
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimisation of the preservation conditions for molecularly imprinted polymer nanoparticles specific for trypsin
Abeer H. M.
Safaryan
*ab,
Adam M.
Smith
b,
Thomas S.
Bedwell
b,
Elena V.
Piletska
 *b,
Francesco
Canfarotta
c and
Sergey A.
Piletsky
b
*b,
Francesco
Canfarotta
c and
Sergey A.
Piletsky
b
aChemistry Department, Science College, Mustansiriyah University, Baghdad, Iraq. E-mail: as1022@le.ac.uk
bDepartment of Chemistry, College of Science and Engineering, University of Leicester, LE1 7RH, UK. E-mail: ep219@le.ac.uk
cMIP Diagnostics Ltd, Colworth Park, MK44 1LQ, Bedford, UK
First published on 21st August 2019
Abstract
The influence of lyophilisation, autoclaving and sonication on the stability and performance of trypsin-specific molecularly imprinted polymer nanoparticles (MIP NPs) has been studied in order to improve their long-term physical stability. Glucose, glycine, sorbitol and trehalose were tested as cryoprotectant agents during the lyophilisation treatment. The effect of lyophilisation and sterilisation on affinity of trypsin-specific NPs was assessed using Biacore 3000 instrument. The results have demonstrated that MIP NPs successfully withstood the lyophilisation and autoclaving conditions without a reduction of their recognition properties and affinity. It is possible to conclude that both tested lyophilisation and sterilisation treatments were suitable for a long-term storage of the prepared MIP NPs and could be used to store MIP NPs in dry state and hence reduce the chance of the bacterial contamination. An effective preservation of the MIP NPs is a crucial requirement for their future applications in the clinical diagnostics and bioimaging.
Introduction
Molecularly imprinted polymers (MIPs) are synthetic materials based on acrylic or methacrylic functional monomers, that are polymerised in the presence of a specific target molecule called the ‘template’ and able of selectively rebinding it. Over the years, MIPs were developed for a wide range of applications, such as analytical separation, micro-extraction and chemical sensing.1–5 A major breakthrough, which was achieved with a development of the solid-phase synthesis of the molecularly imprinted polymer nanoparticles (MIP NPs), has solved most of the drawbacks of the traditional MIPs, particularly, difficulties associated with removal and re-use of the template, and polydispersity of the binding sites.6,7 The nano-sized MIPs have demonstrated many beneficial features, such as aqueous solubility, high stability, compatibility with automatisation, low cost, fast production and possibility to re-use the immobilised template. Demonstrating high specificity and affinity similar to the specificity of natural antibodies, MIP NPs as synthetic recognition elements are already successfully competing with natural receptors (enzymes and antibodies) in sensors, assays and imaging.8–10In order to make the MIP NPs suitable for in vitro and, even more importantly, for in vivo applications, it is necessary to develop the protocol which would allow storing them safely and sterile for a long time. Typically, freshly prepared MIP NPs are collected and stored in aqueous media for immediate use. However, when left in storage even at 4 °C, the aqueous solution of MIP NPs is susceptible to aggregation and bacterial contamination, which is even more significant when stored in the buffers at physiological pH. Furthermore, nanoparticles stored in solution are prone to agglomeration.
Among the most common methods used for safe and long storage of biological compounds are lyophilisation and sterilisation. Lyophilisation, also known as freeze-drying, is a technique used to improve the long-term stability of nanoparticles. In this process, water is removed from a frozen sample by sublimation and desorption under vacuum. In order to protect the material from the stress that occurs during the freezing and drying steps, compounds called cryoprotectants are added to the sample. Cryoprotectants are typically sugars, which are usually added to the aqueous solution of biological molecules in order to protect them from stress of aggregation and desiccation.11–15 Autoclaving, on the other hand, is a sterilisation technique used to prepare nanoparticles for immediate use. In this process water is heated to 121 °C, causing it to boil and increase the pressure inside the chamber resulting in destruction and denaturation of the biological contaminants.16–18
A serine protease trypsin, which catalyses the hydrolysis of peptide bonds at the C-terminal end of lysine and arginine residues, was chosen as a model template for preparation of MIP NPs due its widespread usage in various industrial and research applications for protein digestion and tissue repairs.19,20 The trypsin-specific MIP NPs were subjected to several treatments, e.g. lyophilisation in the presence of several cryoprotectants, filtration and sterilisation.11,12 In order to evaluate the effect of the lyophilisation and sterilisation on the recognition properties of MIP NPs, their affinity towards trypsin was tested before and after each treatment using a SPR platform (Biacore 3000).
Results and discussion
The procedure of the solid-phase synthesis of trypsin-specific MIP NPs was adapted from the protocols of Guerreiro and Poma.21,22 The particle size determined using TEM was 150 ± 0.1 nm (Fig. 1). | ||
| Fig. 1 TEM image of trypsin-specific MIP NPs. | ||
The concentration of MIP NPs solution was about 0.1 ± 0.01 mg mL−1 which was determined by correlation between the optical density and a range of concentrations of nanoparticles measured at 197 nm giving total amount of 10 mg. Four common cryoprotectants (glucose, glycine, sorbitol and trehalose) were tested. It is thought that an observed action of four sugars tested was based on creating a protective layer around the MIP NPs. Glucose is a naturally occurring sugar molecule that can polymerise to form starch or cellulose. Glycine is a simple amino acid found within most biological systems. Under neutral pH the glycine forms a zwitterion, with a positive and negative ends which can loosely interact with each other to form a protective mesh surrounding the nanoparticles. Sorbitol is sugar-based alcohol, which is produced by the reduction of glucose. Sorbitol is used as a cryoprotectant for the preservation of certain foods. The sorbitol likely uses intermolecular bonding such as hydrogen bonding to form a protective matrix around the nanoparticles due to the presence of six hydroxyl groups. Trehalose is a naturally occurring disaccharide composed of two α-glucose monomers. When cells undergo significant dehydration, trehalose turns into a gel, which keeps the organelles within a cell in place preventing cellular damage. When water is re-introduced to the cells, trehalose disperses back and facilitates the rehydration. Fig. 2 shows the effect of lyophilisation process on particle size. For direct comparison, a lyophilisation was carried out using samples with and without cryoprotectants. It is thought that an observed action of all tested sugars is based on creating a protective layer around the MIP NPs.
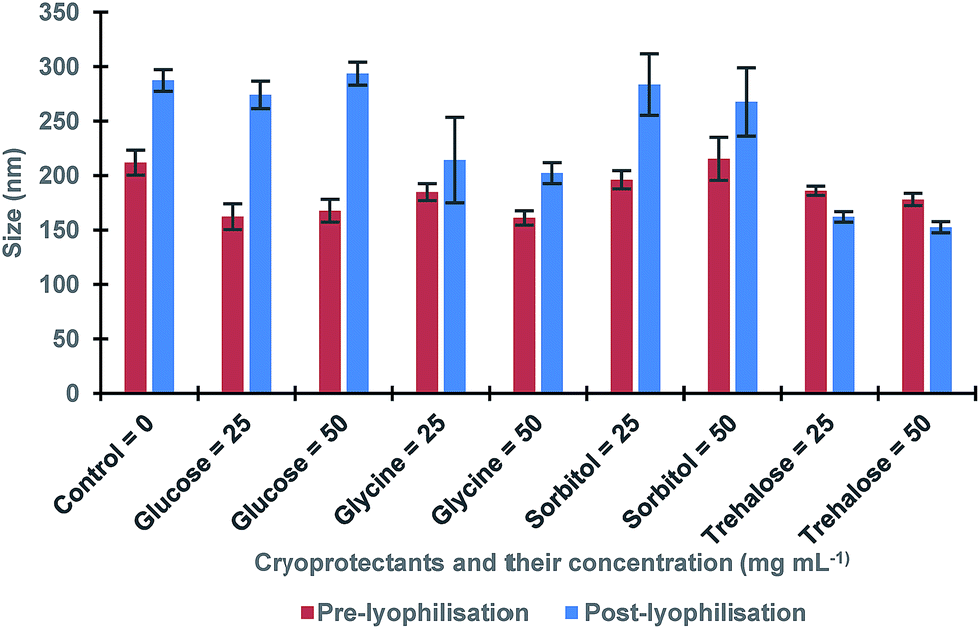 | ||
| Fig. 2 The size of MIP NPs samples before and after lyophilisation in presence of various cryoprotectants and without (control). All samples were measured in triplicates. | ||
The results showed a significant effect of lyophilisation on the size of nanoparticles. There was an increase in the size of post-lyophilised samples without cryoprotectants. These results could occur due to the removal of solvent during lyophilisation that caused the nanoparticles to become clumped together. When glucose was used, a more significant increase in post-lyophilisation size as compared to the control was observed, while glycine and sorbitol gave less increase. Furthermore, trehalose seemed to be the most promising cryoprotectant since it led to the smallest change in the size of the nanoparticles after lyophilisation. In order to optimise the trehalose concentration the lyophilisation in the presence of 5–100 mg mL−1 trehalose was conducted to determine the minimal amount required to generate a protective effect on the nanoparticles (Table 1).23,24
| Trehalose (mg mL−1) | Size pre-lyophilisation (nm) | Size post-lyophilisation (nm) |
|---|---|---|
| 0 | 169.9 ± 7.2 | 234.5 ± 9.6 |
| 5 | 160.2 ± 9.9 | 190.1 ± 6.3 |
| 10 | 162.4 ± 4.7 | 161.0 ± 4.6 |
| 15 | 157.7 ± 6.0 | 160.1 ± 7.7 |
| 20 | 156.6 ± 9.8 | 179.5 ± 9.2 |
| 25 | 186.0 ± 4.2 | 162.1 ± 4.8 |
| 50 | 177.9 ± 5.6 | 152.5 ± 5.1 |
| 100 | 202.7 ± 7.6 | 167.5 ± 6.6 |
According to obtained results, the presence of 10 mg mL−1 of trehalose has resulted in the smallest change of the nanoparticles size before and after the lyophilisation, therefore confirming trehalose as an efficient cryoprotectant even at lower concentration and emphasising the importance of the cryoprotectant concentration optimisation. This finding correlate with publications on the trehalose which is known as an exceptional cryoprotectant. Trehalose is commonly used as protein stabiliser and helps to retain the activity of enzymes in solution as well as in the freeze-dried state.25 It is thought that the mechanism of action of trehalose lies in entrapment of the proteins in the protective “cage” which slows down the water molecules adjacent to the protein and prevents the ice crystallisation.26 It is possible to suggest that the same mechanism is applied to the performance of trehalose for the preservation of MIP nanoparticles. Other benefits of the trehalose as cryoprotectant in comparison with the other tested sugars include its low chemical reactivity, low level of hygroscopicity and absence of internal hydrogen bonding which allows the more flexible formation of hydrogen bonds with nanoparticles during lyophilisation.27,28
The effect of the sonication time on the size of MIP NPs was also studied by sonicating their solution in ultrasonic bath for up to 15 min with and without trehalose in the concentration range 10–20 mg mL−1. It was found that there was no significant difference in the nanoparticles size before and after lyophilisation. The effect of the lyophilisation on fluorescence of the MIP NPs sample at a λex = 490 nm and λem = 510 nm was also investigated. It was found that the process of lyophilisation had minimal effect on the fluorescence properties of the nanoparticles as there was only a small change observed (Fig. 3).
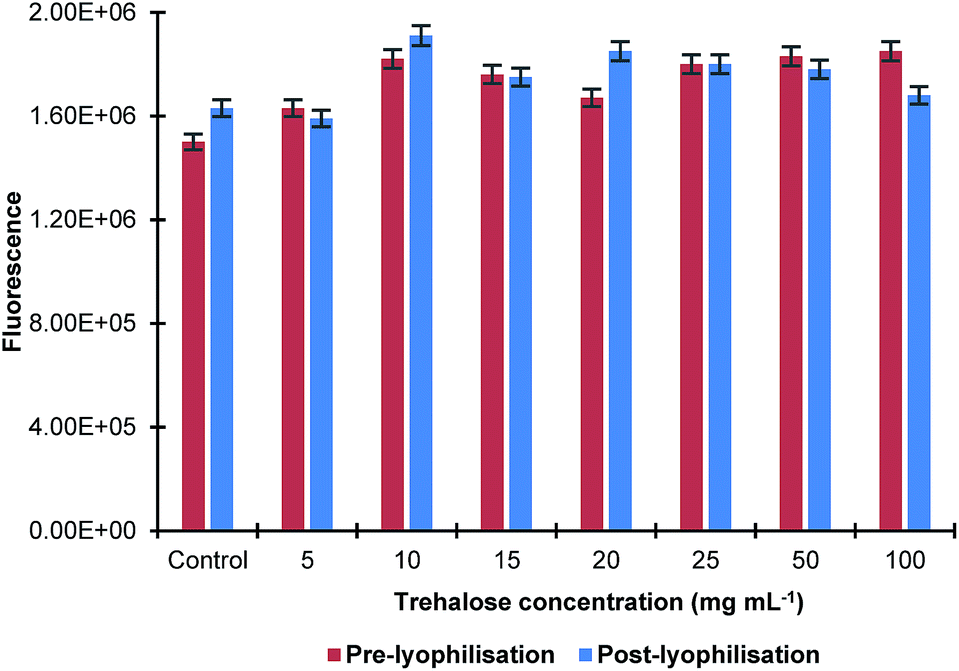 | ||
| Fig. 3 Effect of lyophilisation in the presence of different concentrations of trehalose on fluorescence properties of MIP NPs. All samples were measured in triplicates. | ||
The effect of autoclaving on size of MIP NPs was also studied indicating some small increase in nanoparticle size of approximately 50 nm. This increase is likely a result of the heat of the autoclaving process inducing the nanoparticles to become aggregated. The effect of sterilisation on the MIP NPs absorbance and fluorescence was also studied as it was described earlier. A slight increase in the absorbance of the nanoparticles by 10% was observed after autoclaving. This increase is likely caused by the increased pressure and heat used during the autoclaving process which could cause the nanoparticles to become aggregated, increasing the amount of light absorbed. It was found that the increase of fluorescence was negligible (>2%).
In order to assess the effect of the lyophilisation and sterilisation treatments on MIP NPs recognition properties, SPR study was conducted using Biacore 3000 instrument (GE Healthcare, UK). The self-assembled gold sensor chip (SA) was modified with carboxyl groups and trypsin was immobilised using EDC/NHS coupling as it was described in the Methods section. The KD of the interaction between trypsin and trypsin-specific MIP NPs has been estimated 15.8 nM (Chi2 – 0.03) Fig. 4A.
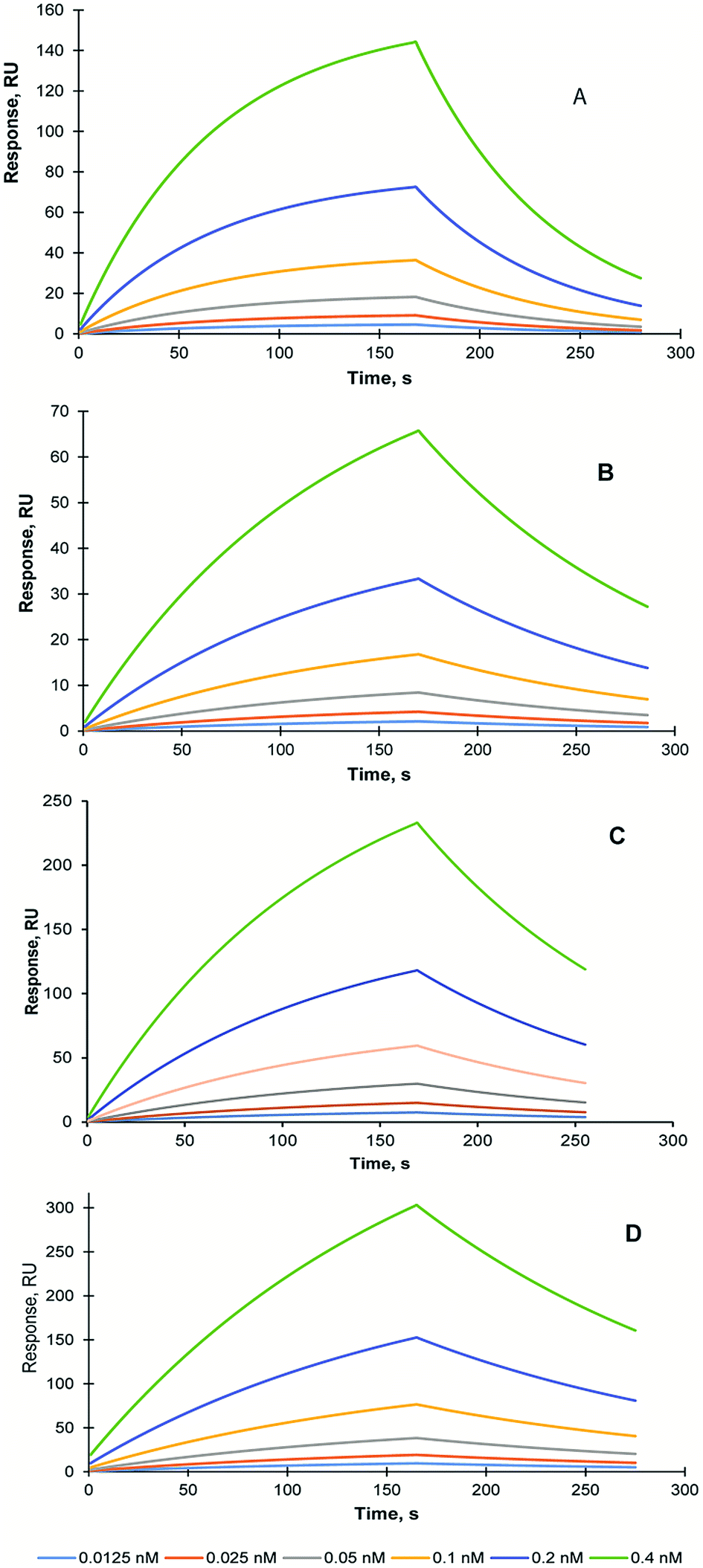 | ||
| Fig. 4 SPR sensorgrams of the molecular interactions between trypsin immobilised on the SA chip and trypsin-specific MIP NPs before treatments (A); trypsin-specific MIP NPs which were subjected to lyophilisation and then reconstituted to the original concentration (B); trypsin-specific MIP NPs which were subjected to lyophilisation in the presence of (10 mg mL−1) trehalose (C); trypsin-specific MIP NPs, which were subjected to autoclaving (D). Solutions of the MIP NPs were injected at concentrations range between 0.0125 nM to 0.4 nM. All SPR experiments were performed in PBS buffer pH 7.4 at 25 °C. | ||
To analyse the stability of the MIP NPs under various treatments, the trypsin-specific MIP NPs were lyophilised with and without trehalose and autoclaved. The affinity of the treated MIP NPs was measured and compared with an affinity of the original (untreated) MIP NPs.
The KD of the interaction between trypsin and lyophilised trypsin-specific MIP NPs has been estimated as 7.1 nM (Chi2 – 1 × 10−5) (Fig. 4B). It was found that affinity of the lyophilised MIP NPs was not affected by the treatment. It could be explained by the effect which lyophilisation might have on the improvement of the dispersion of the nanoparticles after the treatment. It is possible to conclude that lyophilisation treatment is suitable for the long-term storage of the prepared MIP NPs. One of the MIP NPs lyophilisation experiments was performed in the presence of 10 mg mL−1 trehalose (1%). The KD of interaction between trypsin and trehalose-containing lyophilised trypsin-specific MIP NPs has been estimated as 2.4 nM (Chi2 – 2.6 × 10−3) (Fig. 4C). The KD of trypsin-specific MIP NPs was also measured after the autoclaving of the original solution. The KD of interaction between sterilised trypsin-specific MIP NPs has been estimated as 12.2 nM (Chi2 – 2.3 × 10−4) using LB model (Fig. 4D). It is possible to conclude the harsh sterilisation conditions have not affected the affinity and recognition properties of the MIP NPs. Therefore, autoclaving could be used to prolong the storage of the MIP NPs and prevent the bacterial contamination, which might occur when the MIP NPs are stored in water. Both, lyophilisation and autoclaving could extend the shelf-life of the nanoparticles and make them compatible with potential clinical and diagnostics applications.
Conclusions
It was demonstrated that trypsin-specific MIP NPs, which were produced using a solid-phase approach, retained their original physical properties and recognition ability following autoclaving and lyophilisation. The compatibility of these inherently stable synthetic recognition elements with commonly used lyophilisation and sterilisation techniques will further prolong their shelf-life and make them suitable for a variety of diagnostic and imaging applications, which is especially important when cold chain logistics is not available. The methodology described here is applicable for other MIP nanoparticles with similar monomer composition.Methods
Materials
Glass beads SPHERIGLASS® A-Glass 2429 (70–100 μm diameter) obtained from Potters Industries LLC. Acrylic acid (AA), ammonium persulfate (APS), 1,2-bis(trimethoxysilyl)ethane, glutaraldehyde (GA), phosphate buffered saline (PBS) tablets, N-isopropylacrylamide (NIPAM), N,N′-methylene-bisacrylamide (BIS), N-tertbutylacrylamide (TBAm), N,N,N′,N′-tetramethyl-ethylenediamine (TEMED), trypsin from bovine pancreas, glucose, glycine, sorbitol, trehalose, 12-mercaptododecanoic acid, acetone, ethanol, methanol and toluene were purchased from Sigma-Aldrich, UK. 3-Aminopropyltriethoxysilane and sodium cyanoborohydride were obtained from Acros Organics. N-(3-Aminopropyl) methacrylamide hydrochloride (NAPMA) was purchased from PolySciences Inc., UK. Acetonitrile (ACN), sodium hydroxide (NaOH) and sulphuric acid (H2SO4) were obtained from Fisher Scientific (UK). Ultrapure water was produced by a Millipore Milli-Q system (Millipore, Bedford, MA, USA). Double-distilled ultrapure water (Millipore) was used for analysis. All chemicals and solvents were analytical or HPLC grade and were used without further purification. Phosphate buffered saline (PBS) was prepared as directed from PBS buffer tablets (Sigma-Aldrich, Gillingham, UK). N-Fluorescein acrylate was provided by Leicester Biotechnology Group.Preparation of trypsin-modified glass beads
Glass beads (120 g) were boiled in sodium hydroxide (4 M, 200 mL) for 15 minutes; beads were then filtered and rinsed with distilled water (600 mL). The beads were incubated with sulphuric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:1) and then rinsed with distilled water and PBS. Finally, the beads were washed with acetone (200 mL) and then heated at 150 °C for 30 minutes. Afterwards, the beads were incubated in a solution of anhydrous toluene (160 mL) containing 3-aminopropyltriethoxysilane (3.2 mmol) and 1,2-bis (trimethoxysilyl)ethane (1.9 mmol) under nitrogen overnight at 70 °C. The beads were then rinsed with methanol and acetone, and finally dried. The beads were then heated again at 150 °C for 30 min before being stored. For trypsin immobilisation, the beads were incubated in PBS solution (160 mL, 0.1 M) containing glutaraldehyde (11.2 mmol) for 2 hours. The beads were then collected, washed with distilled water (900 mL) and dried. The template immobilisation was performed by incubating the beads in PBS (160 mL, 0.1 M) containing 0.5 mg mL−1 of trypsin in PBS (429.2 nmol) overnight at 4 °C. To reduce the Schiff base, a solution of sodium cyanoborohydride (1 mg mL−1 in PBS) was added and left to react for 30 minutes. Finally, the beads were washed with distilled water, then dried and stored at 4 °C until use.
:water (1:1) and then rinsed with distilled water and PBS. Finally, the beads were washed with acetone (200 mL) and then heated at 150 °C for 30 minutes. Afterwards, the beads were incubated in a solution of anhydrous toluene (160 mL) containing 3-aminopropyltriethoxysilane (3.2 mmol) and 1,2-bis (trimethoxysilyl)ethane (1.9 mmol) under nitrogen overnight at 70 °C. The beads were then rinsed with methanol and acetone, and finally dried. The beads were then heated again at 150 °C for 30 min before being stored. For trypsin immobilisation, the beads were incubated in PBS solution (160 mL, 0.1 M) containing glutaraldehyde (11.2 mmol) for 2 hours. The beads were then collected, washed with distilled water (900 mL) and dried. The template immobilisation was performed by incubating the beads in PBS (160 mL, 0.1 M) containing 0.5 mg mL−1 of trypsin in PBS (429.2 nmol) overnight at 4 °C. To reduce the Schiff base, a solution of sodium cyanoborohydride (1 mg mL−1 in PBS) was added and left to react for 30 minutes. Finally, the beads were washed with distilled water, then dried and stored at 4 °C until use.
Synthesis of trypsin-specific MIP NPs
The polymerisation mixture consisted of NIPAM (39 mg), BIS (2 mg), NAPMA (5.8 mg), fluorescein acrylate (3.5 mg) and AA (2.2 μL) in 100 mL of PBS solution. TBAm (33 mg) was dissolved in 0.4 mL of ethanol and added to monomeric mixture. The mixture was sonicated and purged with nitrogen for 20 minutes. This deoxygenated monomeric solution was added to 60 g of glass beads with immobilised trypsin. In order to initiate polymerisation, a mixture of APS (30 mg) and TEMED (30 μL) in 1 mL of distilled water was added, sealed and left in the dark for 1 h at room temperature. After the polymerisation, the beads were transferred to a 60 mL empty syringe tube fitted with a polystyrene frit (20 μm porosity), and washed with distilled water (600 mL) at room temperature to discard low-affinity materials and unreacted monomers. Then, the high-affinity nanoparticles were eluted using 120 mL of hot ethanol (65 °C). The ethanol in the collected sample was evaporated at 50 °C and replaced with 60 mL of distilled water. To replace ethanol with water and further purify the high affinity MIP NPs, 30 mL of the nanoparticles were centrifuged at 3500 rpm (2200g) for 6 min in the Amicon Ultra-15 regenerated cellulose membrane 50 mL centrifuge filter unit with cut-off of 30000 NMWL using Sigma-16 centrifuge (SciQuip Ltd., UK). Subsequently, the nanoparticles were washed with distilled water (5 × 14 mL) in the same filter unit, then concentrated down to 4 mL and stored at 4 °C until further use.
Characterisation of MIP NPs
The image of MIP NPs was taken using Transmission Electron Microscopy (TEM) using a JEOL JEM-1400 TEM with an accelerating voltage of 120 kV, and digital images were collected using a 16 MP Gatan RIO digital camera.The concentration of the nanoparticles was determined using a UVPC1800 UV-vis spectrophotometer (Shimadzu, UK) at a wavelength of 197 nm and the size of the nanoparticles was determined by dynamic light scattering (DLS) using a Zetasizer Nano (Nano-S) from Malvern Instruments Ltd (Malvern, UK).
The interaction analysis was performed using Biacore 3000 at 25 °C using PBS (pH 7.4) as the running buffer at flow 35 μL min−1. The self-assembled gold sensor chip (SA) was first cleaned in a plasma chamber K1050X (Emitech, UK) and placed in an ethanol solution of 12-mercaptododecanoic acid (2.2 mg mL−1). Before assembly, the sensor chip was rinsed with ethanol and water and dried in a stream of nitrogen. The trypsin solution in PBS (10 μg mL−1) has been injected and immobilised in situ on the chip surface containing carboxyl groups using the EDC/NHS coupling (0.4 and 0.6 mg mL−1, correspondingly). The trypsin-specific MIP NPs were diluted in PBS in the concentration range between 0.0125 nM and 0.4 nM. Sensorgrams have been collected sequentially for all nanoparticles concentrations running in KINJECT mode (injection volume – 100 μL and dissociation time – 120 s). Dissociation constants (KD) were calculated from plots of the equilibrium biosensor response using the BiaEvaluation v4.1 software using a Langmuir Blodgett (LB) algorithm using the AB (absorption) component of the SPR response, which was obtained after the subtraction of the drift and bulk effects.
Lyophilisation of MIP NPs
Glycine, glucose, sorbitol and trehalose were selected as the most commonly used cryoprotectant agents to determine their effect on agglomeration and affinity of the MIP NPs after lyophilisation. Eight samples (1 mL each) were mixed with either 25 mg or 50 mg of glycine, glucose, sorbitol and trehalose, respectively. Each sample was analysed with UV-vis and DLS to determine the pre-lyophilisation size. The samples were frozen in liquid nitrogen, then placed under high vacuum (∼0.4 mbar) in a FreeZone Freeze Drier (Labconco, UK) overnight.11 After lyophilisation each sample was reconstituted with 1 mL of water and analysed using UV-vis spectrophotometer and DLS to evaluate the post-lyophilisation size.Optimisation of trehalose concentration
Eight samples of concentrated nanoparticles (1 mL each) were spiked with different quantities of trehalose (0 mg, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 50 mg and 100 mg) were added. All samples were analysed using UV-vis spectrophotometer and DLS to determine their pre-lyophilisation size. After the lyophilisation, which was done as described above, the samples were reconstituted in 1 mL of water and analysed using UV-vis spectrophotometer and DLS to evaluate their post-lyophilisation size.Optimisation of sonication time before lyophilisation
Sonication of MIP NPs solution containing 10 mg mL−1 and 20 mg mL−1 of trehalose was performed for 0, 5, 10 and 15 min using a ultrasonic bath (Fisher, UK) before and after the lyophilisation to check if ultrasonication of the sample before lyophilisation could reduce agglomeration.Effect of lyophilisation on MIP NPs fluorescence
The MIP NPs were lyophilised as described above and their fluorescence was measured using fluorescence analysis at a λex = 490 nm and in the emission range between 510 nm and 650 nm using a FluoroMax-2 fluorimeter (Horiba, Japan).Autoclaving MIP NPs
A sample of nanoparticles (1 mL) was sterilised by autoclaving at 121 °C by saturated steam process using Classic Prestige Medical Autoclave (Prestige Medical Ltd, UK) for 60 minutes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Natalie Allcock, Core Biotechnology Services, Electron Microscopy Facilities at the University of Leicester for her assistance with TEM imaging.Notes and references
- A. Speltini, A. Scalabrini, F. Maraschi, M. Sturini and A. Profumo, Anal. Chim. Acta, 2017, 974, 1 CrossRef CAS PubMed.
- G. Li and K. H. Row, Sep. Purif. Rev., 2018, 47, 1 CrossRef.
- L. I. Andersson, R. Müller, G. Vlatakis and K. Mosbach, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4788 CrossRef CAS PubMed.
- G. Vlatakis, L. I. Andersson, R. Müller and K. Mosbach, Nature, 1993, 361, 645 CrossRef CAS PubMed.
- S. Scorrano, L. Mergola, M. P. Di Bello, M. R. Lazzoi, G. Vasapollo and R. Del Sole, Int. J. Mol. Sci., 2015, 16, 13746 CrossRef CAS PubMed.
- A. Poma, A. Guerreiro, S. Caygill, E. Moczko and S. Piletsky, RSC Adv., 2014, 4, 4203 RSC.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443 CrossRef CAS PubMed.
- I. Chianella, A. Guerreiro, E. Moczko, J. S. Caygill, E. V. Piletska, I. M. Perez De Vargas Sansalvador, M. J. Whitcombe and S. A. Piletsky, Anal. Chem., 2013, 85, 8462 CrossRef CAS PubMed.
- K. Smolinska-Kempisty, O. Sheej Ahmad, A. Guerreiro, K. Karim, E. Piletska and S. Piletsky, Biosens. Bioelectron., 2017, 96, 49 CrossRef CAS PubMed.
- A. Cecchini, V. Raffa, F. Canfarotta, G. Signore, S. Piletsky, M. MacDonald and A. Cuschieri, Nano Lett., 2017, 17, 2307 CrossRef CAS PubMed.
- G. R. Ramos Yacasi, A. C. Calpena Campmany, M. A. Egea Gras, M. Espina García and M. L. García López, Drug Dev. Ind. Pharm., 2017, 43, 637 CrossRef CAS PubMed.
- L. Wang, Y. Ma, Y. Gu, Y. Liu, J. Zhao, B. Yan and Y. Wang, J. Microencapsulation, 2018, 35, 241 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert and H. Fessi, Int. J. Pharm., 2006, 309, 178 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert and H. Fessi, Eur. J. Pharm. Biopharm., 2006, 63, 87 CrossRef CAS PubMed.
- W. H. Morrison and S. M. Truhlsen, Am. J. Ophthalmol., 1950, 33, 357 CrossRef.
- Y. Darwis and I. W. Kellaway, S.T.P. Pharma Sciences, 2002, 12, 91 CAS.
- N. S. El-Salamouni, R. M. Farid, A. H. El-Kamel and S. S. El-Gamal, Int. J. Pharm., 2015, 496, 976 CrossRef CAS PubMed.
- H. K. Hustoft, H. Malerod, S. R. Wilson, L. Reubsaet, E. Lundanes and T. Greibrokk, A Critical Review of Trypsin Digestion for LC-MS Based Proteomics, INTECH Open Access Publisher, 2012, DOI:10.5772/29326.
- D. Shah and K. Mital, Adv. Ther., 2018, 35, 31 CrossRef CAS PubMed.
- A. Guerreiro, A. Poma, K. Karim, E. Moczko, J. Takarada, I. Perez de VargasSansalvador, N. Turner, E. Piletska, C. S. de Magalhaes, N. Glazova, A. Serkova, A. Omelianova and S. Piletsky, Adv. Healthcare Mater., 2014, 3, 1426 CrossRef CAS PubMed.
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P. F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821 CrossRef CAS PubMed.
- A. M. Lammert, S. J. Schmidt and G. A. Day, Food Chem., 1998, 61, 139 CrossRef CAS.
- L. Jiang, S. Li, J. Jiang, Y. Liang and P. Wang, J. Chem. Eng. Data, 2014, 59, 4021 CrossRef CAS.
- D. Corradini, E. G. Strekalova, H. E. Stanley and P. Gallo, Sci. Rep., 2013, 1218 CrossRef PubMed.
- J. K. Kaushik and R. Bhat, J. Biol. Chem., 2003, 278, 26458 CrossRef CAS PubMed.
- J. H. Crowe, F. A. Hoekstra and L. M. Crowe, Annu. Rev. Physiol., 1992, 54, 579 CrossRef CAS PubMed.
- L. M. Crowe, D. S. Reid and J. H. Crowe, Biophys. J., 1996, 71, 2087 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |