 1707–1711 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, J. Phys. Chem. B, 2005, 109, 14257–14261 CrossRef PubMed.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207–211 CrossRef CAS.
- X. Xia, S. Xie, M. Liu, H. C. Peng, N. Lu, J. Wang, M. J. Kim and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6669–6673 CrossRef CAS PubMed.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2014, 54, 2022–2051 CrossRef PubMed.
- S. K. Meena, S. Celiksoy, P. Schäfer, A. Henkel, C. Sönnichsen and M. Sulpizi, Phys. Chem. Chem. Phys., 2016, 18, 13246–13254 RSC.
- J. Hassinen, V. Liljeström, M. A. Kostiainen and R. H. A. Ras, Angew. Chem., 2015, 127, 8101–8104 CrossRef.
- Y. Lu, J. Y. Huang, C. Wang, S. Sun and J. Lou, Nat. Nanotechnol., 2010, 5, 218–224 CrossRef CAS PubMed.
- C. Zhu, H. C. Peng, J. Zeng, J. Liu, Z. Gu and Y. Xia, J. Am. Chem. Soc., 2012, 134, 20234–20237 CrossRef CAS PubMed.
- G. Dai, B. Wang, S. Xu, Y. Lu and Y. Shen, ACS Appl. Mater. Interfaces, 2016, 8, 13506–13511 CrossRef CAS PubMed.
- S. Yoon, B. Lee, C. Kim and J. H. Lee, Cryst. Growth Des., 2018, 18, 4731–4736 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, L. M. Liz-Marzán and P. R. Lang, J. Phys. Chem. C, 2007, 111, 5020–5025 CrossRef.
- S. Yoon, B. Lee, J. Yun, J. G. Han, J. S. Lee and J. H. Lee, Nanoscale, 2017, 9, 7114–7123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00157c |
| This journal is © The Royal Society of Chemistry 2019 |