
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of polyvinylpyrrolidone (PVP) as a capping and structure-directing agent in the formation of Pt nanocubes
I. A.
Safo
a,
M.
Werheid
 a,
C.
Dosche
a and
M.
Oezaslan
*ab
a,
C.
Dosche
a and
M.
Oezaslan
*ab
aPhysical Chemistry, Carl von Ossietzky University of Oldenburg, 26129 Oldenburg, Germany. E-mail: mehtap.oezaslan@uni-oldenburg.de
bInstitute of Technical Chemistry, Technical University of Braunschweig, 38106 Braunschweig, Germany. E-mail: m.oezaslan@tu-braunschweig.de
First published on 24th June 2019
Abstract
In this work, we have investigated the specific role of PVP and Ag+ ions in the formation of platinum nanocubes (NCs) in polyol synthesis. Various characterization techniques such as transmission electron microscopy (TEM), Fourier transform infrared (FTIR) spectroscopy and X-ray photoelectron spectroscopy (XPS) were employed to unravel the effects of PVP and Ag+ ion concentrations on the monodispersity and particle size of the obtained Pt NCs. Very interestingly, we have already fabricated Pt NCs with similar monodispersity and particle size using only 0.4 M PVP (absence of Ag+ ions). Furthermore, the dispersity of the Pt NCs strongly depends on the initial PVP concentration. This observation underscores the important role of PVP during the NC formation processes by controlling the relative growth rates along the <100> direction with respect to those of the <111>. Time-resolved experiments show that the formation and growth of Pt NCs are much faster in the absence of Ag+ ions than with Ag+ ions, which can be explained by the enhanced growth rate along the <100> direction or/and the suppression of the growth rate along the <111>. Electronic interactions between the chemisorbed pyrrolidone ring of the PVP and Pt surface are revealed from the XPS and FTIR data, showing a negative shift of the binding energy of N 1s and a red shift of the Pt–CO vibration band. From our experimental results, we propose extended formation and growth mechanisms based on PVP as the main structure-directing agent. Our model indicates that the aliphatic chains of PVP forming a multi-layer shell influence the mass transport of precursor ions to the initial Pt seed to control the growth rate of Pt NCs with exposed {100} planes. Altogether, we provide a simple, efficient and resource-friendly synthetic guideline for the preparation of nano-sized Pt NCs with high monodispersity and high purity.
1. Introduction
In the past few decades, shape-controlled metallic nanoparticles have attracted great attention due to their unique magnetic, electronic and catalytic properties.1–5 They are suitable candidates to bridge the material gap between single crystals prepared under ultra-high vacuum conditions and nanoparticles deposited on a support material as industrially relevant catalysts. Large potential lies in the advanced link between the fundamental studies on single crystals and those of nanoparticle catalysts to better understand heterogeneous and electrochemical processes. However, controlling the shape of these metal nanoparticles results in a uniform surface atomic arrangement. The surface structure (together with surface defects such as steps, kinks, and edges) is a very critical parameter to unravel fundamental structure–reactivity correlations for heterogeneous and electrochemical reactions. Shape-controlled platinum nanoparticles have widely been applied as efficient catalysts for electrochemical structure-sensitive reactions such as oxygen reduction reaction (ORR).6 Based on their unique properties, many research groups have focused on the development of simple, robust and reproducible synthetic routes to highly monodisperse metal nanoparticles with well-defined facets such as platinum nanocubes (Pt NCs). Pt NCs have extensively been applied as a suitable nano-sized model catalyst in the fields of heterogeneous and electrochemical catalysis. For instance, Pt NCs with highly exposed {100} facets have shown promising electrocatalytic activities for the ORR.7Recently, several synthetic methods have been developed to prepare shape-controlled metal nanoparticles. Thermodynamic factors (such as the type of capping agent, temperature, additives, etc.) and kinetic factors (such as the reduction rate, addition rate, reaction time, concentration of reactants, etc.) play an important role in the formation of various particle shapes. In particular, it is widely accepted that the formation and growth of the particle shape are a critical interplay between the reduction rate of metal precursor and the relative growth rate along the <100> direction with respect to that of the <111>.8,9 Pt NCs were successfully synthesized for the first time in the early 90s by El-Sayed et al.10,11 using linear polyacrylate as both capping and structure-directing agent. Since then, various research groups have designed synthetic methods under different conditions to further improve the preparation of Pt NCs with controlled monodispersity and particle size.12,13 For example, Teranishi et al.14 have studied the effect of initial polyvinylpyrrolidone (PVP) concentration on the formation of Pt nanoparticles with different particle shapes. In their study, at low PVP concentrations the NCs are formed by a very slow reduction process of Pt precursor using hydrogen gas. Unlike a fast reduction process of the precursor in the presence of initial Pt particles might be accompanied by the transformation of a truncated octahedral shape into a cubic particle shape.
Other synthetic routes such as the polyol method (using ethylene glycol) have been reported, where Br− ions are applied as a structure-directing agent to prepare 10 nm-sized Pt NCs and PVP is mainly used to prevent particle agglomeration.15 In the same polyol method, Song et al.16 have applied Ag+ ions as a main structure-directing agent in place of Br− ions to adjust the particle shape of Pt nanoparticles such as cubes, cuboctahedra and octahedra. Obviously, the introduction of either Ag+ or Br− ions in the polyol method allows fabrication of Pt NCs with high monodispersity under certain conditions.
Although PVP is known to preferentially adsorb on the Pt surface, to the best of our knowledge, its detailed role has not been specified in most of the proposed formation mechanisms of shape-controlled Pt nanoparticles. This raises further questions whether the attributed role of the Ag+ or Br− ions dominates over that of PVP. Our work is inspired by the unspecified role of PVP in the formation mechanism of Pt NCs. PVP is known to be a strong capping agent, thus preventing the aggregation of nanoparticles via the steric effect that is based on its hydrophobic chains.17 An important property of PVP is the presence of pyrrolidone as a functional group, which might bind strongly on the Pt(100) facets. The promotion of these facets might depend on the interactions between the PVP and Pt surface. In addition, the PVP–Pt interactions are controlled by the molecular weight, concentration, alkyl chain lengths or the degree of cross-linking of the PVP used. For instance, Song et al.18 reported that smaller molecular weight of PVP controls the particle shape by selective adsorption on the Ag seed with exposed (100) planes, while steric effects prevail with increasing the molecular weight of PVP. Therefore, longer chains of PVP cause larger steric effects, but interact weakly with Ag NPs due to the thicker multi-layer shell formation.
In this work, we have investigated the actual role of PVP and Ag+ ions by varying their initial concentrations during the synthesis of shape-controlled Pt nanoparticles. Based on our HR-TEM analysis, the monodispersity of Pt NCs was found to depend on the initial concentration of PVP. Hence, we conclude that PVP is the main structure-directing agent for the preparation of Pt NCs. From the Fourier-transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS) results, we uncovered the electronic interactions between the PVP and Pt surface. Finally, we have proposed an extended formation mechanism of Pt NCs featuring PVP as the structure-controlling agent.
2. Experimental part
2.1. Materials
Dihydrogen hexachloroplatinate (H2PtCl6·6H2O, 99.9% trace metal basis, Alfa Aesar), polyvinylpyrrolidone (PVP, Mw = 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma Aldrich), silver nitrate (AgNO3, 99.999% trace metal basis, Sigma Aldrich), and ethylene glycol (EG, 99.8%, Sigma Aldrich) were used without any purification.
000, Sigma Aldrich), silver nitrate (AgNO3, 99.999% trace metal basis, Sigma Aldrich), and ethylene glycol (EG, 99.8%, Sigma Aldrich) were used without any purification.
2.2. Preparation of Pt nanocubes (NCs) under modified synthetic conditions
The polyol method used for the common synthesis of Pt NCs has been reported in our previous work and elsewhere.16,19,20 First, Ar-bubbled ethylene glycol (EG) was used to prepare 0.0625 M Pt precursor solution, 0.4 M PVP solution, and 5 mM AgNO3 solution. 3 mL of degassed EG was heated in an oil bath for 20 min under reflux. 0.5 mL of AgNO3 solution (5 mM) was then added to the boiling EG and heated for 5 min. 0.15 mL of a mixture of 0.4 M PVP and 0.0625 M H2PtCl6·6H2O was injected drop-wise into the AgNO3/EG solution every 30 s over a 16 min period. The initial yellow-orange color of the Pt–PVP-containing solution changed rapidly to dark-brown after adding to the hot AgNO3/EG, indicating the formation of colloidal Pt nanoparticles. In the end, the resulting reaction mixture was heated for 5 min and then cooled down quickly to room temperature. The final product was collected by centrifugation (7.815×g/7890 rpm, 5 min) and washed three times with a mixture of hexane/ethanol (volume ratio of 1:3) and subsequently with a mixture of ethanol/methanol (volume ratio of 1:3). To investigate the role of PVP and Ag+ ions, independent synthetic batches of Pt NCs were prepared by varying (i) the initial PVP concentration (0.2, 0.4 and 0.8 M) with a constant Ag+ concentration of 5 mM and (ii) the initial concentration of Ag+ ions (0, 5 and 10 mM) with a constant PVP concentration of 0.4 M, respectively. Time-resolved experiments for formation processes of NCs were performed with and without Ag+ ions at 0.4 M PVP, in which 2 mL of samples from the reaction solution were taken at periods of 4, 8, and 16 min by using a syringe.
2.3. High resolution transmission electron microscopy (HR TEM)
After washing, a dispersion of Pt NCs was dropped onto a carbon-coated Cu grid and dried in air at room temperature. A JEOL JEM 2100F transmission electron microscope operated at an acceleration voltage of 200 kV was employed. To establish the monodispersity and mean edge length of the Pt NCs obtained, more than 200 particles from the HR TEM micrographs were analyzed by using ImageJ Software (version 1.38).2.4 Energy-dispersive X-ray spectroscopy (EDS)
A Hitachi S-3200N scanning electron microscope (SEM) equipped with a cold filter emitter and an energy-dispersive X-ray spectroscopy (EDS) detector (INCA, Oxford Instruments) was employed to analyze the chemical composition and purity of the ensemble of Pt NCs. The microscope was operated at an acceleration voltage of 20 kV, a beam current of 0.4 nA, and a working distance of 15 mm. A film was prepared by dropping a dispersion of Pt NCs on an Au-sputtered glass surface. The chemical composition was established from the characteristic energy intensities of Kα lines for both Ag and Pt by averaging about 45 scans on several positions of the sample.2.5 X-ray photoelectron spectroscopy (XPS)
An ESCALAB 250 Xi spectrometer (Thermo Fisher) with monochromatized Al Kα (hυ = 1486.68 eV) radiation was employed. A dispersion of Pt NCs in a mixture of 2-propanol and water (1:1) was added onto a gold-evaporated glass surface as the substrate and dried in air. All binding energies were calibrated with the C 1s XPS peak of ubiquitous aliphatic hydrocarbon contamination at 284.8 eV. The high resolution XPS spectrum of Pt 4f was measured with a pass energy of 10 eV, step-size of 0.02 eV, dwell time of 5 ms and an average of 10 scans, while the high resolution XPS spectra for N 1s, Ag 3d, O 1s and Cl 2p were acquired with a pass energy of 20 eV, step-size of 0.02 eV, dwell time of 5 s and an average of 10–20 scans. The measurements were taken at a spot size of 500 μm. The high resolution Pt 4f XPS peaks were deconvoluted using CASA XPS software (version 2.3.15) using a Gaussian–Lorentzian (GL) Tailing (T) line shape with a Shirley background. The peak deconvolution of the N 1s, Ag 3d and O 1s XPS spectra was performed using a Gaussian–Lorentzian peak shape using Avantage software (version 5.982, from Thermo Fisher).
2.6 Fourier-transform infrared spectroscopy (FTIR)
FTIR spectra were recorded on a PerkinElmer 400 spectrometer between 400 and 4000 cm−1 with a resolution of 4 cm−1 by averaging 10 scans. During the spectra acquisition, the sample chamber was flushed continuously with Ar. The samples were prepared by drying the Pt NC dispersions at 80 °C overnight. After mixing with KBr powder, the samples were pressed to form a pellet with an outer diameter of 13 mm and a thickness of 2 mm.3. Results and discussion
3.1. Formation mechanism of Pt NCs
As the initial point, we have adopted the polyol synthesis reported by Song et al.,16 where they have mainly concluded that Ag+ ions serve as a main structure-directing agent for the preparation of shape-controlled Pt nanoparticles. They varied the concentration of Ag+ ions to tune the particle shape ranging from cubes to octahedra. Apart from the LaMer's theory on the formation of Pt nuclei,21 the authors assume that in the presence of Ag+ ions the growth rate along the <100> direction is predominated and/or the growth rate along the <111> direction is concurrently inhibited. The relative change in these crystalline growth rates is influenced by the preferred adsorption behavior of reduced Ag species (formed in EG) on the more active {100} surface of the Pt nuclei. This trend is very likely caused by the higher desorption energy of reduced Ag species on the kinetically stable Pt(100) surface compared to thermodynamically favored Pt(111) as reported from single crystal studies.22 The adsorbed Ag on the Pt(100) seed is continuously exchanged with [PtCl6]2−via a galvanic displacement process, whereby the Pt precursor ions were spontaneously reduced by the oxidation of adsorbed silver to silver chloride. Thus, the growth of the {100} orientation to form shape-controlled Pt nanoparticles such as cubes is more enhanced. To stabilize the dispersion of colloidal Pt NCs, the PVP polymer acts as a capping agent due to its steric properties such as long alkyl chains. We have illustrated the proposed formation mechanism of Pt NCs at low AgNO3 concentrations in Fig. 1. | ||
| Fig. 1 Schematic representation of the Pt nanocube (NC) formation and growth process with low contents of Ag+ ions reported by Song et al.16 | ||
Instinctively, the question arises about the fundamental role of PVP and Ag+ ions in the synthesis and particularly which agent might be ascribed to the formation of shape-controlled nanoparticles. To answer this question, we varied the initial concentrations of PVP and Ag+ ions over a broad range and analyzed the monodispersity, mean edge length and chemical/electronic structure of the obtained Pt NCs by using several characterization techniques such as TEM, XPS and FTIR.
3.2. Characterization of the Pt NCs prepared using the standard protocol
We used the synthetic method for the preparation of Pt NCs reported by Song et al.,16 which is referred to as “standard” protocol throughout the paper. The only modification performed was the previous mixing of Pt precursor and PVP before the PVP–[PtCl6]2− solution was injected drop-wise into the hot AgNO3-containing EG every 30 s over a 16 min period. After adding all reactants, the final concentrations in the reaction solution were 0.013 M H2PtCl6·6H2O, 0.17 M PVP and 0.1 mM AgNO3, respectively.A TEM micrograph of representative Pt NCs in lower magnification is displayed in Fig. 2a. The monodispersity and mean edge size of the Pt NCs were established by analyzing more than 200 particles. We want to emphasize that only cubic nanoparticles with sharp edges and deviation of both edge lengths (a and b) of lower than 10% were considered in this study. Based on these strong criteria, the monodispersity of NCs is about 68% and is, therefore, the major fraction, while other minor shapes are tetrahedral, spherical and irregular particles (not shown). The edge length of the NCs was used as a descriptor for the particle size because of the particle shape. Fig. 2b displays the mean edge length and size distribution of the Pt NCs prepared by using the “standard” protocol. The mean edge length of the obtained Pt NCs is around 6.5 ± 0.5 nm, which is in good agreement with the work reported by Song et al.16 (7.1 ± 0.6 nm). We determined the atomic distance between the lattice fringes from the HR TEM image of a single cubic nanoparticle to be 0.194 nm, which correlates with the interplanar distance of the {200} plane of a face-centered cubic (fcc) Pt structure.
 | ||
| Fig. 2 (a) TEM micrograph of the pristine Pt NCs prepared by using the protocol from Song et al.,16 (b) size distribution of the Pt NCs showing a mean edge length of 6.5 ± 0.5 nm (inset: high resolution TEM micrograph of a single NC), (c) high resolution XPS spectrum of Pt 4f peaks, (d) chemical composition of more than 100 single NCs investigated by using STEM-EDS. | ||
After several washing steps with ethanol/methanol (1:3) the obtained Pt NCs were re-dispersed and dried on a gold-covered glass substrate and analyzed by using XPS. Fig. 2c shows the high resolution XPS spectrum of Pt 4f peak. The Pt 4f XPS spectrum was deconvoluted and fitted with double peaks for Pt 4f7/2 and 4f5/2. From the Pt 4f7/2 peak, two chemical species including metallic Pt0 and Pt2+ were identified at binding energies of 70.9 and 72.6 eV (reference values23,24 of 71.2 ± 0.5 eV and 72.4 ± 0.8 eV for metallic Pt and Pt2+, respectively). The area ratio of the corresponding Pt 4f7/2 peaks for Pt2+ and Pt0 was about 0.3 based on the relatively large particle size of 6.5 nm.
Since AgNO3 was used for the preparation of Pt NCs, we also determined the amount of Ag in the single washed NCs by using the STEM-EDS technique. The EDS analysis of more than 100 single cubic nanoparticles evidences that the obtained NCs consist of around 99 at% Pt and ∼1 at% Ag, respectively (see Fig. 2d). Therefore, the atomic Ag:Pt ratio obtained from the STEM-EDS is around 0.01, and it is much lower than the theoretical ratio of 0.5 used in the synthesis. This observation signifies that most of the residual silver was successfully removed by solvent washing of the Pt NC dispersion.
3.3. Effect of the initial PVP concentration on the formation of Pt NCs
In this work, we investigated whether PVP has a particle size- and particle shape-controlling effect on NC formation. First, we synthesized several batches using the standard protocol (containing only the Pt precursor and Ag+ ions) in the absence of PVP. From several TEM micrographs (not shown), we observed strong agglomeration of Pt NCs without the addition of PVP. Thus, PVP is an indispensable capping agent required in the preparation of well-dispersed NCs.In the next step, we varied the initial PVP content and maintained the Ag+ concentration to determine the effect of PVP concentration on the formation process of Pt NCs. Low magnification TEM micrographs of representative Pt NCs prepared with PVP concentrations of 0.2 M and 0.8 M and with the same AgNO3 content (5 mM) are displayed in Fig. 3a and b. It is visible that numerous polydisperse nanoparticles were produced by using PVP concentrations of 0.2 M and 0.8 M compared to that using the standard protocol (0.4 M PVP, Fig. 3c). In other words, the monodispersity dramatically decreased below 50%. Simultaneously, the number of other shapes like tetrahedral, irregular and spherical nanoparticles increased strongly when using 0.2 M and 0.8 M PVP during the synthesis. The significant loss of monodispersity is illustrated in Fig. 3d. Hence, the highest amount of Pt NCs was fabricated with 0.4 M PVP and 5 mM AgNO3. On the other hand, the edge lengths of the as-prepared NCs were found to be about 7.7 ± 1.1 nm, 6.8 ± 1.2 and 6.1 ± 1.3 nm for 0.2 M, 0.4 M and 0.8 M PVP, respectively, signifying a minor influence of the PVP concentration on the growth process of NCs. This observation is very likely related to the adsorption properties of the PVP polymer as a surfactant on the NC surface.
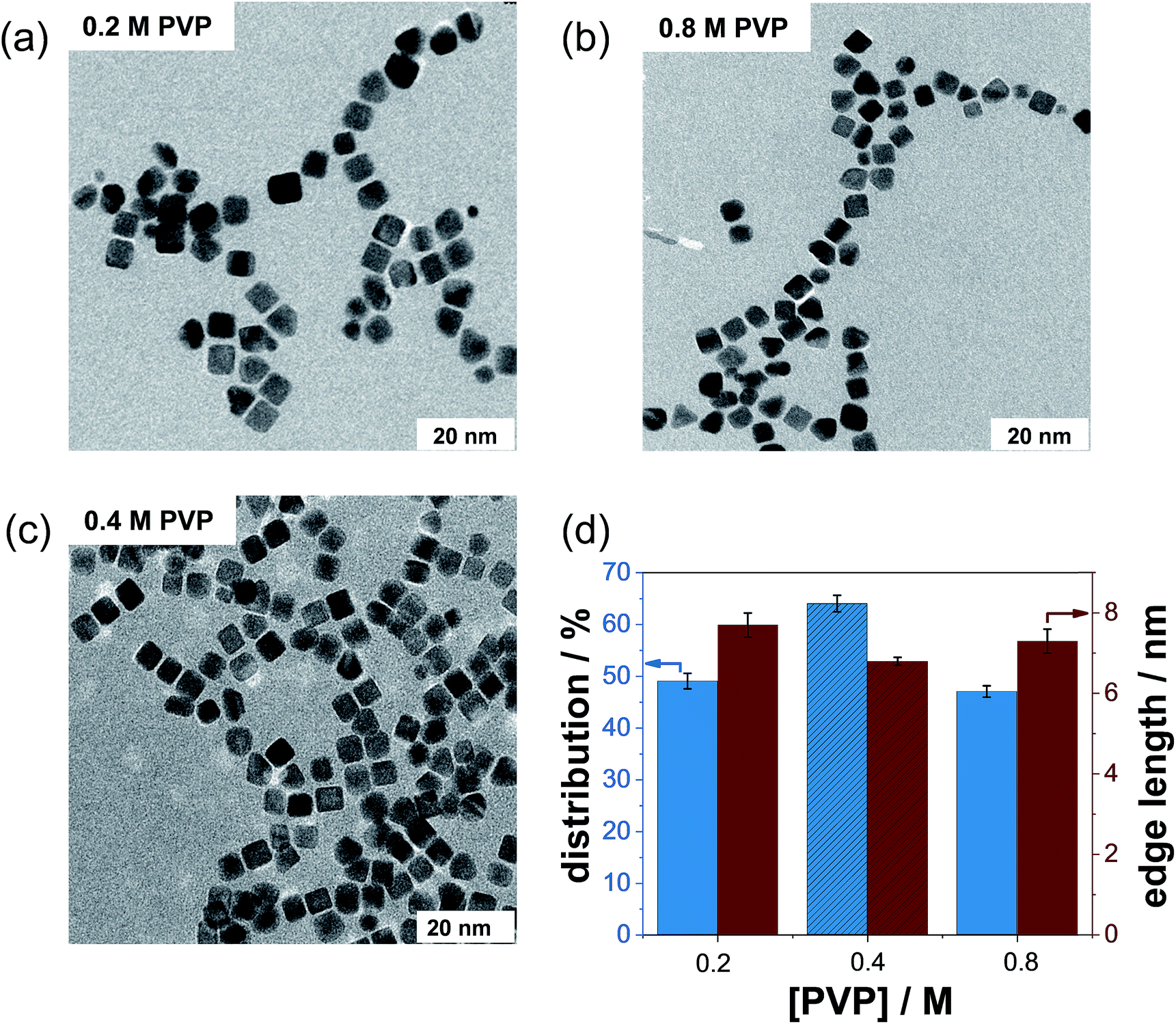 | ||
| Fig. 3 TEM micrographs of representative Pt NCs prepared with (a) 0.2 M PVP, (b) 0.8 M PVP, and (c) 0.4 M PVP (standard protocol), respectively. (d) Distribution of the Pt NCs as a function of the initial PVP concentration (0.2, 0.4, and 0.6 M) in terms of monodispersity and edge length. | ||
3.4. HR TEM, FTIR and XPS analysis of the Pt NCs prepared with various PVP concentrations
A HR TEM micrograph of a single Pt NC after the washing is displayed in Fig. 4. The single NC is partially located at the carbon support on a TEM grid. Very interestingly, the free part of the particle oriented to the vacuum is entirely covered by amorphous carbon which seems to form a multi-layer shell. These layers are highly disordered, signifying an amorphous carbon structure which differs from that of the carbon film of the TEM and carbon support material. We strongly believe that this experimentally observed multi-layer shell very likely stems from the adsorbed PVP polymer as a capping agent surrounding the single washed Pt NCs. Detailed analysis of the thickness of the PVP-containing shell is very difficult due to the limited number of free-standing single NCs found in the HR TEM micrographs. At first glance, it seems that the thickness of the organic shell is not uniform and ranges from sub-nanometers to a few nanometers. We want to stress that even after the washing steps the obtained Pt NCs are entirely surrounded by PVP as an organic multi-layer shell irrespective of the concentrations applied. Only the very weakly adsorbed PVP can be removed by using polar solvents. The effect of different solvents on the effectiveness of the cleaning process for the PVP-capped Pt NCs has been reported in our previous work.25 In short, the highest effectiveness of the cleaning process was obtained with ethanol/methanol (volume ratio of 1:3) used in this study. To corroborate this observation and to provide further information on the interactions of PVP with Pt surface atoms, we performed detailed FTIR and XPS investigations.
 | ||
| Fig. 4 HR TEM micrograph of a single Pt NC surrounded by amorphous carbon very likely based on residual PVP as an organic multi-layer shell (denoted with long-dashed lines), while the border of the carbon support is symbolized using short-dashed lines. | ||
FTIR spectroscopy is an analytical technique capable of revealing the nature of electronic interactions between PVP and Pt NCs. We measured several FTIR spectra of the Pt NCs synthesized with 0.2, 0.4, and 0.8 M PVP. Reference spectra of pure PVP and as-treated PVP were also recorded. The as-treated PVP is referred to as PVP which was boiled in ethylene glycol without the addition of Pt precursor and Ag+ ions and further purification. Fig. 5a shows a series of FTIR spectra of Pt NCs prepared with various PVP contents and pure and as-treated PVP between 1000 and 4000 cm−1. Starting with the FTIR spectrum of pure PVP, the absorption band located around 1644 cm−1 can be ascribed to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the pyrrolidone group. In addition, the CH stretching modes can be assigned to five overlapping signals: asymmetric CH2 stretching (chain: 2983 cm−1, ring: 2954 cm−1), symmetric CH2 stretching (chain: 2919 cm−1; ring: 2885 cm−1) and ternary CH (2852 cm−1).17,26 The bands at 1423 cm−1 and 1371 cm−1 also correspond to the CH deformation modes from the CH2 group. In addition, absorption bands at 1279 cm−1 which are related to C–N bending vibration from the pyrrolidone structure could be identified. It is noted that absorption bands of amines at around 3400–3500 cm−1 were not observed because PVP is a bi-substituted amide. For the PVP which was treated in ethylene glycol in the absence of Pt precursor and Ag+ ions, the CO stretching band is located at 1660 cm−1 compared to that for pure PVP (1644 cm−1). The blue shift of the CO stretching band clearly signifies a chemical change of the PVP structure during the synthesis. This observation will be important in terms of understanding the FTIR spectra of Pt NCs.
O in the pyrrolidone group. In addition, the CH stretching modes can be assigned to five overlapping signals: asymmetric CH2 stretching (chain: 2983 cm−1, ring: 2954 cm−1), symmetric CH2 stretching (chain: 2919 cm−1; ring: 2885 cm−1) and ternary CH (2852 cm−1).17,26 The bands at 1423 cm−1 and 1371 cm−1 also correspond to the CH deformation modes from the CH2 group. In addition, absorption bands at 1279 cm−1 which are related to C–N bending vibration from the pyrrolidone structure could be identified. It is noted that absorption bands of amines at around 3400–3500 cm−1 were not observed because PVP is a bi-substituted amide. For the PVP which was treated in ethylene glycol in the absence of Pt precursor and Ag+ ions, the CO stretching band is located at 1660 cm−1 compared to that for pure PVP (1644 cm−1). The blue shift of the CO stretching band clearly signifies a chemical change of the PVP structure during the synthesis. This observation will be important in terms of understanding the FTIR spectra of Pt NCs.
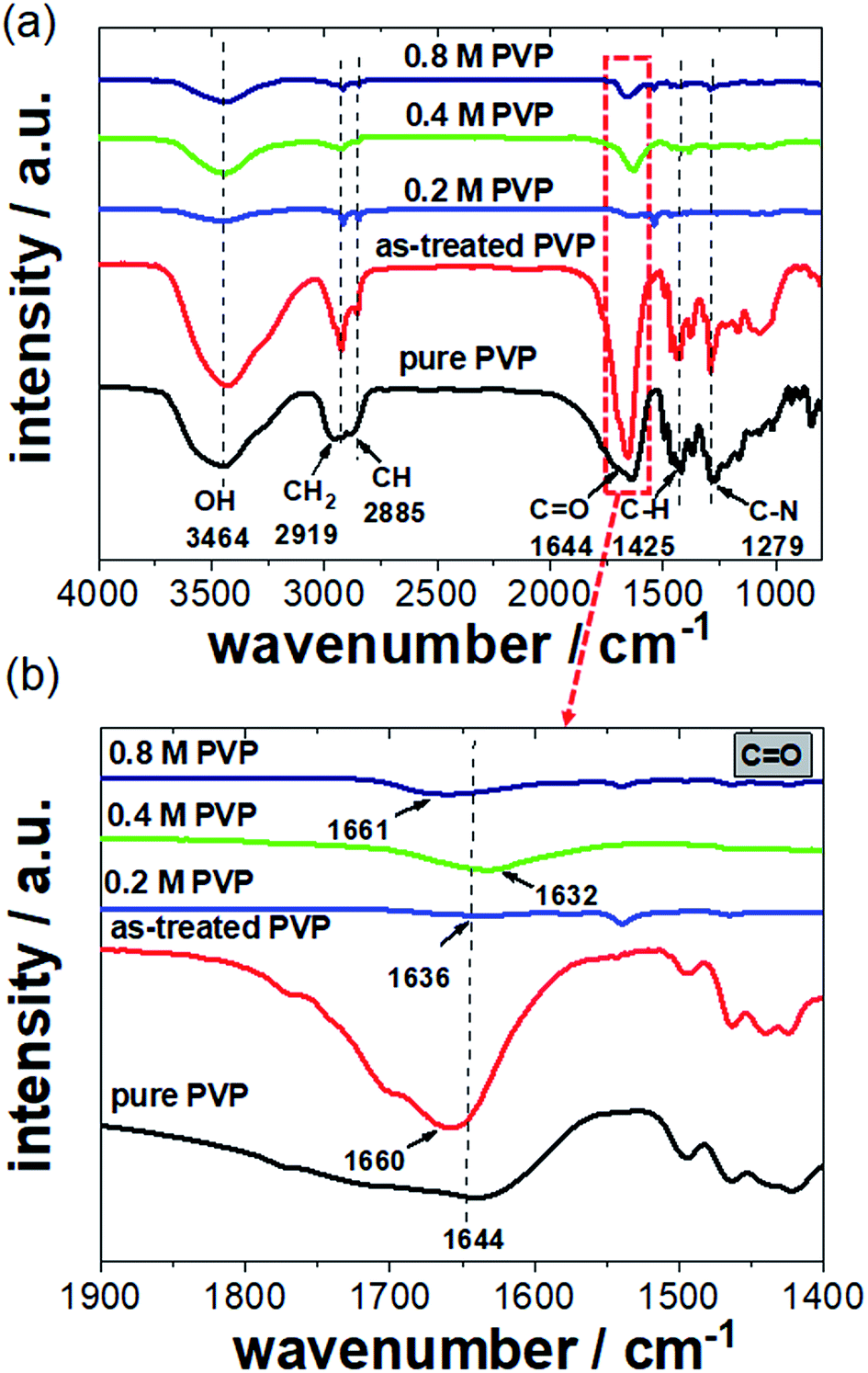 | ||
| Fig. 5 (a) Survey FTIR spectra of washed Pt NCs prepared with various PVP contents as well as those of pure and as-treated PVP in the range of 900–4000 cm−1. (b) Zoom-in view of the FTIR spectra showing the CO–Pt interaction between 1450 and 1850 cm−1. | ||
In the case of the Pt NC samples washed with a polar solvent, the intensities of the absorption bands from the residual PVP are not very high compared to those for pure PVP due to the successful removal of weakly adsorbed PVP. However, the main absorption bands for NCs like CO (1644 cm−1), C–N (1270 cm−1) and CH2/CH (25843–2918 cm−1) groups could still be identified in the FTIR spectra.25 We found that the washed Pt NCs prepared with 0.2, 0.4, and 0.8 M PVP contents show a clear CO stretching band at around 1636, 1632 and 1661 cm−1, respectively.
More interestingly, the CO stretching band is shifted to lower wavenumbers in the presence of Pt NCs particularly for those prepared with 0.2 and 0.4 M PVP. This red shift arises from the strong chemisorption of the CO on the surface of Pt NCs, which decreases the electron density in the carbonyl bond and thus the vibration energy. The extent of the red shift is very likely attributed to the strength of the interaction between the PVP and Pt surface. In this case, 0.4 M PVP seems to interact strongly with the Pt surface. Contrarily, at high PVP concentrations such as 0.8 M a blue shift of the CO band was observed. It is obvious that the position of the CO stretching band for 0.8 M PVP (1661 cm−1) is very similar to that for the as-treated PVP (1660 cm−1) shown in Fig. 5b. We strongly believe that the observed stretching band of the CO group for Pt NCs with 0.8 M PVP mainly corresponds to the excess of PVP which had not been removed by washing three times. Due to its high concentration, the retained PVP forms a thicker layer which surrounds the Pt NCs. Therefore, the observed blue shift of the CO stretching band for the Pt NC sample prepared with 0.8 M PVP is attributed to the excess of PVP. Similar values for the carbonyl group have been reported by Teranishi et al.,14 where high PVP concentrations were applied for the preparation of Pt nanoparticles.
X-ray photoelectron spectroscopy (XPS) as a surface-sensitive technique was used to establish the chemical composition and oxidation state of elements present in the obtained NCs. First, the high resolution Pt 4f XPS spectra of all NC samples show similar chemical behavior, signifying the contribution between Pt0 species in the particle core of NCs and the Pt2+ species at the particle surface (see Fig. 2c). No significant changes of Pt0:Pt2+ ratios were observed because the particles prepared with various PVP concentrations exhibit similar particle sizes. More interestingly, we clearly confirmed the strong chemisorption of PVP on the Pt NC surface based on the high resolution XPS spectra of the N 1s, O 1s, and C 1s peaks. For accurate surface analysis of the obtained NCs, we only focused on the high resolution XPS spectrum of the N 1s, because the O 1s and C 1s components may originate from the PVP and/or residual organic solvents from the washing.
Fig. 6 shows the high resolution N 1s XPS spectra of the Pt NC samples prepared with 0.2, 0.4, and 0.8 M PVP concentrations as well as of the pure PVP and PVP treated in ethylene glycol (EG) under reflux in the absence of Pt precursor and Ag+ ions and without purification as reference materials. In the high resolution XPS spectrum, only one N 1s peak for the pure PVP at a maximum binding energy of 398.8 eV and a full-width at half-maximum (FWHM) of 1.6 eV was detected which can be ascribed to the N atom in the pyrrolidone group of PVP polymer. This large N 1s signal is clearly visible in all XPS spectra for washed NCs, revealing the presence of weakly adsorbed PVP at the Pt surface. However, this N 1s signal is shifted to a higher binding energy at ∼399.8 eV. To confirm the cause of this shift, we repeated the synthesis with only PVP in ethylene glycol under reflux without Pt or Ag precursors. Interestingly, we observed a similar shift of the N 1s signal (399.9 eV) for the as-treated PVP. We suggest that PVP undergoes structural changes during the synthesis at around 180 °C via protonation of the nitrogen from ethylene glycol or resonance stabilization. The observed binding energies are similar to those for secondary amines and electron deficient amide groups.27,28
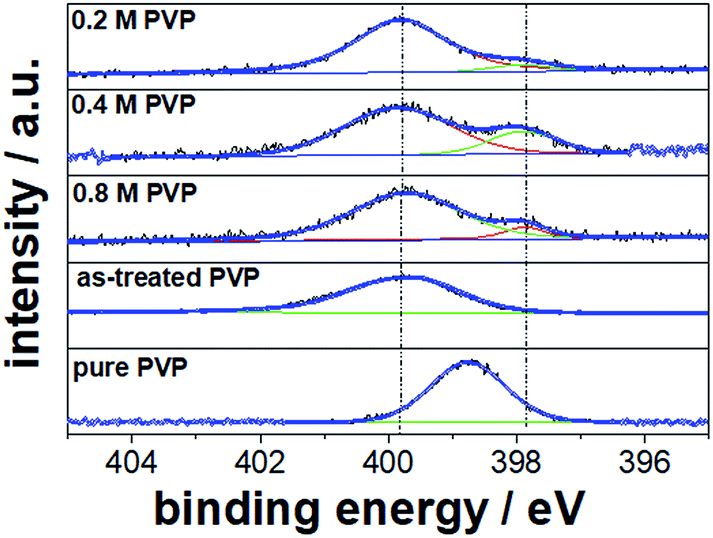 | ||
| Fig. 6 High resolution N 1s XPS spectra for Pt NCs prepared with 0.2, 0.4, and 0.8 M PVP concentrations. Pure PVP and PVP treated in ethylene glycol under reflux are taken as reference materials. | ||
Apart from the large N 1s peak at ∼399 eV, an additional N 1s peak shifted to lower binding energies was clearly detected for the washed Pt NCs prepared with 0.2, 0.4 and 0.8 M PVP concentrations. We suggest that this N 1s peak at lower binding energies corresponds to the strongly chemisorbed PVP on the Pt surface originating from the electronic interactions (electronic donor–acceptor). In fact, similar N 1s binding energy shifts have recently been reported for PVP-capped Pt nanoparticles in other studies.29 From our relative quantification with respect to Pt 4f, we obtained about 6–10 at% strongly chemisorbed N from the PVP on the particle surface. In agreement with the FTIR results, strong interactions between the chemisorbed pyrrolidone of PVP and the Pt surface are uncovered in the present work.
3.5. Effect of initial Ag+ ion concentration on the formation of Pt NCs
To investigate if silver is the main structure-directing agent, we prepared several batches of NCs with 0, 1.1, and 2.2 wt% Ag+ ion concentrations, while the concentration of PVP was maintained at 0.4 M.Fig. 7a–c show the TEM micrographs of representative Pt NCs prepared with three different Ag+ concentrations. Very interestingly, the TEM micrograph of the Pt NCs prepared in the absence of Ag+ ions exhibits particles with a well-defined cubic shape and only a few nanoparticles with tetrahedral and irregular shapes are produced (see Fig. 7a). This result clearly differs from the observation of Song et al.,16 where the monodispersity of cubic Pt nanoparticles drastically reduces without the addition of 1.1 wt% AgNO3. The reason why we do not observe a decrease of the quality of Pt NCs in the PVP-assisted polyol synthesis (absence of Ag+ ions) is very likely related to the strong interactions between the PVP and [PtCl6]2−. The previous mixing of these solutions might initiate a ligand exchange between the Cl− and PVP in [PtCl6]2−. Teranishi et al.14 have reported that the interactions between PVP–[PtCl6]2− are stronger than those between PVP–Pt nanoparticles by using UV-vis. Based on the ligand-to-metal charge-transfer, the observed difference in the peak maxima in the UV-vis spectra is very likely attributed to the ligand exchange of PVP and Cl−, resulting in a slight expansion of the ligand field splitting of the Pt 5d orbital. In other words, the coordination of N and/or O atoms of pyrrolidone of PVP to Pt4+ is stronger than that of Cl− to Pt4+. However, the particles synthesized with an Ag+ content of 2.2 wt% show clearly round and irregular shapes (see the TEM micrograph in Fig. 7b).
 | ||
| Fig. 7 TEM micrographs of representative Pt NCs prepared with (a) 0 wt%, (b) 2.2 wt%, and (c) 1.1 wt% Ag+ concentrations, respectively. (d) Distribution of the Pt NCs as a function of the initial Ag+ ion concentration in terms of monodispersity and edge length. | ||
As an important result from the TEM data, Fig. 7d presents the distribution and edge length of the obtained Pt NCs as a function of the Ag+ ion concentration obtained after analyzing more than 200 particles. We observed that the monodispersity is around 63 ± 4%, 68 ± 5% and 45 ± 6% for 0, 1.1, and 2.2 wt% Ag+ ion contents used in the synthesis, respectively. Evidently, nearly the same amount of NCs (within the error range) is produced in the absence of Ag+ ions and with 1.1 wt% Ag+. Therefore, the addition of Ag+ ions has no significant enhancement effect on the monodispersity of NCs obtained from the ethylene glycol synthesis. Moreover, an increase in the Ag+ concentration up to 2.2 wt% even decreases the amount of NCs formed. This brings up the question if the attributed role of the Ag+ ions as a structure-directing agent still holds. The average edge lengths of the NCs prepared with 0, 1.1, and 2.2 wt% Ag+ ion contents were determined to be 6.3 ± 1.2, 6.8 ± 1.1 and 7.5 ± 1.4 nm, respectively. Thus, we can sum up that the edge lengths are similar within the experimental error range (±1.1 nm) for the NCs prepared with and without Ag+ ions.
In summary, Ag+ ions have no significant effect on the formation of particle shape and particle size of Pt NCs.
The chemical composition of the Pt NC-containing films was established using SEM-EDS. Here, we focused on the NCs synthesized with high Ag+ ion concentration (2.2 wt%). We found that the content of Pt and Ag in the NCs was estimated to be 98 ± 2 at% and 2 ± 1 at%, respectively. The estimated Ag:Pt ratio of 0.02 for the NCs is therefore much lower than the theoretical ratio used during the synthesis (Ag:Pt = 0.9), which indicates that most of the initial Ag atoms were successfully removed after washing. The question often arises whether the residual Ag is incorporated in the Pt lattice of the NCs. Recently, we showed that no additional reflexes of Ag-containing crystal phases or/and no shift of the reflexes (e.g. by insertion of Ag atoms in the Pt lattice) were detected in the XRD profiles of pristine Pt NCs in the context of inaccuracy due to the reflex broadening for small crystallites.20 In contrast, the STEM-EDS analysis of around one hundred single cubic nanoparticles with a reliable statistic shows an Ag content of below 1 at%. Altogether, the entire exclusion of Pt–Ag alloy formation is very difficult for these nano-materials.
To analyze the chemical state of the residual Ag in the washed Pt NCs, we carried out XPS investigations. After washing, the NCs were re-dispersed in a mixture of ethanol/water (volume ratio of 1:1), dropped onto a gold-evaporated glass substrate and dried in air to form a dense film. Fig. 8 shows the high resolution XPS spectra of Ag 3d5/2 and 3d3/2 for the NC-containing films prepared with 1.1 and 2.2 wt% Ag+ ion concentrations. Silver foil was taken as the reference material. The reference XPS spectrum of the Ag 3d core level clearly confirms the presence of metallic silver (binding energy of 368.3 eV for 3d5/2 Ag), which is in excellent agreement with the literature (368.2 eV).30 In addition, the XPS peaks for the Ag 3d core level are typically narrow and asymmetric in shape as displayed in Fig. 8.
 | ||
| Fig. 8 High resolution XPS spectra of Ag 3d for the Pt NC-containing films prepared with 1.1 and 2.2 wt% Ag+ concentrations. Ag foil was added as the reference material. | ||
However, for our samples prepared with an initial Ag+ ion content of 1.1 and 2.2 wt%, the XPS spectra show a clear shift to lower binding energies at 367.3 eV for the Ag 3d5/2 component and the peaks appear broader and more symmetric. This chemical shift of about 0.9 eV and the corresponding peak shape/symmetry are likely attributed to the presence of Ag oxide and chloride as the residues of NC dispersion. It is noted that the chemical states of AgxO are difficult to analyze due to the similarity of the binding energies of Ag2O and AgO.30 From our peak deconvolution and fitting process, we determined the atomic Ag:Pt ratios to be 0.04 and 0.07 for the NC-containing films prepared with 1.1 and 2.2 wt% AgNO3, respectively. These ratios are slightly lower than the theoretical Ag:Pt ratios (0.06 for 1.1 wt% and 0.11 for 2.2 wt% AgNO3), indicating that residual silver oxide or chloride needs to further leach out from the Pt NC-containing dispersion. For instance, the washing process has to be repeated several times despite the risk of losing particles. Therefore, the general absence of AgNO3 is enormously beneficial to prepare highly monodisperse and pure Pt NCs.
3.6. Time-resolved formation and growth process of Pt NCs with and without Ag+ ions
HR TEM investigations were carried out to monitor the formation and growth processes of Pt NCs as a function of the reaction time with and without Ag+ ions at a constant PVP concentration of 0.4 M. More than 100 nanoparticles were evaluated from the HR TEM micrographs. A selection of HR TEM micrographs at different formation stages of Pt NCs after 4, 8, and 16 min of reaction time is illustrated in Fig. 9. Based on the HR TEM analysis, the monodispersity and edge length of Pt NCs were established and are summarized in Fig. 9d (with Ag+ ions) and Fig. 9h (without Ag+ ions). Again, the PVP concentration in both experiments was the same. | ||
| Fig. 9 Series of TEM micrographs to probe the formation and growth processes of Pt NCs as a function of the reaction time (4, 8, and 16 min) with the addition of Ag+ ions (a–c) and without Ag+ ions (e–g) at a constant PVP concentration of 0.4 M. Analysis of the monodispersity and particle size of the obtained Pt NCs in the presence (d) and absence (h) of Ag+ ions. | ||
In the first few minutes of reaction time, the monodispersity of Pt NCs is very low (around 10%) independent of if Ag+ ions are present or not. The initially formed Pt nanoparticles mainly show a spherical particle shape. It is noted that the mean particle size of spherical Pt nanoparticles is smaller in the absence of Ag+ ions, indicating the slower reduction rate of the [PtCl6]2− precursor. The slower reduction rate is very likely related to the ligand exchange between PVP and Cl−. With increasing reaction time, the growth behavior of the almost octahedral Pt nanoparticles is significantly altered and more and more cubic nanoparticles appear. More precisely, a reaction time of 8 min yields a monodispersity of ∼28% with Ag+ ions and ∼42% without Ag+ ions, respectively. It is obvious that the formation of Pt NCs is much faster in the absence of Ag+ ions than that with Ag+ ions. The increased formation of Pt NCs is related to the enhanced growth rate along the <100> direction or/and the suppression of the growth rate along the <111>. This direct comparison highlights the unique role of PVP as a structure-directing agent for the preparation of monodisperse Pt NCs in the polyol synthesis. We suggest that the continuous growth of the Pt NCs is very likely associated with the ad/desorption behavior of PVP. The gradual metal deposition mainly occurs if the surface of the Pt seed is accessible and free. Therefore, knowledge about the interaction of PVP with the Pt surface and its arrangement/configuration in the adsorbed state is very critical to fabricate nano-sized Pt NCs with high monodispersity.
Finally, after 16 min of reaction time the monodispersity of Pt NCs achieved is around ∼65% with and without the addition of Ag+ ions. The mean edge lengths of the Pt NCs are similar independent of the presence of Ag+ ions. We can sum up that the quality of Pt NCs in terms of monodispersity and edge length is mainly controlled by PVP rather than Ag+ ions.
3.7. Proposed formation mechanism and growth of Pt NCs
Our work illustrates that the formation of Pt NCs depends on the initial PVP concentration. In the absence of Ag+ ions, the PVP already acts as a main structure-directing agent. Based on the electronic interactions and steric effects between PVP and Pt4+/Pt0, we have developed a modified model of the formation of NCs as shown in Fig. 10. Generally, the formation of particle shape is controlled by the reduction rate of the metal precursor and the relative growth rate along the <100> direction with respect to that of the <111>.8,9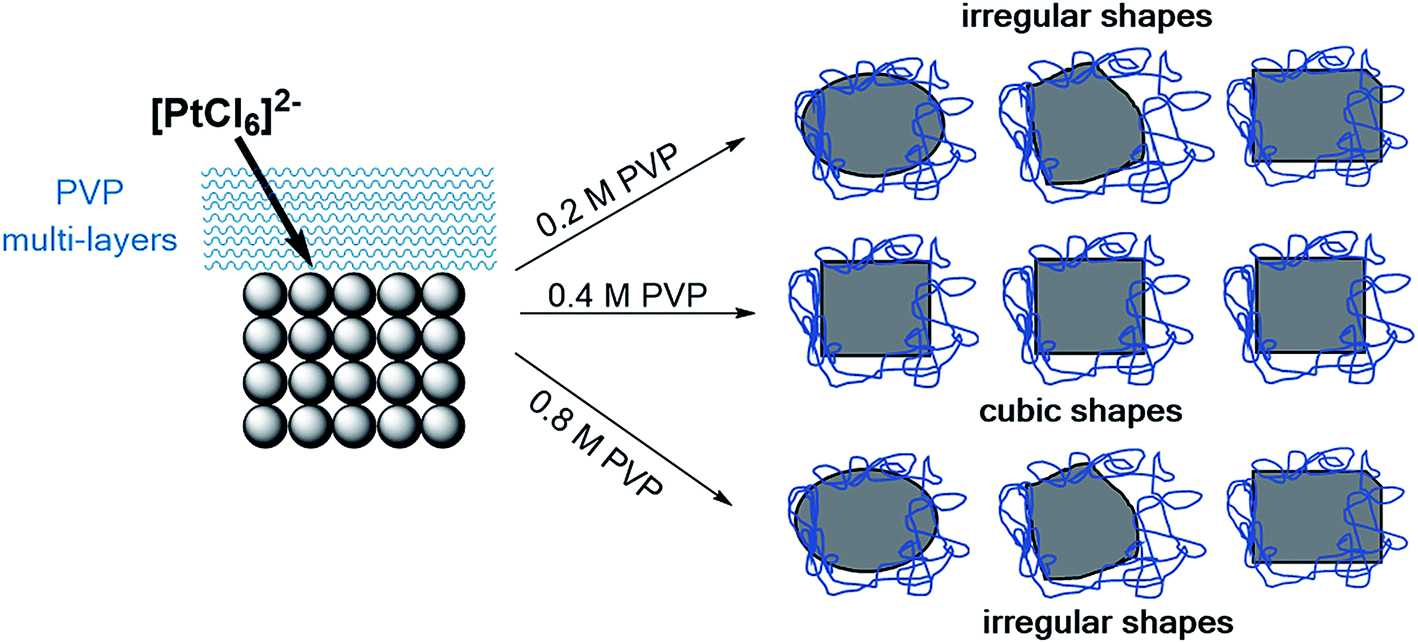 | ||
| Fig. 10 Proposed mechanism for the formation and stability of Pt NCs based on PVP as a main-structure directing agent. | ||
In the initial stage, a ligand exchange between PVP and Cl− modifies the reduction rate of the [PtCl6]2− precursor during the polyol synthesis. This phenomenon is based on the slight expansion of the ligand field splitting of Pt 5d orbital, where the coordination of N and/or O atoms of pyrrolidone of PVP to Pt4+ is stronger than that of Cl− to Pt4+. This observation can be confirmed by using UV vis absorption spectroscopy and was described elsewhere.14 We suggest that due to the strong interaction, the reduction rate of Pt4+ to Pt0 to form nuclei becomes slower. It is noted that the reaction time for all synthesis experiments was very similar in this work. The processes of shape formation and/or transformation of the initial Pt seeds towards cubic nanoparticles are shown in Fig. 9. Time-resolved TEM data reveal that the growth rate along the <100> direction of the Pt seed is mainly controlled by PVP rather than Ag+ ions. In the PVP-assisted polyol synthesis we could point out that the interactions between PVP and Pt nanoparticles are very strong that even intensive washing with a polar solvent is not capable of removing the adsorbed PVP without losing the particle shape.25 Therefore, we have focused on the interactions between the PVP and Pt surface with exposed {100} planes. Our data revealed that the pyrrolidone group of the PVP strongly chemisorbs on the Pt surface which leads to the stabilization of the {100} facet, while the aliphatic chains of the PVP point away from the Pt surface and act as a steric barrier. This model originates from our data, where the electronic interactions between the PVP and Pt surface are found out by the red shift of the CO vibration bands in the FTIR spectra (Fig. 5) and by the shift of the N 1s peak to lower binding energies in the XPS spectra (Fig. 6).
The aliphatic chains of PVP polymer can form a multi-layer shell or polymer shell which is able to control the flux of the initial [PtCl6]2− ions towards the (100) seed. The thickness of the PVP polymer shell, which is controlled by its concentration, strongly influences the mass transport of initial [PtCl6]2− ions and thus the relative growth rate along the <100> direction with respect to that of the <111>. This represents the growth stage of Pt NCs. We want to stress that a large excess of PVP with respect to the Pt precursor was applied in all experiments. Surprisingly, despite the high PVP concentration a change in monodispersity was found. We suggest that the configuration and arrangement of PVP change with its initial concentration used in the reaction solution and strongly modify the ligand exchange and flux of the [PtCl6]2− ions to reduce on the initial Pt(100) seed. This might have an effect on the uniformity and thickness of the multi-layer shell surrounding the Pt NCs. In addition, the structural changes of PVP via possible protonation during heating in EG (see Fig. 6) need to be considered. Computational calculations are needed to provide insights into the configuration and arrangement of PVP at different concentrations and how these manipulate the [PtCl6]2− ion flux. Analogous studies have already been reported for PVP-mediated growth of Ag nano-materials.31
Furthermore, the edge length of the Pt NCs is not influenced by the Ag+ ions as well as by the PVP content. This observation is very likely related to the strong steric effect of PVP which is still present even if its concentration is low or high.
With respect to our new synthesis, an optimal concentration of PVP is required. We observed the highest monodispersity of Pt NCs for 0.4 M PVP without the use of silver. Our new synthesis route allows the fabrication of monodisperse and clean Pt NCs at sizes of smaller than 10 nm. This makes the entire synthesis much more simple, efficient and sustainable. In addition, the concerns over the insertion of other metals like Ag into the Pt lattice are not raised with this new synthetic route. Successful strategies to remove the PVP from the Pt surface which might serve as site-blocking species in electrochemical and catalytic applications have been reported in our previous work.20,25
4. Conclusion
In this work, we investigated the attributed role of PVP and Ag+ ions as capping and structure-directing agents, respectively, in the formation mechanism and growth of Pt NCs. We prepared several independent batches of Pt NCs by varying the initial PVP (0.2, 0.4 and 0.8 M) and Ag+ ion (0, 1.1 and 2.2 wt%) concentrations. Our comprehensive TEM study reveals that the formation of NCs strongly depends on the initial PVP concentration. Although in the literature high monodispersity of Pt NCs has been reported with 0.4 M PVP and 1.1 wt% AgNO3, we also obtained similar monodispersity and particle size of NCs even in the absence of Ag+ ions, highlighting the remarkable role of PVP in the formation and growth processes HR TEM the Pt(100) seed. On the other hand, the increase of the Ag+ ion concentration up to 2.2 wt% rather decreases the monodispersity and yields polydisperse nanoparticles.The analysis reveals that the washed Pt NCs are entirely covered with an organic multi-layer shell formed by the aliphatic chains of PVP. To unravel the electronic interactions between PVP and Pt NCs, we performed FTIR and XPS investigations. From the FTIR data, we confirmed strong chemisorption of PVP on the Pt surface via the carbonyl group of the pyrrolidone. We suggest that the aliphatic chains form a multi-layer shell surrounding the Pt particles to provide a steric effect. In addition, the XPS analysis shows electronic interactions between the chemisorbed PVP and Pt surface. Both spectroscopic techniques highlight that the chemisorption of PVP on the metallic surface occurs from the pyrrolidone group of PVP. We can conclude that PVP strongly interacts with the Pt surface atoms to stabilize the {100} facets, whereas the aliphatic chains form a multi-layer shell surrounding the initial Pt nuclei. The growth of the NCs is tuned by the flux and deposition processes of the initial Pt precursor ions which are strongly controlled by the thickness of the PVP polymer shell. Therefore, PVP is capable of adjusting the relative growth rate along the <100> direction with respect to that of the <111> to enhance the formation of Pt NCs.
Our study paves a way for the straightforward synthesis of monodisperse Pt NCs with high purity (absence of Ag+) by using only PVP. The simplification of the entire synthesis makes this process more simple, robust and resource-friendly.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Federal Ministry of Education and Research (BMBF, FKZ 03SF0539) is gratefully acknowledged. Furthermore, the funding for the JEOL JEM2100F TEM and ESCALAB 250 Xi XPS spectrometer by the DFG (INST 184/106-1 FUGG and INST 184/144-1 FUGG) is acknowledged. Our appreciation goes to Dr Volker Steenhoff of DLR – Institute of Network Energy Systems, Oldenburg, Germany, for his assistance with the FTIR measurements. I. A. S. thanks Kölner Gymnasial – und Stiftungsfonds, Cologne-Germany, for his scholarship. We also thank Rieke Meinen and Jan-Steffen Haverkamp for their experimental assistance in the lab and in particular Dr Peter Broekmann (University of Bern, Switzerland) for the discussion.References
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed.
- Y. Kang, P. Yang, N. M. Markovic and V. R. Stamenkovic, Nano Today, 2016, 11, 587–600 CrossRef CAS.
- J. Liu, X. Fan, X. Liu, Z. Song, Y. Deng, X. Han, W. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2017, 9, 18856–18864 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143–164 CrossRef CAS.
- N. M. Marković, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1996, 100, 6715–6721 CrossRef.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed.
- J. M. Petroski, Z. L. Wang, T. C. Green and M. A. El-Sayed, J. Phys. Chem. B, 1998, 102, 3316–3320 CrossRef CAS.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, A. Henglein and M. A. El-Sayed, Chem. Mater., 1996, 8, 1161–1163 CrossRef CAS.
- J. M. Petroski, T. C. Green and M. A. El-Sayed, J. Phys. Chem. A, 2001, 105, 5542–5547 CrossRef CAS.
- Y. J. Kang, X. C. Ye and C. B. Murray, Angew. Chem., Int. Ed., 2010, 49, 6156 CrossRef CAS PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- T. Teranishi, M. Hosoe, T. Tanaka and M. Miyake, J. Phys. Chem. B, 1999, 103, 3818–3827 CrossRef CAS.
- C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816–5822 CrossRef CAS PubMed.
- H. Song, F. Kim, S. Connor, G. A. Somorjai and P. Yang, J. Phys. Chem. B, 2005, 109, 188–193 CrossRef CAS PubMed.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- Y.-J. Song, M. Wang, X.-Y. Zhang, J.-Y. Wu and T. Zhang, Nanoscale Res. Lett., 2014, 9, 17 CrossRef PubMed.
- N. V. Long, N. D. Chien, T. Hayakawa, T. Matsubara, M. Ohtaki and M. Nogami, J. Exp. Nanosci., 2012, 7, 133–149 CrossRef.
- I. A. Safo and M. Oezaslan, Electrochim. Acta, 2017, 241, 544–552 CrossRef CAS.
- J. Polte, CrystEngComm, 2015, 17, 6809–6830 RSC.
- U. Strüber, A. Kastner and J. Küppers, Thin Solid Films, 1994, 250, 101–110 CrossRef.
- F. Şen and G. Gökaǧaç, J. Phys. Chem. C, 2007, 111, 5715–5720 CrossRef.
- C. Kim, M. Min, Y. W. Chang, K.-H. Yoo and H. Lee, J. Nanosci. Nanotechnol., 2010, 10, 233–239 CrossRef CAS PubMed.
- I. A. Safo, C. Dosche and M. Oezaslan, Z. Phys. Chem., 2018, 232, 1319 CAS.
- Y. Borodko, S. E. Habas, M. Koebel, P. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052–23059 CrossRef CAS PubMed.
- E. T. Kang, K. G. Neoh and K. L. Tan, Polym. J., 1989, 21, 873 CrossRef CAS.
- R. Dziembaj and Z. Piwowarska, Synth. Met., 1994, 63, 225–232 CrossRef CAS.
- L. Qiu, F. Liu, L. Zhao, W. Yang and J. Yao, Langmuir, 2006, 22, 4480–4482 CrossRef CAS PubMed.
- A. M. Ferraria, A. P. Carapeto and A. M. Botelho do Rego, Vacuum, 2012, 86, 1988–1991 CrossRef CAS.
- X. Qi, T. Balankura, Y. Zhou and K. A. Fichthorn, Nano Lett., 2015, 15, 7711–7717 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |