
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum dot cellular uptake and toxicity in the developing brain: implications for use as imaging probes†
Mengying
Zhang
a,
Brittany P.
Bishop
 b,
Nicole L.
Thompson
b,
Kate
Hildahl
b,
Binh
Dang
b,
Olesya
Mironchuk
d,
Nina
Chen
e,
Reyn
Aoki
b,
Vincent C.
Holmberg
*abc and
Elizabeth
Nance
*abfg
b,
Nicole L.
Thompson
b,
Kate
Hildahl
b,
Binh
Dang
b,
Olesya
Mironchuk
d,
Nina
Chen
e,
Reyn
Aoki
b,
Vincent C.
Holmberg
*abc and
Elizabeth
Nance
*abfg
aMolecular Engineering & Sciences Institute, University of Washington, Seattle, WA 98195-1652, USA. E-mail: eanance@uw.edu; holmvc@uw.edu
bDepartment of Chemical Engineering, University of Washington, Seattle, WA 98195-1750, USA
cClean Energy Institute, University of Washington, Seattle, WA 98195-1653, USA
dDepartment of Bioengineering, University of Washington, Seattle, WA 98195-5061, USA
eDepartment of Biology, University of Washington, Seattle, WA 98195-1800, USA
fCenter on Human Development and Disability, University of Washington, Seattle, WA 98195-7920, USA
gDepartment of Radiology, University of Washington, Seattle, WA 98195-7117, USA
First published on 30th July 2019
Abstract
Nanometer-sized luminescent semiconductor quantum dots (QDs) have been utilized as imaging and therapeutic agents in a variety of disease settings, including diseases of the central nervous system. QDs have several advantages over traditional fluorescent probes including their small size (5–10 nm), tunable excitation and emission spectra, tailorable surface functionality, efficient photoluminescence, and robust photostability, which are ideal characteristics for in vivo imaging. Although QDs are promising imaging agents in brain-related applications, no systematic evaluation of QD behavior in brain-relevant conditions has yet been done. Therefore, we sought to investigate QD colloidal stability, cellular uptake, and toxicity in vitro, ex vivo, and in vivo in the brain environment. We found that QD behavior is highly dependent on surface functionality and that treatment of cultured organotypic whole hemisphere (OWH) slices with QDs results in dose-dependent toxicity and metallothionein increase, but no subsequent mRNA expression level changes in inflammatory cytokines or other oxidative stress. QDs coated with poly(ethylene glycol) (PEG) were protected from aggregation in neurophysiologically relevant fluids and in tissue, allowing for greater penetration. Importantly, QD behavior differed in cultured slices as compared to monolayer cell cultures, and behavior in cultured slices aligned more closely with that seen in vivo. Irrespective of surface chemistry and brain-relevant platform, non-aggregated QDs were primarily internalized by microglia in a region-dependent manner both in slices and in vivo upon systemic administration. This knowledge will help guide further engineering of candidate QD-based imaging probes for neurological application.
Introduction
Nanoscale probes are an emerging cutting-edge tool for both disease process detection and neurological imaging.1–4 Various engineered nanoparticles have shown advantages over conventional contrast agents by providing protection from clearance and demonstrating the ability to overcome barriers to drug delivery, such as the blood brain barrier (BBB).5,6 Luminescent semiconductor nanocrystals, or quantum dots (QDs), have especially attracted the interest of researchers due to their tunable excitation/emission spectra and tailorable surface functionality, as well as their robust photostability and efficient luminescence, which make them amendable for in vivo animal imaging.7–9 In particular, cadmium selenide (CdSe) QDs are a well-understood, model system that has been widely studied and optimized over the past thirty years, with tunability across the entire visible spectrum. Moreover, recent developments have led to breakthroughs in photostability and QD efficiency, resulting in core–shell QDs with nearly ideal luminescence characteristics.10,11 Because of these advantageous attributes, CdSe QDs have become a common material for use in a variety of different biological applications, including multi-modal imaging and studies of lymphatic basins12 and live cells.13 Furthermore, the small hydrodynamic size (10–20 nm) of functionalized QDs has been reported to be suitable for neuron and glial cell labeling and tracking.14,15 QD applications in the brain have been widely explored in both fundamental research as well as in clinical investigation,16–23 correlating with the interest in using functionalized QDs as drug-delivery vehicles or targeted-imaging biomarkers for central nervous system (CNS) diseases. Previous literature has shown that chemically modified QDs can deliver drugs and peptides to the developing brain.24 Recently, the use of QDs to monitor signal transmission in neurons17,25 opened a new field for QD-based functional probes in the brain.However, there currently lacks a comprehensive assessment of QD behavior in the brain, which could hinder further investigation and translation of QD-based neurological imaging probes. Toxicity is a common concern for QDs, due to either the leaching of inorganic ions from the QD crystal,26 or loss of QD colloidal stability and subsequent micrometer-scale aggregate formation upon administration, leading to inflammation and granuloma formation.2,3,27 Moreover, different QD surface coatings can have different cell- or tissue-targeting effects,28 and lead to diverse cell cytotoxicity,29 with varying degrees of injury and in vivo biodistribution.30 It has also been reported that various terminal end groups on functionalized QDs can result in a variable level of inflammatory responses.31 Despite the focus on QD engineering, a failure to adequately consider the physiological environment has also limited the application of QD-based nanoparticles.32 A systematic evaluation of QDs focused on both nanoparticle engineering and physiological variance in the tissue environment is therefore essential.
Hence, we investigated QD stability, cellular uptake, and toxicity with three surface end groups in relevant brain environments, including in vitro, in cultured brain slices (ex vivo), and in vivo. An array of efficient, red-emitting (λmax > 600 nm), aqueous-dispersible QDs with three custom surface functionalities were targeted: 3-mercaptopropionic acid (MPA), poly(ethylene glycol) (PEG)-5000k-methoxy (PEG-OMe), and (PEG)-5000k-hydroxyl (PEG-OH). A core–shell CdSe/CdS QD architecture was selected in order to maximize luminescence efficiency,10,33–35 while care was taken to keep the overall diameter of the inorganic nanocrystal below 8 nm, such that the overall hydrodynamic diameter of the functionalized QDs would be below ∼20 nm (accounting for a ∼6 nm-thick ligand corona). We have previously shown that this size range facilitates efficient transport within the brain environment.14,15 CdSe nanocrystal cores were synthesized with a protective shell of CdS to facilitate efficient luminescence (quantum efficiencies of up to 60% were achieved), and custom, red-emitting core–shell QDs were then generated via functionalization with the desired ligand and transfer to aqueous media. Numerous replicates of the optimized core–shell synthesis were carried out in order to produce sufficient quantities of functionalized QDs for the in vitro, ex vivo, and in vivo studies described in this study. QD colloidal stability and aggregation kinetics were evaluated in phosphate buffered saline (PBS) and artificial cerebrospinal fluid (aCSF). QD cellular uptake was measured in organotypic brain slices, and QD toxicity was monitored in both in vitro microglia cells and organotypic brain slices, along with changes in expression levels of cytokine and oxidative stress markers. QD cellular localization was evaluated following intraperitoneal (i.p.) administration in a newborn rat model with hallmarks of neuroinflammation, and real-time imaging was utilized to show QD internalization and trafficking in cells. The combination of in vitro, ex vivo, and in vivo models allows for investigation of QD–cell interactions in a three-dimensional (3D) environment that captures the complexity of the brain microenvironment.36,37 Importantly, incorporating the effect of the brain microenvironment enables the development of QD-based imaging probes that could target regions of interest in the CNS and alter localization and cellular interaction based on the intended outcome.
Results and discussion
Physicochemical characterization of core–shell CdSe/CdS QDs
The physicochemical properties of the core–shell CdSe/CdS QDs were characterized as a function of surface functionality in 10 mM NaCl (Table 1). Details of the QD synthesis and surface functionalization are provided in the Materials and methods section. QD-MPA particles exhibited hydrodynamic diameters of 11.3 ± 1.7 nm, with a surface charge (zeta potential, ζ-potential) of −30.1 ± 5.5 mV, confirming the presence of negatively-charged surface carboxylate groups. QD-PEG-OH and QD-PEG-OMe particles exhibited hydrodynamic diameters of 23.9 ± 2.0 nm and 17.3 ± 0.8 nm respectively, corroborating the attachment of 5000 Da PEG, which added an additional 5–6 nm to the hydrodynamic diameter of the particle, as compared to the non-PEGylated QD-MPA particles. PEG-functionalized QDs with an inert (methoxy) pendant functional group (QD-PEG-OMe) exhibited surface potentials of −8.3 ± 3.7 mV, while PEG-functionalized QDs with pendant hydroxyl functional groups (QD-PEG-OH) exhibited zeta potentials of −13.1 ± 1.5 mV. Since PEG is an uncharged hydrophilic polymer, PEG-functionalized QDs exhibited a more near-neutral net surface charge, as compared to the carboxylate-functionalized QD-MPA particles (−30.1 ± 5.5 mV).| Particle | Surface functionality | Size ± SEOM | ζ-potential ± SEOM |
|---|---|---|---|
| QD-MPA | MPA | 11.3 ± 1.7 | −30.1 ± 5.5 |
| QD-PEG-OH | PEG-OH | 23.9 ± 2.0 | −13.1 ± 1.5 |
| QD-PEG-OMe | PEG-OMe | 17.3 ± 0.8 | −8.3 ± 3.7 |
Both CdSe nanocrystal cores and core–shell CdSe/CdS QDs have a roughly spherical morphology (Fig. 1), with CdSe nanocrystal cores exhibiting diameters of 3.4 ± 0.4 nm and final core–shell CdSe/CdS QDs exhibiting diameters of 6.7 ± 0.5 nm (Fig. 1B), excluding the outer ligand corona. The spectral profiles of the QDs (Fig. 1) remained largely unperturbed, irrespective of which surface ligation was selected. Additional physicochemical evidence of successful surface functionalization via attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy can be found in the (ESI Fig. 1†). Both PEGylated QD samples exhibit all of the expected characteristic FTIR peaks,38–40 with the QD-MPA samples exhibiting clear carboxylate (COO−) stretches at 1560 and 1410 cm−1,41,42 indicating full deprotonation of the MPA ligands.
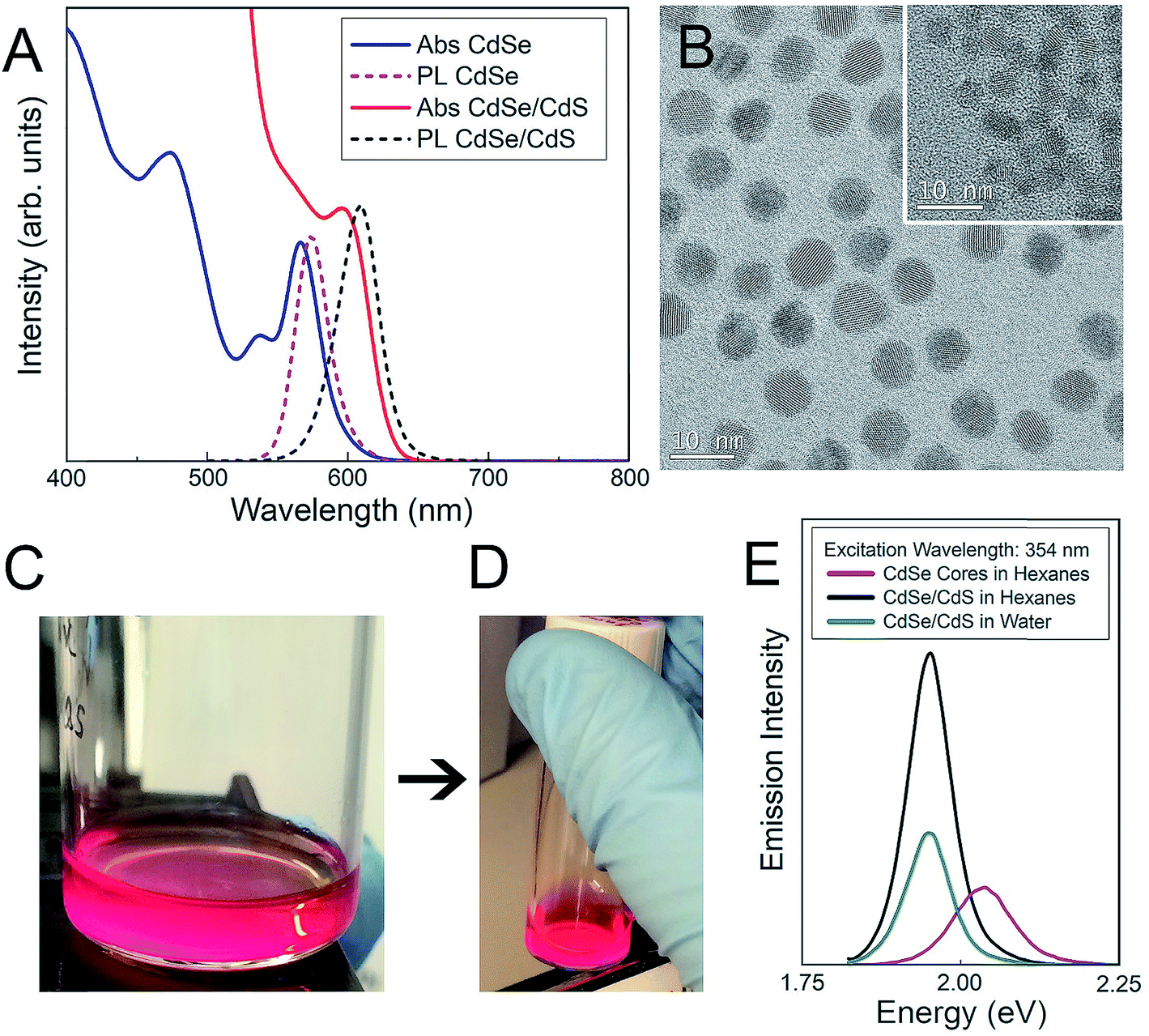 | ||
| Fig. 1 Surface functionalization and characterization of CdSe–CdS core–shell QDs. (A) Absorbance and photoluminescence of CdSe nanocrystal cores (blue and pink) and final core–shell CdSe/CdS QDs (red and black) with 60% quantum yield. (B) TEM images of final 6.7 ± 0.5 nm-diameter core–shell QDs and initial 3.4 ± 0.4 nm-diameter nanocrystal cores (inset). (C, D) Images of QD dispersions under UV excitation (C) before and (D) after surface functionalization and transfer to aqueous media. (E) Relative emission intensity of initial nanocrystal cores (pink), and final core–shell QDs before (black) and after (green) surface functionalization and transfer to aqueous media. | ||
Colloidal stability of QDs in neurophysiologically relevant fluids
QD-based nanoparticle probes must be both stable and monodisperse to be effective as biomarkers, particularly for applications in the brain. As such, we conducted both long-term and short-term QD stability studies under neurophysiologically relevant conditions. We first monitored the hydrodynamic diameter of the core–shell CdSe/CdS QDs with different surface functionalities in 1× PBS and aCSF at 0 h, 4 h, and 24 h at 37 °C, using dynamic light scattering (DLS) to evaluate any long-term tendency toward aggregation in brain-related fluids at physiological temperatures. Hydrodynamic diameters measured from dispersions of QD-MPA particles increased dramatically from 20 nm to more than 200 nm over 24 h in 1× PBS, while QD-PEG-OH and QD-PEG-OMe retained an unchanging hydrodynamic diameter (20–30 nm) over the entire measurement period (Fig. 2A). When transferred to aCSF – a more complex and more brain-representative media – QD-MPA particles tended to aggregate immediately (hydrodynamic diameter > 1500 nm) (Fig. 2B). In contrast, QD-PEG-OMe particles were relatively stable in aCSF over the entire 24 hour period, while QD-PEG-OH showed a statistically increased hydrodynamic diameter at 24 h but remained below 100 nm in total hydrodynamic size (Fig. 2B).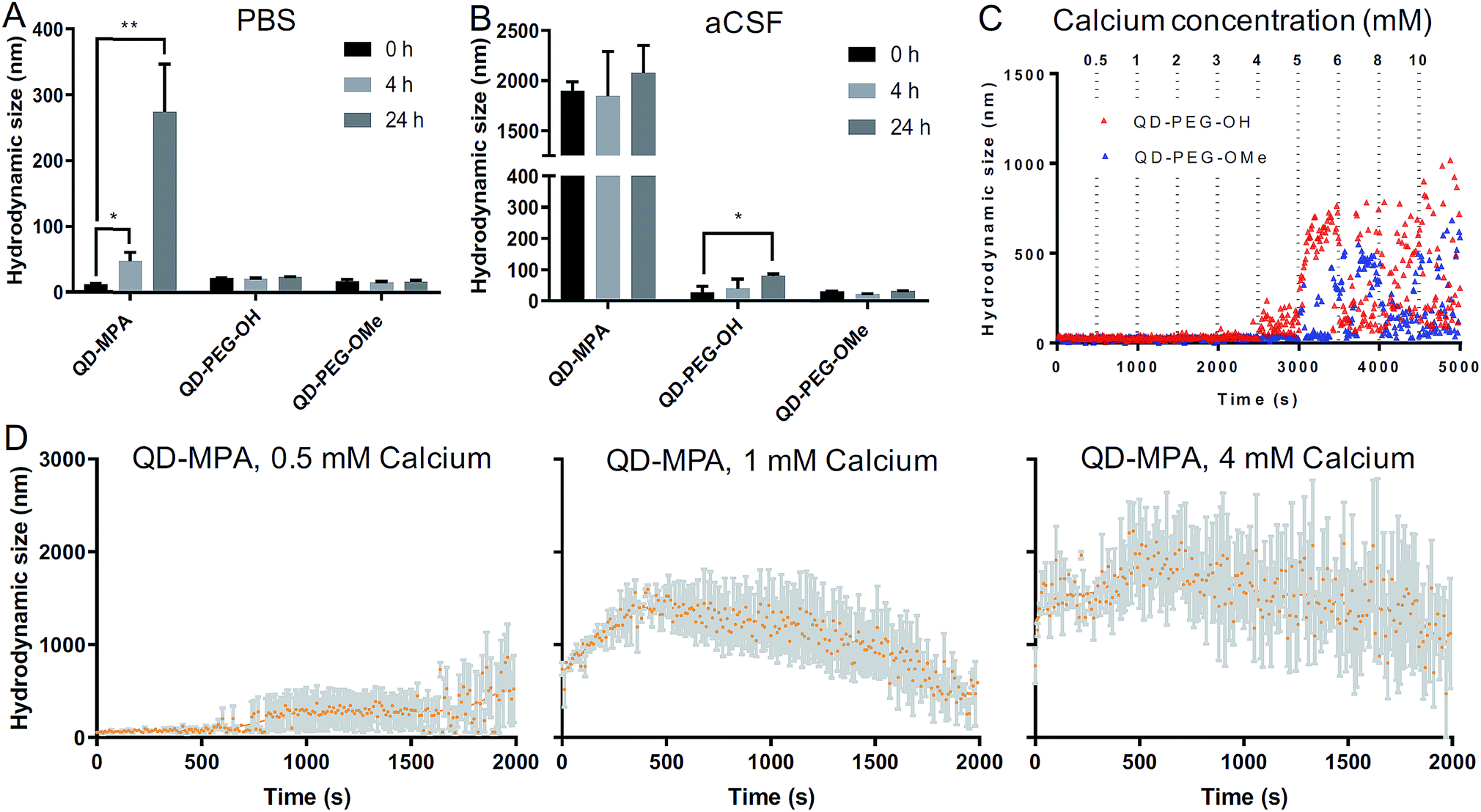 | ||
| Fig. 2 Colloidal stability of QDs at 37 °C in neurophysiological-relevant fluids. (A) QD hydrodynamic diameter at 37 °C in 1× PBS at 0, 4, 24 h (n = 3 measurements per particle type). (B) QD hydrodynamic diameter at 37 °C in aCSF at 0, 4, 24 h (n = 3 measurements per particle type). (C) Initial aggregation kinetics of QD-PEG-OH and QD-PEG-OMe at 37 °C in aCSF with incremental addition of CaCl2 every 500 s, up to a 10 mM Ca2+ concentration. (D) Initial aggregation kinetics of QD-MPA at 37 °C in CaCl2 solution with 0.5, 1, and 4 mM Ca2+ over a period of 2000 s (n = 3 measurements per particle type). Trendlines showing initial QD-MPA aggregation were generated using a LOWESS regression in GraphPad. | ||
To further explore media components that can give rise to rapid aggregation, we then evaluated the initial aggregation profiles of each functionalized QD in aCSF with different concentrations of calcium over a period of 30 min. Calcium ions play a significant role in the brain as a universal messenger of extracellular signals in a variety of cells, and pathological changes in calcium homeostasis could cause alterations in neuronal function and have been associated with brain aging and Parkinson's disease.43,44 Calcium is a prevalent component in CSF and has been shown to lead to significant aggregation of colloidal particles.45 As such, we investigated how QD stability changes in aCSF with different Ca2+ concentrations in order to gain preliminary insight into nanoparticle behavior in the brain microenvironment. QD-MPA was observed to aggregate in aCSF with Ca2+ concentrations as low as 0.5 mM and exhibited increased rates of aggregation with 1 mM and 4 mM Ca2+ concentrations (Fig. 2C and D). Sedimentation was observed after initial QD-MPA aggregation, where particles aggregated to sizes greater than 1000 nm and then sedimented to the bottom of cuvette, resulting in a decrease in DLS signal which manifests as a drop in the apparent hydrodynamic diameter reported by the instrument. When Ca2+ levels were increased, QD-MPA aggregation kinetics shifted from gradual aggregation to much more rapid aggregation followed by particle sedimentation (Fig. 2D, ESI Fig. 2A†), demonstrating a more severe instability of QD-MPA in an environment with a stronger ionic strength. Decreasing the system temperature slowed the aggregation kinetics of QD-MPA, although aggregation was still clearly present (ESI Fig. 2B and C†). In contrast, both QD-PEG-OH and QD-PEG-OMe remained stable at room temperature and at physiological temperature in 1× PBS and aCSF for up to 24 h, supporting the use of PEG functionalization to provide steric stabilization.46 QD-PEG-OH was stable up to a Ca2+ concentration of 4 mM, but exhibited significant aggregation above 5 mM Ca2+ (Fig. 2C). QD-PEG-OMe showed a similar trend, with stable dispersions up to a 5 mM Ca2+ concentration with signs of aggregation at higher concentrations. Considering that the Ca2+ concentration in the human brain is on average 2 mM, these data suggest that the application of PEGylated QD particles to the brain is viable.
Importantly, in aCSF we see no dissolution of QDs into appreciable amounts of Cd ions. Using inductively coupled plasma mass spectroscopy (ICP-MS) analysis, we measured Cd-ion concentrations following incubation for 0 and 24 h in aCSF. Cd-ion concentrations were stable for all QDs across the experimental time frame, and remained less than 10 parts per billion (ppb) (ESI Fig. 3A†). We also incubated 0.1 μM QDs in aCSF at 37 °C for 24 h, and measured the luminescence intensity of the QD suspensions at 0 h, 1 h, 4 h, and 24 h of incubation (ESI Fig. 3B†). The photoluminescence (PL) of the QD-MPA particles remained stable over 24 h, while the PL intensity of the PEGylated QDs decreased over time. This likely occurs because the well-dispersed PEGylated QDs are completely exposed to the aCSF environment, while the non-PEGylated QDs (QD-MPA) aggregate quickly (Fig. 2A), resulting in a lower initial PL intensity, but a less rapid decrease in PL intensity since the QD-MPA particles contained within the aggregate are protected from aCSF exposure. Independent of the change in PL, all confocal imaging studies have clear QD detection at low laser power with high signal to noise in the in vitro cell culture, ex vivo brain slices, and in vivo tissue sections.
Both long-term and short-term QD stability studies indicated a significantly different aggregation pattern for non-PEGylated QDs (QD-MPA) as compared to PEGylated QDs (QD-PEG-OH and QD-PEG-OMe). PEGylated QDs were found to be stable at different temperatures and in different neurophysiologically relevant fluids, while non-PEGylated QDs were very sensitive to changes in media conditions, with decreased stability at higher Ca2+ concentrations or longer incubation time.
Ex vivo QD cellular uptake and time-lapse internalization imaging
A comprehensive understanding of cellular uptake is critical when discussing the interpretation of QD-based biomarkers. Thus, the cellular uptake of QDs in organotypic whole hemisphere (OWH) slices was evaluated as a function of surface functionality, brain cell type, and brain region. Independent of surface chemistry, all QDs were found localized primarily in microglia in the neonatal rat brain, particularly in the corpus callosum and hippocampal regions (Fig. 3A). Microglia – the innate immune cells of the brain – become activated in response to injury and uptake invading foreign bodies by phagocytosis.37 Our cellular uptake findings not only further support previous observations that QDs are preferentially taken up by microglial cells,18 but also indicate that there is region-specific microglial uptake of QDs, as seen in our organotypic slice and in vivo studies. Our observations that QDs preferentially localize in the corpus callosum could be due to the abundance of proliferative and phagocytic amoeboid microglia cells in the corpus callosum in the developing brain.47 | ||
| Fig. 3 Iba1+ microglia and lysosome uptake of QDs in ex vivo OWH slices. (A) Representative 40× magnification images of QD-MPA, QD-PEG-OH, and QD-PEG-OMe distribution and Iba1+ cellular uptake in the cortex, corpus callosum, and hippocampus in P14 Sprague-Dawley (SD) rat OWH slices (300 μm thickness). QDs (red, all images) were found internalized into Iba1+ microglia (green, all images), especially in the corpus callosum and hippocampus regions. Representative high-magnification images of QD-MPA, QD-PEG-OH, and QD-PEG-OMe localization inside Iba1+ cells are shown as insets. Cell nuclei were stained with DAPI and display blue luminescence. (B) Representative 40× magnification images of QD-PEG-OH (red) and QD-PEG-OMe (red) internalization into LysoTracker-stained lysosomes (green) in the corpus callosum region in a live OWH slice after 2 h of QD exposure. | ||
QD-MPA, as compared to QD-PEG-OH and QD-PEG-OMe, had significant aggregation in the extracellular space (ECS) in the OWH slice model (ESI Fig. 4†), consistent with the aggregation observed in aCSF and PBS. In this case, the tendency of QD-MPA toward aggregation inhibited successful distribution to all the microglial cells spread throughout the tissue. Confocal images acquired at 60× magnification and 4-fold zoom enabled a closer look at QD internalization in microglia at the single-cell level. Although several QD-MPA aggregates were still observed to uptake in the microglia, the QD-PEG-OH and QD-PEG-OMe uptake was much more distinctive and diffuse in the microglial cytosol (Fig. 3A inset). This observation led us to further explore the intracellular destination of the internalized QDs.
Upon staining live OWH slices with LysoTracker, we clearly observed both PEG-OH and PEG-OMe QD localization with lysosomes 120 min after QD introduction (Fig. 3B, Movie 1 of QD-PEG-OH and Movie 2† of QD-PEG-OMe). Our observation of PEGylated QDs colocalizing with lysosomes supports the previously identified endocytic mechanism of QD cellular uptake and the degradation of QDs in the lysosomes of glial cells.48,49 Importantly, the QDs were also found to be stable in low-pH environments (ESI Fig. 5†), equivalent to those found in lysosomes.50 Additional studies involving the manipulation of entry pathways and intracellular trafficking through the use of inhibitors are needed for deeper understanding of QD intracellular trafficking, and eventual exocytosis or breakdown, in glial cells.
The internalization of QDs into microglia was further demonstrated by time-lapse imaging in OWH slices, where QDs were observed to uptake into CD11b+ microglia in the corpus callosum within the first 4 h of exposure, irrespective of surface functionality (Movies 3–5†). To our knowledge, these live-tissue, time-lapse imaging studies provide the first direct observations of real-time QD internalization in ex vivo brain slices, and further confirm microglial targeting of QDs (Movies 3–5†). QD-MPA rapidly formed large aggregates in the ECS, which prevented further internalization into microglia (Movie 3†), although non-aggregated QD-MPA did show some microglial uptake. In order to understand whether QD trafficking into microglia is surface functionality dependent, we quantified the fraction of Iba1+ cells containing QDs in the corpus callosum region – the region where we saw the highest levels of QD internalization into cells. It was observed that the fraction of Iba1+ cells that uptake QDs after 24 h of QD exposure was in the range of 55% to 66% and was not significantly different between the three QD surface functionalities (ESI Fig. 6†). Representative cross-sectional 240× z-stack images of QD internalization also show localization with Iba-1+ microglial cells in brain slices (ESI Fig. 7†). Previous literature has shown that aggregates greater than 1000 nm in size preclude uptake into microglia and directly impact cellular responses.51 However, if only non-aggregated QDs are considered, the extent of microglial QD internalization was independent of surface functionality.
Although rarely observed in our study, on occasion some QDs also interacted with neurons, mostly in the cortex and hippocampus region (Fig. 4). QDs were not observed to interact with or be internalized into oligodendrocytes (ESI Fig. 8†). The distribution pattern of all QDs in neurons (Fig. 4) is similar to previous studies done in organotypic hippocampal cultures, showing a potential interaction with the neuronal membrane and along the synapse.16 QD interactions with neurons may be due to the negative charge on the surface, which leads to an association with the electrophysiological activity of neuronal cells.17 Dante et al. observed that QDs stained along a synapse and associated with the neuronal membrane, when administered at a low dose (10 nM) on primary neuron cultures, and Walters et al. observed that QDs interact with neurons in a sparse and distributed pattern.16,17 Sophisticated engineering of QD ζ-potential should be taken into consideration, as Dante et al. found that strong detectable interactions with neurons were only observed for QDs with zeta potentials less than −20 mV,17 while Walters et al. illustrated that positively-charged QDs interacted specifically with oligodendrocytes.16 It is worth mentioning that previous literature presents contradictory results, indicating that QDs were internalized into one or other cell types to different degrees – results that might be conflicting due to the different conditions used in each study.16–18 The source of the study platform – either cell culture or organotypic brain slice – can result in the differentiation of QD–cell interactions due to differences in cytoarchitecture network or accurate ECS representation,52 as we have demonstrated in the present study. The emphasis of our study on QD–cell interaction in side-by-side comparisons of in vitro, ex vivo, and in vivo models demonstrates that the chosen model platform can clearly impact the outcome of a particular investigation. For example, the presence of tissue barriers could lead to a difference in QD accessibility to cells, and the stability of particles in various fluids could alter the biological identity of the QDs when applied to the different models. This knowledge helps to understand QD–cell interaction in a more systematic way, and emphasizes the importance of carefully considering the biological microenvironment when interpreting particle behavior.
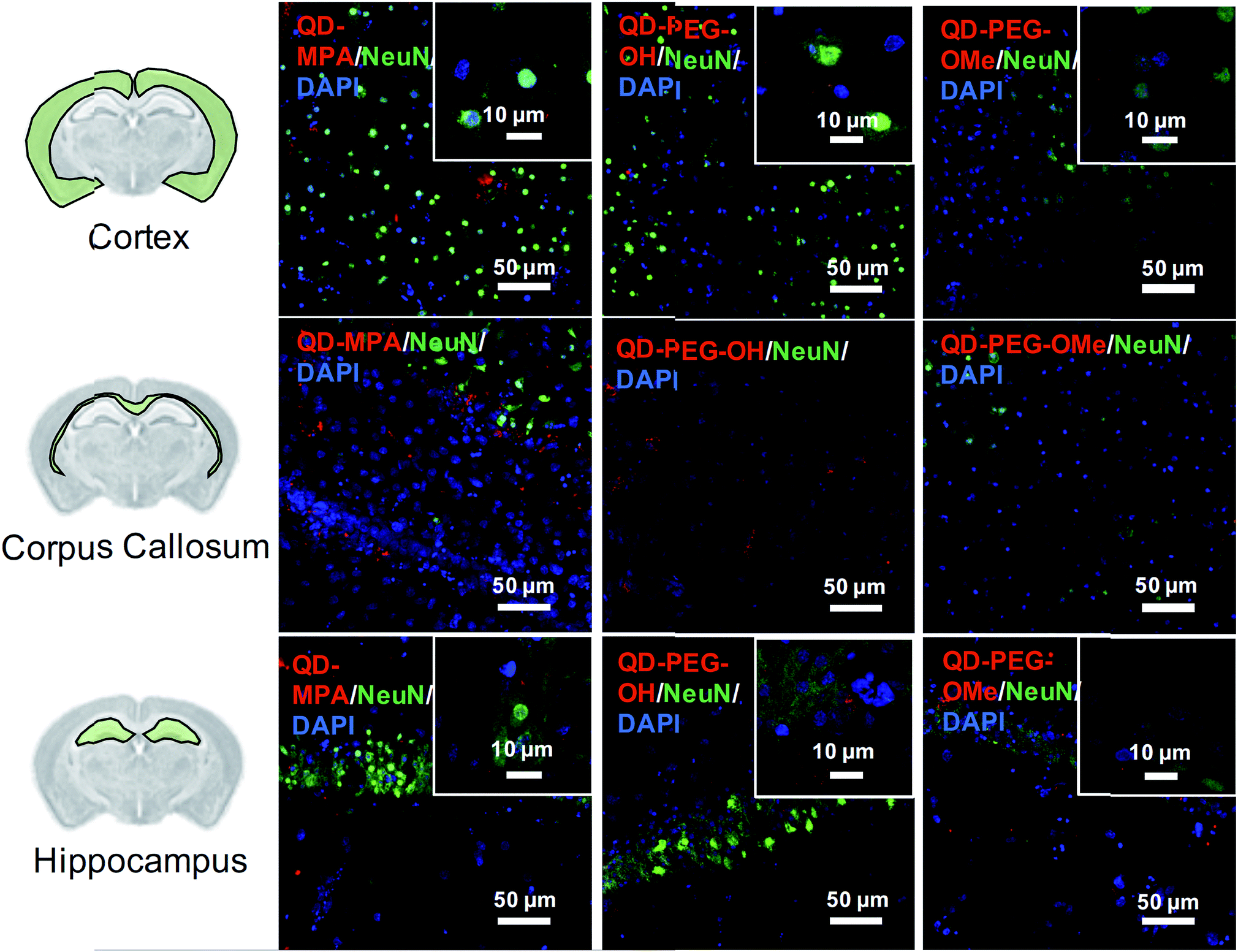 | ||
| Fig. 4 NeuN+ neuron uptake of QDs in ex vivo OWH slices. Representative 40× magnification images of QD-MPA, QD-PEG-OH, and QD-PEG-OMe (red, all images) distribution and interaction with NeuN+ neurons (green, all images) in the cortex, corpus callosum, and hippocampus in P14 SD rat OWH slices (300 μm thickness). Representative high-magnification images of QD-MPA, QD-PEG-OH, and QD-PEG-OMe interaction with NeuN+ cells are displayed as insets. Cell nuclei were stained with DAPI (blue). | ||
QD-induced toxicity, oxidative stress, inflammation and metal detoxification in OWH slices as a function of surface functionality
The cytotoxicity of QDs has long raised concerns about the application of QDs in biomedical applications. To use QDs in a highly complex functional organ, such as the brain, their toxicity needs to be carefully and fully investigated.53 However, it is difficult to draw clear conclusions from reported studies thus far because of the use of numerous different cell lines, which each have varying thresholds for QD-induced toxicity and only represent certain types of brain cells rather than the whole integrated neurological network.54,55 By using 300 μm-thick organotypic brain slices, the toxicity of QDs can be examined in a representative 3D heterogeneous environment. To study the amount of QD-induced toxicity in OWH slices, we first evaluated whole-slice cytotoxicity caused by the QDs by measuring cell death after 24 h of QD exposure, as a function of QD surface functionality and QD concentration. To note, QD-MPA was not included in the OWH slice toxicity assessment due to the observed rapid aggregation in biological fluids and subsequent hindered penetration and cellular uptake in OWH slices. The overall toxicity of PEGylated QDs was found to be dose-dependent (Fig. 5). At 0.01 μM and 0.1 μM concentrations, PEGylated QD-induced cytotoxicity was below 10%, and there was no significant difference between the non-treated (NT) group and either of the two PEG functionalities (PEG-OH and PEG-OMe). When the dose was increased to 1 μM, QD-induced cytotoxicity increased to around 15% for QD-PEG-OMe and slightly above 10% for QD-PEG-OH. The cytotoxicity of 1 μM QD-PEG-OMe was significantly higher than the NT group, starting at 4 h post-treatment, and remained statistically higher at 8 h and 24 h. The relatively low toxicity of QD-PEG-OH and QD-PEG-OMe at low concentration is likely imparted due to the PEG coating, which has been previously shown to effectively decrease cytotoxicity and maintain tissue viability.56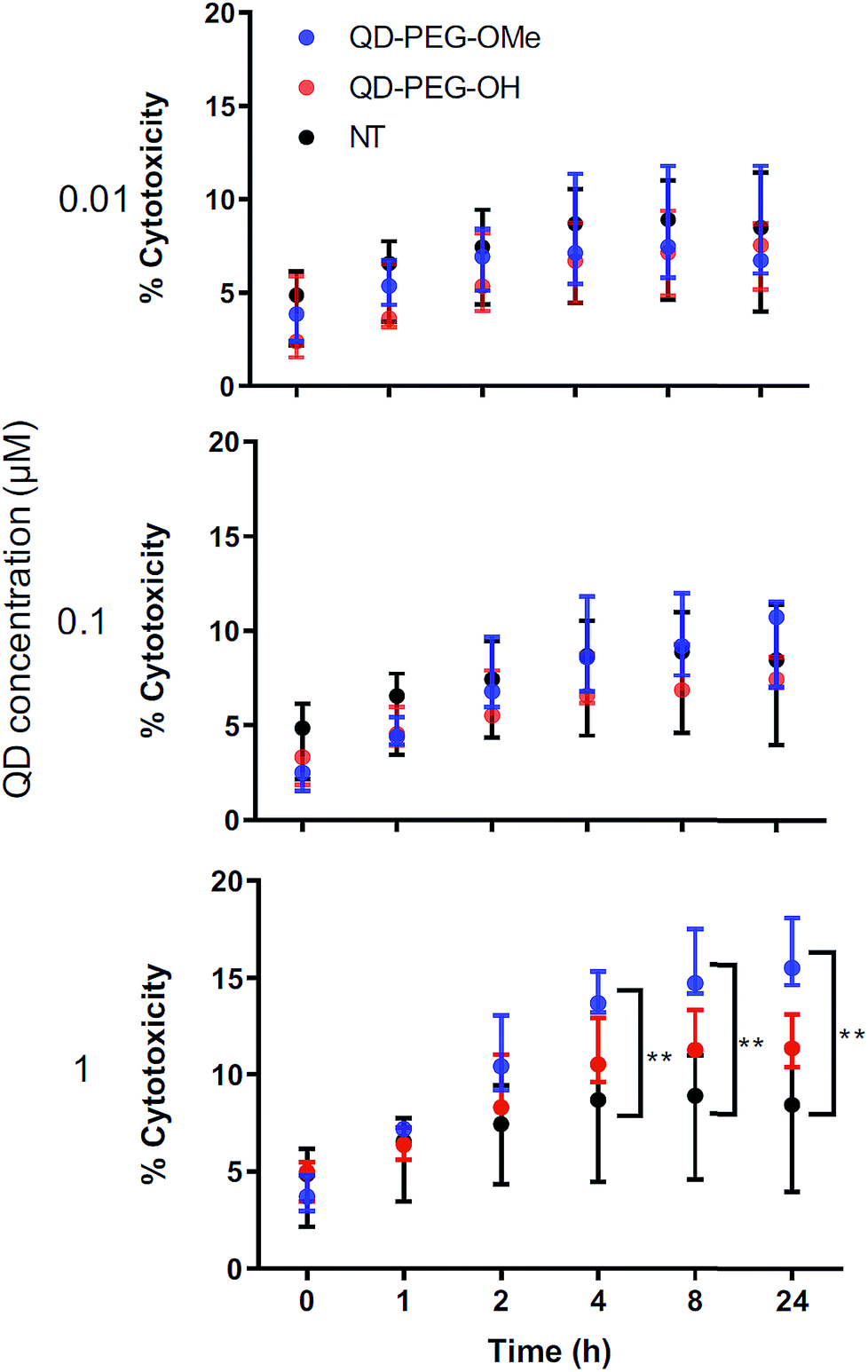 | ||
| Fig. 5 Dose-dependent toxicity of QD-PEG-OH and QD-PEG-OMe in ex vivo OWH slices. Treatment of QD-PEG-OH (red) and QD-PEG-OMe (blue) at 0.01, 0.1, and 1 μM concentrations in P14 rat organotypic brain slices for 24 h. QD toxicity was determined by LDH assay at 0, 1, 2, 4, 8, and 24 h as follows. For each QD concentration, n = 3 slices per QD per concentration condition were evaluated; for NT group (black), n = 5 slices. | ||
QD toxicity in the CNS can be due to multiple mechanisms, including intracellular QD breakdown29,49,54,55,57 and subsequent release of carcinogenic Cd2+ ions.26 QD-induced inflammation or oxidative stress can also contribute to cell death and a cascade of function loss. QDs with different functional groups were reported to induce different levels of proinflammatory effects in vitro on human lung epithelial cell lines as well as in vivo in mouse lung tissue following direct administration through intratracheal instillation.31,58 For example, metallothionein proteins (MT, Mt for mRNA), which can sequester Cd ions and limit oxidative stress, have shown increased expression after QD treatment in kidney or lung cells.54,59–61 However, no studies have been performed for the activation of MT in the brain following QD treatment. To investigate QD-induced inflammation, metal detoxification (Fig. 6), and oxidative stress (ESI Fig. 9†) in OWH slices, we also evaluated time-dependent mRNA profiles in response to QD exposure. MT-1, MT-2A, and MT-3 (different isoforms of MT) are protective against metal ion toxicity and oxidative stress, and are found in the CNS.62,63 Strikingly, at 0.1 μM QD concentration, QD-PEG-OH and QD-PEG-OMe induced a significant 7.4- and 13.2-fold increase of Mt1 expression, respectively, in OWH slices after 6 h of incubation, and the Mt1 increase remained 3.5-fold higher after 24 h of QD-PEG-OMe incubation (Fig. 6A). The significant increase in Mt1 mRNA expression 6 h after 0.1 μM QD exposure reflects the cellular response to activate metal detoxification pathways for Cd2+ exposure, which occurs rapidly upon localization in lysosomes, as observed in our study. The lysosomal localization could induce lysosomal dysfunction and oxidative stress, as previously suggested.64
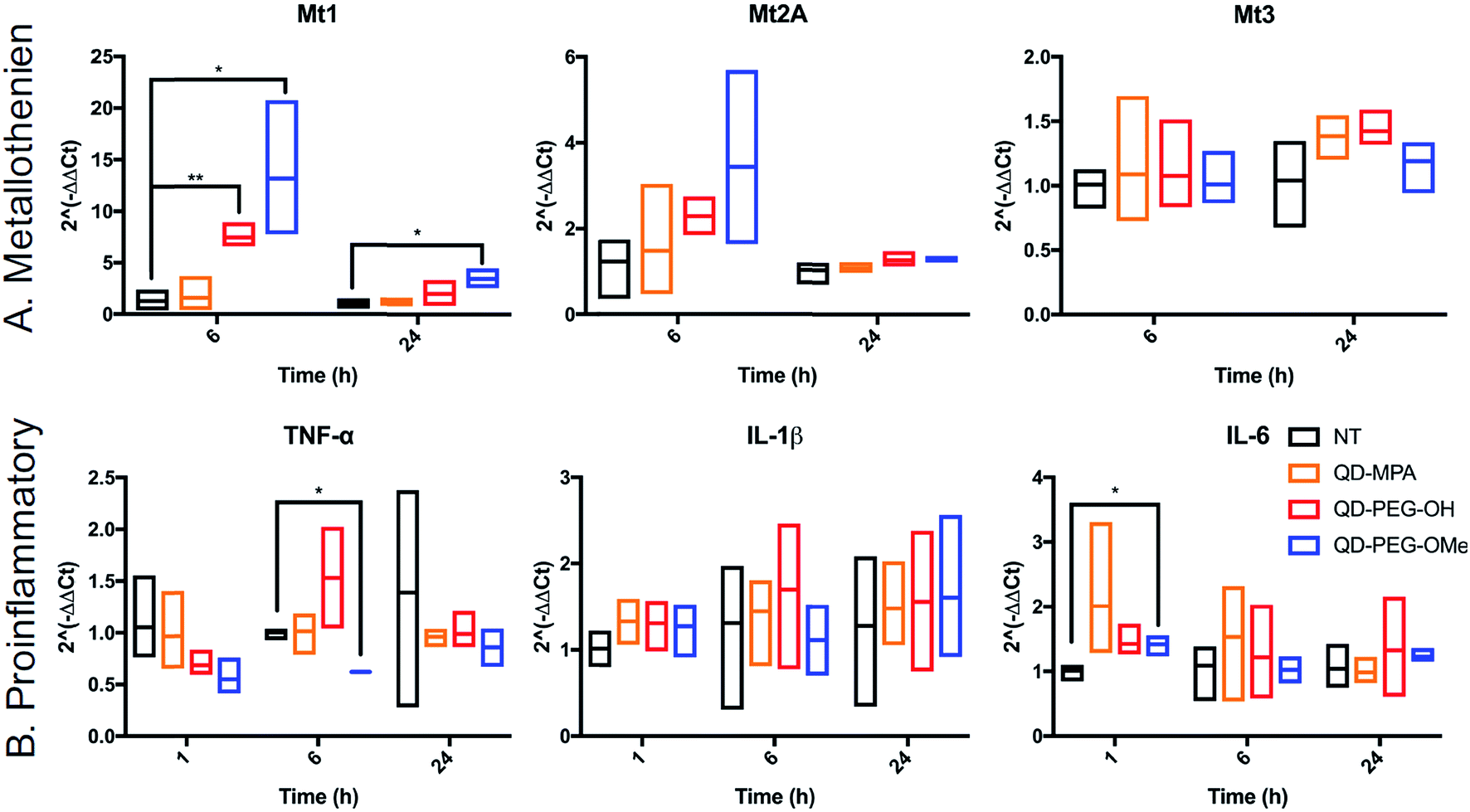 | ||
| Fig. 6 Time-dependent mRNA profiles of QD-induced proinflammatory and metallothionein markers in OWH slices. Fold-changes in mRNA expression were measured at 1 h, 6 h, and 24 h of QD-MPA (orange), QD-PEG-OH (red), and QD-PEG-OMe (blue) exposure at 0.1 μM concentration in OWH slices. The fold-changes were measured for (A) metallothionein isoforms Mt1, Mt2A, and Mt3 and (B) proinflammatory cytokines IL-6, TNF-α, and IL-1β, and compared to NT slices (black). For 1 h, 6 h, and 24 h time points, n = 3 groups and n = 3–6 slices per group were evaluated for each experimental sample (except for the QD-PEG-OMe 6 h proinflammatory sample, where n = 2 groups with n = 6 slices in total were evaluated). All data are reported as median values with 95% confidence intervals. | ||
The pattern of surface functionality-dependent Mt1 increase is aligned with the pattern of surface functionality-dependent LDH release. QD-PEG-OMe induced the most significant expression-fold changes in Mt1, as compared to other QDs, and also had the highest LDH release. When the metal is in overloading conditions, the MT pathway becomes overwhelmed and can no longer effectively scavenge metals, resulting in cell death,65 as we demonstrate with higher concentration (1 μM) QD-PEG-OMe exposure. Meanwhile, Mt2A and Mt3 did not show a significant increase in expression at any time point up to 24 h (Fig. 6A). Mt3 is mainly present in neurons63 and would be less likely to be activated based on our observations that QDs internalize primarily in microglia. Although Mt2A showed a non-significant trend towards an increase in expression for QD-PEG-OMe, the expression increase is insignificant because Mt2A prefers binding Zn while Mt1 prefers Cd,66 the main component of our QDs used in this study. To the best of our knowledge, our study is the first study to reveal QD-induced MT pathway activation in brain cells.
Interestingly, QD exposure resulted in almost no statistically significant inflammatory or other oxidative stress responses at the mRNA level. IL-6, TNF-α, and IL-1β are pro-inflammatory cytokines and are central to neuroinflammatory processes.67 BAX and Casp-3 are involved in neuronal death, oxidative stress, and apoptosis during brain pathology.68,69 IL-10 and TGF-β1 are both anti-inflammatory cytokines that play an important role in altering the function of microglia,70 and are highly influential in the developing brain.67 Only QD-PEG-OMe showed a statistically significant 2.0-fold increase in IL-6 expression after 1 h incubation and a statistically significant 0.6-fold decrease in TNF-α expression after a 6 h incubation (Fig. 6B). These changes were only present during early time points after initial QD exposure. Previous literature has suggested that PEGylated QDs can induce higher expression of proinflammatory cytokines, which was strongly associated with the functional groups at the end of PEG chain, leading to selective endocytosis.31,58 In our studies, 0.1 μM QD concentrations did not induce statistically significant cytotoxicity in OWH slices, activated a metal detoxification pathway, and led to a minimal inflammatory response in OWH slices over a 24 h period. This concentration was chosen to be representative of what cells would maximally be exposed to in vivo following systemic administration. Therefore, it is our conclusion that PEGylated QDs with pendant hydroxyl or methoxy groups used at this concentration exhibit microglia-dominant uptake and are not inducing inflammation or toxicity to brain cells within this particular timeframe. To further elucidate potential mechanisms of cellular response to QDs, higher doses could be tested in OWH slices to determine changes in mRNA expression, and further analysis on protein expression levels could be evaluated.
In vitro QD toxicity and cellular uptake on BV-2 cells
The influence of the extracellular matrix (ECM) barrier on QD penetration and tissue distribution was observed in our ex vivo slice studies, which further impacted QD toxicity and cellular uptake. The LDH assay indicated similar cytotoxicity of QD-MPA across 0.01–1 μM QD concentrations on OWH slices (ESI Fig. 10†), which did not exhibit toxicity when compared with healthy NT slices. This result is different from what has been reported in previous literature, where non-PEGylated QDs induce more immediate and significant cell death, while PEGylated QDs show minimal toxicity in vitro57 due to the “stealth” ability of PEG. Consequently, we sought to understand the effect of QDs on microglial cells when cells are directly exposed to QDs and no penetration barrier is present. When applied directly to microglia, 0.01 μM QD-MPA induced significant cell death starting at 1 h, as compared to a NT group treated only with 1× PBS. The viability of QD-MPA-treated cells was observed to drop below 50% before some recovery at 24 h (Fig. 7). QD-PEG-OH, however, did not show any statistically significant decrease in cell viability, as compared with NT, except at 6 h (Fig. 7). QD-MPA were immediately taken-up into BV-2 cells at the 1 h time point, with additional uptake continuing throughout the duration of the 24 h study (ESI Fig. 11†). However, PEGylated QDs were not observed to uptake into BV-2 cells at any time point. This indicates that, when applied to an in vitro cell platform, PEG coatings did preclude particles from toxicity and cellular uptake. Considering the rapid instability of QD-MPA in biologically relevant fluids and their aggregation on the surface of OWH slices (ESI Fig. 2 and 4†), QD-MPA are unlikely to overcome the penetration barriers introduced via the presence of the brain extracellular space and extracellular matrix in OWH slices.6,36 This inability to penetrate tissue limits the interaction with cells throughout the tissue, whereas in the in vitro monolayer, QD-MPA had direct access to all cells. Due to the instability of QD-MPA, these particles are unlikely to be suitable for biomedical application or in vivo study.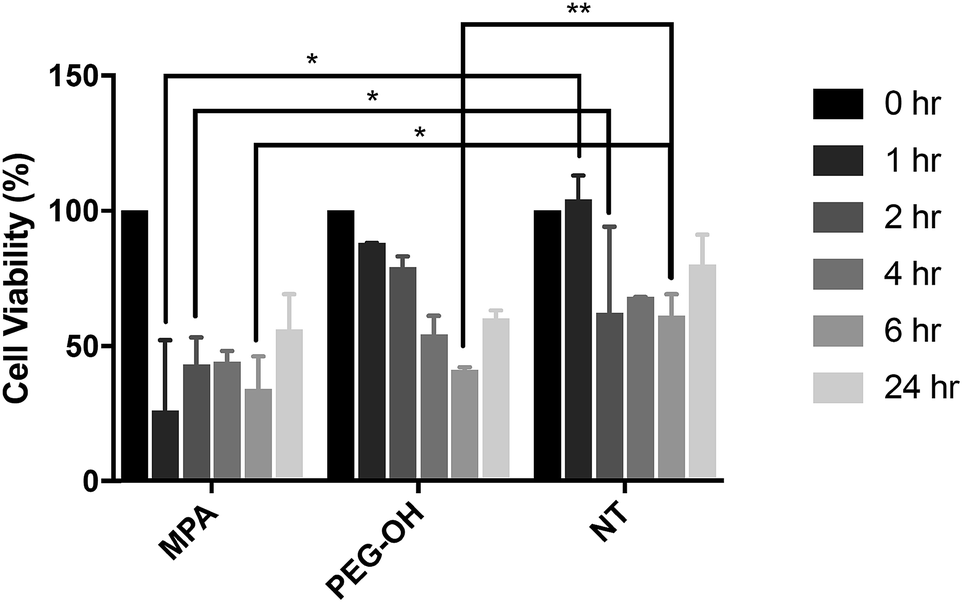 | ||
Fig. 7 Toxicity of QDs on in vitro BV-2 cells. Treatment of QD-MPA and QD-PEG-OH at 0.01 μM concentration on in vitro BV-2 cells for 1, 2, 4, 6, and 24 h, with reported % cell viability. For each condition, n = 3 wells were evaluated with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well when plated. 000 cells per well when plated. | ||
In vivo QD cellular uptake in mglur5 neuroinflammatory rat model
Lastly, we utilized the metabotropic glutamate receptor 5 (mglur5) neuroinflammatory rat model to evaluate QD uptake in vivo. In addition to studying QD behavior in vitro and ex vivo, we sought to explore how these findings translated to an in vivo environment in both normal and neuroinflamed tissue, through the use of a transgenic mglur5 model. mglur5 plays an important role in neuroinflammation,71 especially with respect to microglia-associated inflammation and neurotoxicity.72mglur5 knock-out mice have been used recently as a model of neuroinflammatory diseases with hallmarks of Fragile X syndrome, schizophrenia, and autism spectrum disorder (ASD).73 We utilized mglur5 age-matched and litter-matched wild-type (WT) and knockout (KO) rats to investigate QD behavior in vivo following systemic injection. We detected QD-PEG signal in the brain, at both 4 h and 24 h time points, after intraperitoneal (i.p.) injection in postnatal day (P) 7 WT and KO rat pups. However, the QD signal was minimal, due to the majority of the sub-20 nm QD-PEG being cleared rapidly.74 Although the total quantity of QDs in the brain was low, we identified colocalization of QDs in microglia in various regions. QD-PEG-OH particles were found colocalized with Iba1+ microglia in both the WT and KO brain in the cortex, corpus callosum (Fig. 8), hippocampus, and periventricular region (PVR) (ESI Fig. 12†), with the majority of accumulation in the corpus callosum region at 4 h post-injection (Fig. 8). At 24 h post-injection, allowing for longer in vivo circulation, QD-PEG-OH were still found internalized in Iba1+ microglia (ESI Fig. 13†), consistent with our observations in the OWH slices. Our findings in the WT neonatal brain – where we see comparable QD uptake to the KO brain – are supported by previously published work where QDs were observed to be present in brain vessels or epithelial cells or even to have permeated into brain parenchyma after systemic administration in healthy rodents.75–77 Although the mechanism of QD uptake in the healthy brain is still unknown, current findings suggest QD uptake occurs via transcytosis-mediated transport through the BBB and cellular uptake via phagocytosis and pinocytosis.75 The main regions of QD localization in our study included the corpus callosum and PVR, which are near to ventricles or nearby large blood vessels networks. Consequently, these areas experience high levels of fluid exchange, which can contribute to the distribution of QDs and internalization by cells in this region of the developing brain.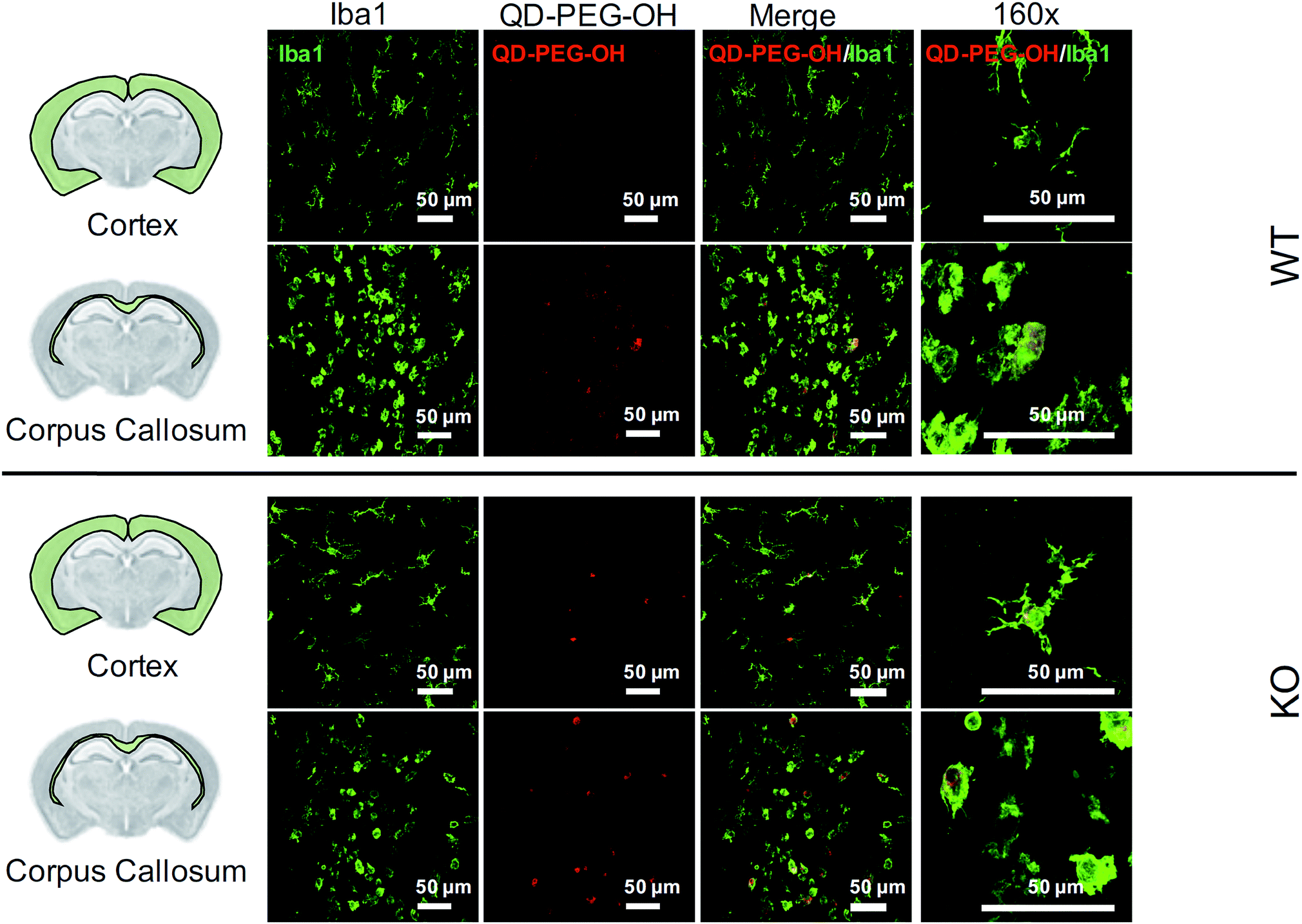 | ||
| Fig. 8 QD cellular uptake in P7 rats, 4 h post-administration. Representative images of QD-PEG-OH (red, all images) colocalization with Iba1+ microglia cells (green, all images) in the cortex and corpus callosum regions in P7 mglur5 WT and KO pup brains, 4 h after i.p. administration. 40× magnification images with 4-fold zoom (160×) are presented in the rightmost column to show QD-PEG-OH internalization in cells. | ||
Conclusions
The implementation of engineered nanoparticles as biomarkers for CNS disease is a challenging task, requiring a comprehensive understanding of both nanoparticle engineering and the disease environment in which the nanoparticle will be applied. To develop QD-based biomarkers for application to the brain, it is necessary to better understand QD behavior in the brain environment. Here we comprehensively assessed the interaction of functionalized, core–shell CdSe/CdS QDs with the brain microenvironment, including QD colloidal stability, toxicity, and cellular uptake in vitro, ex vivo, and in vivo. Importantly, stability, toxicity, and cellular uptake of QDs were dependent on one another, and dependent on the model used to evaluate the effect. We found that surface chemistry strongly influenced QD behavior in the brain. PEG-coatings improved QD stability in complex neurophysiological-relevant fluids, induced low cytotoxicity in brain slices, and led to stable, diffuse cellular uptake. QDs were preferentially taken up into microglia, especially in the corpus callosum, which was further confirmed in vivo. Pegylated QDs induced an increase in Mt expression in the OWH slices as a function of time and surface chemistry, but did not induce inflammatory and other oxidative stress pathways. Non-PEGylated QDs, however, destabilized rapidly when exposed to brain-relevant fluids, which prevented penetration into brain tissue. We also found that administration of functionalized QDs can result in dose-dependent toxicity in brain slices. Lastly, with a goal of reconciling seemingly contradictory findings in literature, our results clearly demonstrate the importance of considering the specific model system that is used to evaluate QD behavior.Materials and methods
Core–shell CdSe/CdS QD synthesis and characterization
:Se (190 mg of Se dissolved in 4 mL of trioctylphosphine) was rapidly injected. Aliquots were taken as the nanocrystals grew, and UV-vis absorption spectroscopy was used to monitor nanocrystal diameter by tracking the position of the 1S3/21Se excitonic absorption feature.79 Once the desired nanocrystal diameter was achieved, the heating mantle was removed, and the mixture was allowed to cool. When the temperature of the flask reached 100 °C, 10 mL of toluene was injected in order to facilitate nanocrystal purification. Once cooled to room temperature, the nanocrystal cores were purified by adding antisolvent (ethanol) to the reaction mixture until the point of opalescence, followed by rapid centrifugation, and dispersion of the resulting nanocrystal precipitate in clean toluene – a process which was repeated three times.
:1000 in 10 mM sodium chloride (NaCl), pH 7.4. Size and polydispersity (PDI) measurements were performed at 23 °C at a scattering angle of 173°, and ζ-potential was determined by laser Doppler anemometry (LDA). The measured hydrodynamic diameter and QD surface charge from different batches were then combined to determine average values for each surface functionality. FTIR spectra for MPA, PEG-OMe, and PEG-OH-functionalized CdSe/CdS QDs were obtained using a Bruker Alpha IR spectrometer equipped with a platinum attenuated total reflectance (ATR) accessory and a diamond crystal sampling module. Prior to FTIR analysis, the QD-PEG-OMe and QD-PEG-OH samples were placed under vacuum in a rotary evaporator until completely dry, and then were re-dispersed into chloroform. The MPA-functionalized QDs were purified via the addition of acetone, followed by centrifugation and dispersion of the precipitated QDs into methanol. Spectra were recorded under a nitrogen atmosphere after forming thin films of material on the diamond ATR crystal. QD thin films were formed by drop-casting the chloroform or methanol particle dispersions directly onto the surface of the ATR crystal and allowing the solvents to evaporate completely.
Animals
This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985). All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Washington, Seattle, Washington. For healthy newborn pups, time-mated pregnant female Sprague-Dawley rats (virus antibody free CD® (SD) IGS, Charles River Laboratories) were purchased and arrived on estrous day 17 (E17). For pups with neuroinflammation, we used a transgenic rat model generated by Sigma Advanced Genetic Engineering (SAGE) labs through knocking out of the metabotropic glutamate receptor (mglur5) on a Sprague Dawley background. mglur5 knockout (KO), wild-type (WT), and heterozygous (HET) pups were bred by HET × HET crosses. All dams used in these studies were housed individually and allowed to acclimate to their environment for a minimum of five days prior to delivering (E22). Day of birth was defined as postnatal day 0 (P0). Litters containing both sexes were cross-fostered and culled to 12 animals on P4, and genotypes for mglur5 pups were determined by real-time PCR (Bio-Rad) on P4. Each litter was housed under standard conditions with an automatic 12 h light/dark cycle, temperature range of 23–26 °C, and access to standard chow and autoclaved tap water ad libitum. Pups were checked daily for health. Both genders were used for all studies.Organotypic whole hemisphere (OWH) slice culture preparation
OWH slices were prepared based on modification to a previously published paper to maintain in vivo-like conditions.37 OWH slices were prepared from healthy P14 newborn SD rats. Neonatal rats were decapitated under aseptic conditions after euthanasia by a pentobarbital overdose >100 mg kg−1. The brain was quickly removed under sterile conditions, dissected into two hemispheres, and sectioned immediately into 300 μm-thick brain slices using a McIlwain tissue chopper (Ted Pella, Inc., USA). For each hemisphere, six consecutive slices at the level of bregma were separated in dissection medium (3.2 g glucose/500 mL HBSS, 1% of penicillin). The separated brain slices were transferred onto sterile 30 mm-diameter, 0.4 μm-pore-size cell culture membrane inserts (Millipore, Billerica, MA) in six-well tissue culture plates. To evaluate slice viability throughout the experimental window, supernatant aliquots were obtained to measure lactate dehydrogenase (LDH), as described below. For the LDH assay, cellular uptake, and time-lapse imaging studies, one OWH slice was placed on each membrane insert; for the quantitative reverse transcription polymerase chain reaction (RT-qPCR) study, three OWH slices were placed on each membrane insert and the volume of media was adjusted accordingly. Each well was pre-filled with 1 mL of slice culture medium (200 mL MEM, 200 mL HBSS, 4 mL Glutamax, and 1% penicillin) pre-warmed to 37 °C. The slices were maintained in a sterile incubator (ThermoFisher) at 37 °C with constant humidity, 95% air, and 5% CO2 overnight and randomized to treatment groups prior to each study.QD dose selection
We estimated the dose used in in vitro and ex vivo studies based on the dose of nanoparticles that would be directly exposed to the brain after intravenous (i.v.) injection in vivo. There is limited published data on QD biodistribution in the neonatal rat brain. Based on previous research with other sub-40 nm diameter nanoparticles,80–82 brain uptake corresponds to a nanomaterial dose ranging from ∼0.2 μg to 30 μg, depending on the material and method of quantification. The selection of QD dose of 0.01–1 μM corresponds to ∼0.12–12 μg, which is in this range. Therefore, the doses used in this study represent an in vivo exposure level for investigation in in vitro and ex vivo environments.Characterization of QD stability in brain-related biological fluid as a function of surface functionality
:DI water = 1:9) was adjusted to pH 2, 4, 7, and 9 by titrating with 0.2 M HCl. QD-PEG-OMe particles were diluted into 0.1× aCSF with different pH values to a final concentration of 0.1 μM. The suspended particles were incubated at 37 °C for up to 24 h. 1 mL samples of suspension were measured by DLS at 0 and 24 h to obtain particle hydrodynamic diameters (n = 3). The pH of each QD suspension was measured at 0 h and 24 h to validate no change in pH during the study.
Evaluation of QD toxicity in OWH slices as a function of surface functionality and dose by lactate dehydrogenase (LDH) assay
QDs were added to brain slices in a dose-dependent manner (0.01, 0.1 and 1 μM) for 24 h, and viability of each whole brain slice was evaluated. Media beneath pre-cut, 300 μm-thick OWH slices on membrane inserts was changed to fresh, pre-warmed slice culture medium. The QD-MPA, QD-PEG-OH and QD-PEG-OMe were diluted to 1, 0.1, and 0.01 μM by 1× PBS, and 100 μL of suspended particles were carefully dropped onto each slice. Slices without QD treatment served as the non-treated control group (NT), and slices receiving 1% Triton X-100 were considered as the 100%-cell-death, maximum-release group (Tx). The slices were then incubated at 37 °C with constant humidity, 95% air, and 5% CO2 for 24 h. At the end of the exposure time, fresh medium was replaced, and supernatant aliquots were taken at 0, 1, 2, 4, 8, and 24 h timepoints. NT and Tx groups were run for each QD-treated study. Supernatants at each time point were utilized to determine cell death with an LDH assay kit, according to manufacturer instructions (Cayman, USA). The percentage of LDH released in each whole-hemisphere brain slice was quantified by measuring the absorbance intensity of the formazan reactive product, subtracting the intensity of NT groups, and normalizing by the intensity of Tx groups. (n = 3)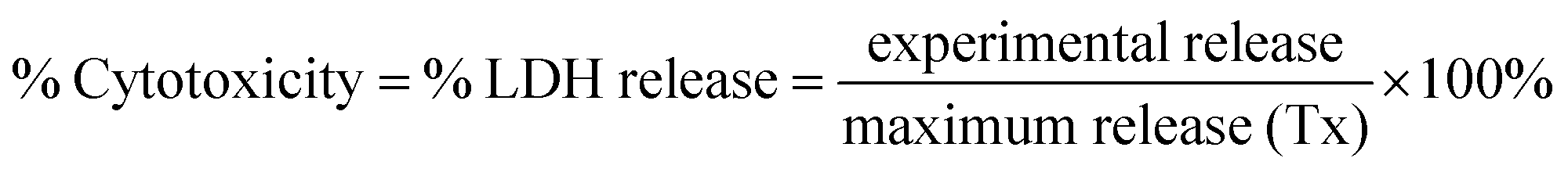
Evaluation of QD-induced oxidative stress, inflammation, and metal detoxification in OWH slices by RT-qPCR
Brain slices were treated with 0.1 μM QDs for 1 h, 6 h, and 24 h, and evaluated by RT-qPCR for mRNA expression of inflammatory cytokines, oxidative stress, and metal detoxification. Each experiment was done on three to six 300 μm-thick OWH slices in order to obtain enough RNA (>250 ng μL−1) post-extraction. P14 brains were sliced as described above, and three OWH slices containing hippocampal regions were placed on each membrane insert. The media beneath brain slices and membrane inserts was changed to fresh, pre-warmed slice culture medium prior to addition of QDs. The QD-MPA, QD-PEG-OH and QD-PEG-OMe were diluted to 0.1 μM by 1× PBS and 300 μL of suspended particles were carefully dropped on top of each slice. The NT control group was prepared using 300 μL of 1× PBS in place of colloidal QD particles. At the end of the exposure time, slices were washed with 300 μL of 1× PBS, removed from the culture dish, collected in RNAlater (Invitrogen) and stored at −80 °C. The RNA extraction, cDNA reverse transcription, qPCR assay and analysis were done following MIQE guidelines.83 RNA extraction was performed using TRIzol (Invitrogen) and chloroform (Sigma). After thawing the slices, the RNAlater was removed and 1 mL TRIzol was added to the slices. The slices were homogenized using a Pasteur pipet, and 0.2 mL chloroform was added per 1 mL TRIzol to separate RNA, DNA, and proteins. The RNA phase was collected, mixed with isopropyl alcohol (Honeywell) and centrifuged for precipitation, and washed with 75% ethanol. The RNA concentration was measured by using a NanoDrop instrument (ThermoFisher), and only RNA samples with A260/A280 ratio above 1.8 and A260/A230 ratio above 1.9 were used for the next step. Single-stranded complementary DNA (cDNA) was reverse-transcribed from 2 μg total-RNA-per-sample with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer instruction. Thermal cycling conditions were as follows: 60 min at 37 °C and 5 min at 95 °C. The cDNA samples were stored at 4 °C temporarily after the thermal cycling and transferred to −20 °C before qPCR. Prior to qPCR assay, multiple reference genes were tested on a custom 96-well plate (Bio-Rad) and GAPDH was selected as the stable reference gene in rat brain tissue for the qPCR study. RNA quality assay was performed to ensure that RNA degradation was minimal, and a positive PCR control assay was done to ensure the sample did not contain reaction-inhibiting factors. 25 nmol primers were ordered from Integrated DNA Technologies (sequences provided in Table 2) and diluted in DEPC-treated water (Invitrogen) to 3 μM. qPCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) based on manufacturer instruction. Briefly, DNA samples, primers, and iTaq Universal SYBR Green Supermix were mixed in wells in a Hard Shell 96-Well PCR Plate (Bio-Rad) and sealed by Microseal ‘B’ PCR Plate Sealing Film (Bio-Rad). Thermal cycling conditions for qPCR were as follows: 30 s at 95 °C, 40 cycles at 95 °C for 5 s, and 60 °C for 30 s. Gene expression was quantified with the ΔΔCt method (threshold cycle, amount of target = 2−ΔΔCt), and normalized to the reference gene GAPDH.| Gene | Abbreviation | Accession number | Forward primer | Reverse primer |
|---|---|---|---|---|
| Glyceraldehyde-3-phosphate dehydrogenase | GAPDH | NM_017008.4 | GTC GGT GTG AAC GGA TTT | TGT AGT TGA GGT CAA TGA AGG |
| Tumor necrosis factor | TNF-α | NM_012675.3 | CCT CAG CCT CTT CTC ATT C | GGA ACT TCT CCT CCT TGT T |
| Interleukin 6 | IL-6 | NM_012589.2 | GGA GAC TTC ACA GAG GAT AC | GCC ATT GCA CAA CTC TTT |
| Transforming growth factor, beta 1 | TGF-β1 | NM_021578.2 | GAA CCA AGG AGA CGG AAT AC | GGG ACT GAT CCC ATT GAT TT |
| Caspase 3 | Casp-3 | NM_012922.2 | CGC ACC CGG TTA CTA TTC | GCA TGA ATT CCA GCT TGT G |
| BCL2 associated X | BAX | NM_017059.2 | GCT ACA GGG TTT CAT CCA | CCA TGT TGT TGT CCA GTT C |
| Interleukin 10 | IL-10 | NM_012854.2 | ACG CTG TCA TCG ATT TCT | GGC CTT GTA GAC ACC TTT |
| Interleukin 1 beta | IL-1β | NM_031512.2 | TTC GAC AGT GAG GAG AAT G | GAT GCT GCT GTG AGA TTT G |
| Metallothionein 1 | Mt1 | NM_138826.4 | CACCGTTGCTCCAGATT | AGCAGCAGCTCTTCTTG |
| Metallothionein 2A | Mt2A | NM_001137564.1 | CAGCGATCTCTCGTTGAT | GAGCAGGATCCATCTGTG |
| metallothionein3 |
Mt3 | NM_053968.3 | TCCTACTGGTGGTTCCT | GCACACTTCTCACATCCT |
QD penetration and cellular uptake in OWH slices
Antibody stains were utilized to evaluate co-localization of QDs in neurons, microglia, and oligodendrocytes in OWH brain slices. Media beneath pre-cut 300 μm-thick brain slices on membrane inserts was changed to fresh, pre-warmed slice culture medium prior to QD addition. To study QD penetration into OWH slices, QDs were diluted to 1 μM by 1× PBS, and 100 μL of suspended particles were carefully dropped on top of each slice. To evaluate cellular uptake, QD-MPA, QD-PEG-OH and QD-PEG were diluted to 0.1 μM by 1× PBS, and 100 μL of suspended particles were carefully dropped on top of each slice. After incubating the slices with QDs at 37 °C with constant humidity, 95% air, and 5% CO2 for 24 h, the slices were fixed by 4% PFA or formalin for 1 h. Slices were then washed with 1× PBS twice. Each brain slice was carefully cut out of the surrounding membrane using forceps and scalpel. To assess QD penetration, slices were imaged using a Nikon A1 confocal microscope (Nikon Instruments Inc, USA) at 20× magnification with z-stack at 5 μm-per-step, and presented in 3D-projection view. To assess QD cellular uptake, slices were stained with primary antibodies for neurons (NeuN, Abcam ab190195, USA), microglia (Iba1, Wako 019-19741, USA), and oligodendrocytes (Olig2, Abcam ab109186, USA), followed by Alexa Fluor 488 secondary antibody (Life Technologies, USA) and DAPI for cell nuclei. Briefly, slices were incubated with 500 μL primary antibody for 6 h with 3% TritonX and 6% serum, followed by 500 μL secondary antibody for 2 h with 3% Triton X. Z-stack images at 40× and 60× were taken using a Nikon A1 confocal microscope, and maximum-intensity images were formed in regions of interest, including the cortex, corpus callosum, and hippocampus.Image analysis was performed in order to quantify the fraction of microglia cells containing QDs in each organotypic brain slice. Brain slices were treated with QDs and stained using anti-Iba1+ for microglia, as described above, with three slices per QD type. For each slice, 2 × 2 confocal tile-scan images were acquired at 40× magnification, in three regions of interest per slice, for each of the nine slices. Iba1+ cells in the corpus callosum were counted, and Iba1+ cells colocalized with QD+ signals were considered to be QD-containing microglia.
Time-lapse imaging of QD cellular and intracellular internalization
Time-lapse imaging of QD internalization into microglia in organotypic brain slices was carried out using a Nikon A1 confocal microscope. In preparation for live tissue staining, each brain slice was carefully cut out of the surrounding membrane using forceps and scalpel, and transferred to a 6-well plate with staining solution. To stain the cells, brain slices were incubated with 500 μL FITC CD11b (BioLegend, USA) at 1:200 ratio in staining media (125 mL HBSS, 250 mL MEM, 125 mL horse serum, 5 mL GlutaMAX, 5 mL penicillin) for 6 h at 37 °C, 5% CO2 and 40% humidity in the dark. After incubation, slices were gently washed twice in staining media. 0.5 μM QD-MPA, QD-PEG-OH, and QD-PEG-OMe were dropped on an uncoated glass-bottom microwell dish (MatTek Corp., USA), and the brain slice was immediately placed on top of the QD solution and imaged. Time-lapse imaging was carried out in an environmental chamber at 37 °C and 5% CO2. Prior to imaging, the environmental chamber was pre-equilibrated for 30 min. The corpus callosum was located, and images were acquired every 10 min at 40× for 3 h and compiled into video format.
To evaluate QD intracellular internalization, OWH slices were first incubated with 300 μL LysoTracker (ThermoFisher) on the membrane insert and 1 mL beneath the insert at 1 μM to stain lysosomes. After incubating for 1 h at 37 °C, 5% CO2, and 40% humidity in the dark, slices were gently washed and removed from the membrane. 1 μM QD-PEG-OH and QD-PEG-OMe were dropped on an uncoated glass-bottom microwell dish (MatTek Corp., USA), and the brain slice was immediately placed on top of the QD solution and imaged. Time-lapse images or z-stack images at 40× were taken every 2–30 min using a Nikon A1 confocal microscope at the corpus callosum region. Maximum-intensity images were formed from the z-stack images.
In vitro QD toxicity and cellular uptake in BV-2 cells by MTT assay and confocal microscopy
The murine microglia cell line BV-2 was purchased from ATCC (CRL-2469). BV-2 cells were cultured in high-glucose DMEM supplemented with 10% FBS, 1% glutamine, and 1% 100 U per mL penicillin-streptomycin at 37 °C in a 5% CO2 atmosphere. After reaching 70–80% confluency, BV-2 cells were passaged and seeded in a new plate. Media was changed every two days. 10000 BV-2 cells were seeded in 96-well plates and cultured for two days prior to the experiment (n = 3 wells per experimental group). Once cells reached 70% confluency, 0.01 μM QD-MPA, QD-PEG-OH, QD-PEG-OMe or 1× PBS were added to cells and incubated at 37 °C and 5% CO2 for 1, 2, 4, 6, and 24 h. The 0 h time point represents a baseline of only BV-2 cells, without the addition of QDs. At each time point, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (ThermoFisher) was performed on 1 plate of cells according to manufacturer instructions to assess cell viability. Additionally, separate plates of cells had 4% formaldehyde added for fixation and staining. Staining of Iba1+ was performed with the above-mentioned primary and secondary antibodies. Stained BV-2 cells were imaged with a Nikon A1 confocal microscope at 10× magnification.
In vivo QD cellular uptake in mglur5 model
Newborn mglur5 KO, and age-matched, litter-matched WT rats were systemically (i.p.) administrated 25 mg kg−1 of QD-PEG-OH on P7 (n = 3 pups per group). Animals were euthanized at 4 h or 24 h post-administration and perfused with 1× PBS. Half of the brain was fixed in formalin for immunohistochemistry (IHC). Staining of Iba1 was performed with the above-mentioned primary and secondary antibodies. Z-stack images at 40× were taken with a Nikon A1 confocal microscope, and maximum-intensity images were formed in regions of interest, including the cortex, corpus callosum, hippocampus, and PVR, similar to the OWH slice study.Statistical analysis
Data were displayed using the mean value ± the standard error of the mean (SEOM) for the colloidal stability study, and using the median value ± 95% confidence intervals for the in vitro cell study, ex vivo OWH slice study, RT-qPCR, and toxicity study. Statistical analysis was performed using the unpaired t-test without correction. Statistical significance was determined under type I error at p < 0.05. Any difference corresponding to p < 0.05 (*) and p < 0.01 (**) was marked on the graph. The change in hydrodynamic size of QD-MPA, QD-PEG-OH, and QD-PEG-OMe during the 2000 second aggregation kinetics study was smoothed using a locally weighted scatterplot smoothing (LOWESS) approach with a window size of 10. All statistical analyses were conducted using GraphPad Prism 7.01 (GraphPad Software Inc).Author contributions
MZ and EN designed the stability, toxicity, uptake, and real-time imaging studies. BPB, NLT, and VCH designed and synthesized the QD emitter platforms and performed the structural and photophysical characterization experiments. BPB and RA performed and optimized the QD functionalization experiments, and we thank David Ollodart and Gary Boon for assisting with these efforts. MZ and BPB performed the cadmium dissolution and photoluminescent stability studies, and we thank Dr Cole DeForest for use of his SpectraMax M5 Microplate Reader. BPB performed the FTIR analysis and we thank Dr Alexandra Velian for use of her FTIR spectrometer. MZ performed the pH dependence study for QD stability. We thank Soohyung Lee for assisting BPB and MZ with ICP-MS measurements and analysis. MZ and KH performed the OWH slice toxicity and mRNA studies. OM and MZ performed all in vitro characterization studies for DLS, QD stability, and QD aggregation kinetics. NC and MZ performed the in vitro BV-2 culture studies. MZ and BD performed the OWH slice studies to evaluate cell-specific QD uptake. MZ and EN performed the in vivo studies in the mglur5 model and KH managed the mglur5 colony, including breeding and genotyping for this study. MZ performed all statistical analysis. EN and VCH directed the research. MZ, BPB, VCH, and EN wrote and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based in part upon work supported by the University of Washington Interdisciplinary Center for Exposures, Diseases, Genomics and Environment of the National Institutes of Health under award number: P30ES007033, and funding from the Burroughs Wellcome Fund Career Award at Scientific Interfaces. Part of this work was conducted at the Photonics Research Center and the Research Training Testbed facility at the University of Washington, which are supported in part by funds from the Clean Energy Institute and the Washington Research Foundation, and at the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure site at the University of Washington which is supported in part by the National Science Foundation (grant ECC-1542101), the University of Washington, the Molecular Engineering & Sciences Institute, and the Clean Energy Institute.References
- O. M. Koo, I. Rubinstein and H. Onyuksel, Role of nanotechnology in targeted drug delivery and imaging: a concise review, Nanomedicine, 2005, 1(3), 193–212 CrossRef CAS PubMed.
- D. P. Cormode, T. Skajaa, Z. A. Fayad and W. J. Mulder, Nanotechnology in medical imaging: probe design and applications, Arterioscler., Thromb., Vasc. Biol., 2009, 29(7), 992–1000 CrossRef CAS PubMed.
- S. Chapman, M. Dobrovolskaia, K. Farahani, A. Goodwin, A. Joshi, H. Lee, T. Meade, M. Pomper, K. Ptak, J. Rao, R. Singh, S. Sridhar, S. Stern, A. Wang, J. B. Weaver, G. Woloschak and L. Yang, Nanoparticles for cancer imaging: The good, the bad, and the promise, Nano Today, 2013, 8(5), 454–460 CrossRef CAS PubMed.
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Levy, Nanoparticles for imaging, sensing, and therapeutic intervention, ACS Nano, 2014, 8(4), 3107–3122 CrossRef CAS PubMed.
- A. Neuwelt, N. Sidhu, C. A. Hu, G. Mlady, S. C. Eberhardt and L. O. Sillerud, Iron-based superparamagnetic nanoparticle contrast agents for MRI of infection and inflammation, AJR, Am. J. Roentgenol., 2015, 204(3), W302–W313 CrossRef PubMed.
- C. Curtis, M. Zhang, R. Liao, T. Wood and E. Nance, Systems-level thinking for nanoparticle-mediated therapeutic delivery to neurological diseases, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9(2), e1422 Search PubMed.
- Y. Xing and J. Rao, Quantum dot bioconjugates for in vitro diagnostics & in vivo imaging, Cancer Biomarkers, 2008, 4(6), 307–319 CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Quantum dot bioconjugates for imaging, labelling and sensing, Nat. Mater., 2005, 4(6), 435–446 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Quantum dots for live cells, in vivo imaging, and diagnostics, Science, 2005, 307(5709), 538–544 CrossRef CAS PubMed.
- O. Chen, J. Zhao, V. P. Chauhan, J. Cui, C. Wong, D. K. Harris, H. Wei, H. S. Han, D. Fukumura, R. K. Jain and M. G. Bawendi, Compact high-quality CdSe-CdS core-shell nanocrystals with narrow emission linewidths and suppressed blinking, Nat. Mater., 2013, 12(5), 445–451 CrossRef CAS PubMed.
- J. Zhou, M. Zhu, R. Meng, H. Qin and X. Peng, Ideal CdSe/CdS Core/Shell Nanocrystals Enabled by Entropic Ligands and Their Core Size-, Shell Thickness-, and Ligand-Dependent Photoluminescence Properties, J. Am. Chem. Soc., 2017, 139(46), 16556–16567 CrossRef CAS PubMed.
- H. Kobayashi, Y. Hama, Y. Koyama, T. Barrett, C. A. Regino, Y. Urano and P. L. Choyke, Simultaneous multicolor imaging of five different lymphatic basins using quantum dots, Nano Lett., 2007, 7(6), 1711–1716 CrossRef CAS PubMed.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Long-term multiple color imaging of live cells using quantum dot bioconjugates, Nat. Biotechnol., 2003, 21(1), 47–51 CrossRef CAS PubMed.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots, Nat. Biotechnol., 2003, 21(1), 41–46 CrossRef CAS PubMed.
- S. Pathak, E. Cao, M. C. Davidson, S. Jin and G. A. Silva, Quantum dot applications to neuroscience: new tools for probing neurons and glia, J. Neurosci., 2006, 26(7), 1893–1895 CrossRef CAS PubMed.
- R. Walters, I. L. Medintz, J. B. Delehanty, M. H. Stewart, K. Susumu, A. L. Huston, P. E. Dawson and G. Dawson, The Role of Negative Charge in the Delivery of Quantum Dots to Neurons, ASN Neuro, 2015, 7(4) DOI:10.1177/1759091415592389.
- S. Dante, A. Petrelli, E. M. Petrini, R. Marotta, A. Maccione, A. Alabastri, A. Quarta, F. De Donato, T. Ravasenga, A. Sathya, R. Cingolani, R. Proietti Zaccaria, L. Berdondini, A. Barberis and T. Pellegrino, Selective Targeting of Neurons with Inorganic Nanoparticles: Revealing the Crucial Role of Nanoparticle Surface Charge, ACS Nano, 2017, 11(7), 6630–6640 CrossRef CAS PubMed.
- S. S. Minami, B. Sun, K. Popat, T. Kauppinen, M. Pleiss, Y. Zhou, M. E. Ward, P. Floreancig, L. Mucke, T. Desai and L. Gan, Selective targeting of microglia by quantum dots, J. Neuroinflammation, 2012, 9, 22 CAS.
- R. G. Thorne and C. Nicholson, In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(14), 5567–5572 CrossRef CAS PubMed.
- J. D. Marshall and M. J. Schnitzer, Optical strategies for sensing neuronal voltage using quantum dots and other semiconductor nanocrystals, ACS Nano, 2013, 7(5), 4601–4609 CrossRef CAS PubMed.
- G. Xu, S. Mahajan, I. Roy and K. T. Yong, Theranostic quantum dots for crossing blood-brain barrier in vitro and providing therapy of HIV-associated encephalopathy, Front. Pharmacol., 2013, 4, 140 Search PubMed.
- Q. Zhang, Imaging single synaptic vesicles in mammalian central synapses with quantum dots, Methods Mol. Biol., 2013, 1026, 57–69 CrossRef CAS PubMed.
- G. Dawson, Quantum dots and potential therapy for Krabbe's disease, J. Neurosci. Res., 2016, 94(11), 1293–1303 CrossRef CAS PubMed.
- R. Agarwal, M. S. Domowicz, N. B. Schwartz, J. Henry, I. Medintz, J. B. Delehanty, M. H. Stewart, K. Susumu, A. L. Huston, J. R. Deschamps, P. E. Dawson, V. Palomo and G. Dawson, Delivery and tracking of quantum dot peptide bioconjugates in an intact developing avian brain, ACS Chem. Neurosci., 2015, 6(3), 494–504 CrossRef CAS PubMed.
- A. L. Efros, J. B. Delehanty, A. L. Huston, I. L. Medintz, M. Barbic and T. D. Harris, Evaluating the potential of using quantum dots for monitoring electrical signals in neurons, Nat. Nanotechnol., 2018, 13(4), 278–288 CrossRef CAS PubMed.
- G. Gobe and D. Crane, Mitochondria, reactive oxygen species and cadmium toxicity in the kidney, Toxicol. Lett., 2010, 198(1), 49–55 CrossRef CAS PubMed.
- P. P. Adiseshaiah, J. B. Hall and S. E. McNeil, Nanomaterial standards for efficacy and toxicity assessment, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2(1), 99–112 CAS.
- M. Rehberg, M. Praetner, C. F. Leite, C. A. Reichel, P. Bihari, K. Mildner, S. Duhr, D. Zeuschner and F. Krombach, Quantum dots modulate leukocyte adhesion and transmigration depending on their surface modification, Nano Lett., 2010, 10(9), 3656–3664 CrossRef CAS PubMed.
- A. Hoshino, K. Fujioka, T. Oku, M. Suga, Y. F. Sasaki, T. Ohta, M. Yasuhara, K. Suzuki and K. Yamamoto, Physicochemical properties and cellular toxicity of nanocrystal quantum dots depend on their surface modification, Nano Lett., 2004, 4(11), 2163–2169 CrossRef CAS.
- Y. Tang, S. Han, H. Liu, X. Chen, L. Huang, X. Li and J. Zhang, The role of surface chemistry in determining in vivo biodistribution and toxicity of CdSe/ZnS core-shell quantum dots, Biomaterials, 2013, 34(34), 8741–8755 CrossRef CAS PubMed.
- Y. Zhang, H. Pan, P. Zhang, N. Gao, Y. Lin, Z. Luo, P. Li, C. Wang, L. Liu, D. Pang, L. Cai and Y. Ma, Functionalized quantum dots induce proinflammatory responses in vitro: the role of terminal functional group-associated endocytic pathways, Nanoscale, 2013, 5(13), 5919–5929 RSC.
- E. Blanco, H. Shen and M. Ferrari, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol., 2015, 33(9), 941–951 CrossRef CAS PubMed.
- A. Monguzzi, D. Braga, M. Gandini, V. C. Holmberg, D. K. Kim, A. Sahu, D. J. Norris and F. Meinardi, Broadband up-conversion at subsolar irradiance: triplet-triplet annihilation boosted by fluorescent semiconductor nanocrystals, Nano Lett., 2014, 14(11), 6644–6650 CrossRef CAS PubMed.
- S. J. Kress, F. V. Antolinez, P. Richner, S. V. Jayanti, D. K. Kim, F. Prins, A. Riedinger, M. P. Fischer, S. Meyer, K. M. McPeak, D. Poulikakos and D. J. Norris, Wedge Waveguides and Resonators for Quantum Plasmonics, Nano Lett., 2015, 15(9), 6267–6275 CrossRef CAS PubMed.
- H. Zhang, K. Cheng, Y. M. Hou, Z. Fang, Z. X. Pan, W. J. Wu, J. L. Hua and X. H. Zhong, Efficient CdSe quantum dot-sensitized solar cells prepared by a postsynthesis assembly approach, Chem. Commun., 2012, 48(91), 11235–11237 RSC.
- E. A. Nance, G. F. Woodworth, K. A. Sailor, T. Y. Shih, Q. Xu, G. Swaminathan, D. Xiang, C. Eberhart and J. Hanes, A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue, Sci. Transl. Med., 2012, 4(149), 149ra119 Search PubMed.
- F. Zhang, E. Nance, Y. Alnasser, R. Kannan and S. Kannan, Microglial migration and interactions with dendrimer nanoparticles are altered in the presence of neuroinflammation, J. Neuroinflammation, 2016, 13(1), 65 CrossRef PubMed.
- V. C. Holmberg, M. R. Rasch and B. A. Korgel, PEGylation of carboxylic acid-functionalized germanium nanowires, Langmuir, 2010, 26(17), 14241–14246 CrossRef CAS PubMed.
- M. Hiroatsu and M. Tatsuo, Intrachain Force Field and Normal Vibrations of Polyethylene Glycol, Bull. Chem. Soc. Jpn., 1968, 41(8), 1798–1808 CrossRef.
- H. Matsuura and T. Miyazawa, Frequency Distribution and Neutron Scattering of Polyethylene Glycol Chain, J. Chem. Phys., 1969, 50(2), 915–919 CrossRef CAS.
- J. Coates, Interpretation of Infrared Spectra: A Practical Approach, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons Ltd., Chichester, 2000, pp. 10881–10882. Search PubMed.
- J. Oomens and J. D. Steill, Free carboxylate stretching modes, J. Phys. Chem. A, 2008, 112(15), 3281–3283 CrossRef CAS PubMed.
- P. Gareri, R. Mattace, F. Nava and G. De Sarro, Role of calcium in brain aging, Gen. Pharmacol., 1995, 26(8), 1651–1657 CrossRef CAS PubMed.
- J. N. Guzman, J. Sanchez-Padilla, D. Wokosin, J. Kondapalli, E. Ilijic, P. T. Schumacker and D. J. Surmeier, Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1, Nature, 2010, 468(7324), 696–700 CrossRef CAS PubMed.
- C. Curtis, D. Toghani, B. Wong and E. Nance, Colloidal stability as a determinant of nanoparticle behavior in the brain, Colloids Surf., B, 2018, 170, 673–682 CrossRef CAS PubMed.
- A. M. Smith, H. Duan, M. N. Rhyner, G. Ruan and S. Nie, A systematic examination of surface coatings on the optical and chemical properties of semiconductor quantum dots, Phys. Chem. Chem. Phys., 2006, 8(33), 3895–3903 RSC.
- E. A. Ling and W. C. Wong, The origin and nature of ramified and amoeboid microglia: a historical review and current concepts, Glia, 1993, 7(1), 9–18 CrossRef CAS PubMed.
- D. Maysinger, M. Behrendt, M. Lalancette-Hebert and J. Kriz, Real-time imaging of astrocyte response to quantum dots: in vivo screening model system for biocompatibility of nanoparticles, Nano Lett., 2007, 7(8), 2513–2520 CrossRef CAS PubMed.
- J. Lovric, H. S. Bazzi, Y. Cuie, G. R. Fortin, F. M. Winnik and D. Maysinger, Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots, J. Mol. Med., 2005, 83(5), 377–385 CrossRef PubMed.
- S. Ohkuma and B. Poole, Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents, Proc. Natl. Acad. Sci. U. S. A., 1978, 75(7), 3327–3331 CrossRef CAS PubMed.
- A. Nagy, A. Zane, S. L. Cole, M. Severance, P. K. Dutta and W. J. Waldman, Contrast of the biological activity of negatively and positively charged microwave synthesized CdSe/ZnS quantum dots, Chem. Res. Toxicol., 2011, 24(12), 2176–2188 Search PubMed.
- C. Humpel, Organotypic brain slice cultures: A review, Neuroscience, 2015, 305, 86–98 CrossRef CAS PubMed.
- T. Wu, T. Zhang, Y. Chen and M. Tang, Research advances on potential neurotoxicity of quantum dots, J. Appl. Toxicol., 2016, 36(3), 345–351 CrossRef CAS PubMed.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, The cytotoxicity of cadmium-based quantum dots, Biomaterials, 2012, 33(5), 1238–1244 CrossRef CAS PubMed.
- E. Chang, N. Thekkek, W. W. Yu, V. L. Colvin and R. Drezek, Evaluation of quantum dot cytotoxicity based on intracellular uptake, Small, 2006, 2(12), 1412–1417 CrossRef CAS PubMed.
- R. Jevprasesphant, J. Penny, R. Jalal, D. Attwood, N. B. McKeown and A. D'Emanuele, The influence of surface modification on the cytotoxicity of PAMAM dendrimers, Int. J. Pharm., 2003, 252(1–2), 263–266 CrossRef CAS.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. Munoz Javier, H. E. Gaub, S. Stolzle, N. Fertig and W. J. Parak, Cytotoxicity of colloidal CdSe and CdSe/ZnS nanoparticles, Nano Lett., 2005, 5(2), 331–338 CrossRef CAS PubMed.
- C. C. Ho, H. Chang, H. T. Tsai, M. H. Tsai, C. S. Yang, Y. C. Ling and P. Lin, Quantum dot 705, a cadmium-based nanoparticle, induces persistent inflammation and granuloma formation in the mouse lung, Nanotoxicology, 2013, 7(1), 105–115 CrossRef CAS PubMed.
- S. Tang, Y. Wu, C. N. Ryan, S. Yu, G. Qin, D. S. Edwards and G. D. Mayer, Distinct expression profiles of stress defense and DNA repair genes in Daphnia pulex exposed to cadmium, zinc, and quantum dots, Chemosphere, 2015, 120, 92–99 CrossRef CAS PubMed.
- D. F. Simon, R. F. Domingos, C. Hauser, C. M. Hutchins, W. Zerges and K. J. Wilkinson, Transcriptome sequencing (RNA-seq) analysis of the effects of metal nanoparticle exposure on the transcriptome of Chlamydomonas reinhardtii, Appl. Environ. Microbiol., 2013, 79(16), 4774–4785 CrossRef CAS PubMed.
- A. Nagy, J. A. Hollingsworth, B. Hu, A. Steinbruck, P. C. Stark, C. Rios Valdez, M. Vuyisich, M. H. Stewart, D. H. Atha, B. C. Nelson and R. Iyer, Functionalization-dependent induction of cellular survival pathways by CdSe quantum dots in primary normal human bronchial epithelial cells, ACS Nano, 2013, 7(10), 8397–8411 CrossRef CAS PubMed.
- S. Sharma, A. Rais, R. Sandhu, W. Nel and M. Ebadi, Clinical significance of metallothioneins in cell therapy and nanomedicine, Int. J. Nanomed., 2013, 8, 1477–1488 CrossRef PubMed.
- N. Thirumoorthy, A. Shyam Sunder, K. Manisenthil Kumar, M. Senthil Kumar, G. Ganesh and M. Chatterjee, A review of metallothionein isoforms and their role in pathophysiology, World J. Surg. Oncol., 2011, 9, 54 CrossRef CAS PubMed.
- K. Peynshaert, S. J. Soenen, B. B. Manshian, S. H. Doak, K. Braeckmans, S. C. De Smedt and K. Remaut, Coating of Quantum Dots strongly defines their effect on lysosomal health and autophagy, Acta Biomater., 2017, 48, 195–205 CrossRef CAS PubMed.
- I. Sabolic, D. Breljak, M. Skarica and C. M. Herak-Kramberger, Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs, BioMetals, 2010, 23(5), 897–926 CrossRef CAS PubMed.
- E. Artells, O. Palacios, M. Capdevila and S. Atrian, Mammalian MT1 and MT2 metallothioneins differ in their metal binding abilities, Metallomics, 2013, 5(10), 1397–1410 RSC.
- B. Becher, S. Spath and J. Goverman, Cytokine networks in neuroinflammation, Nat. Rev. Immunol., 2017, 17(1), 49–59 CrossRef CAS PubMed.
- J. H. Su, G. Deng and C. W. Cotman, Bax protein expression is increased in Alzheimer's brain: correlations with DNA damage, Bcl-2 expression, and brain pathology, J. Neuropathol. Exp. Neurol., 1997, 56(1), 86–93 CrossRef CAS PubMed.
- X. Yang, S. Yang, J. Zhang, L. Xue and Z. Hu, Role of Caspase 3 in neuronal apoptosis after acute brain injury, Chin. J. Traumatol., 2002, 5(4), 250–253 CAS.
- M. Sawada, A. Suzumura, H. Hosoya, T. Marunouchi and T. Nagatsu, Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia, J. Neurochem., 1999, 72(4), 1466–1471 CrossRef CAS PubMed.
- J. Drouin-Ouellet, A. L. Brownell, M. Saint-Pierre, C. Fasano, V. Emond, L. E. Trudeau, D. Levesque and F. Cicchetti, Neuroinflammation is associated with changes in glial mGluR5 expression and the development of neonatal excitotoxic lesions, Glia, 2011, 59(2), 188–199 CrossRef PubMed.
- K. R. Byrnes, B. Stoica, D. J. Loane, A. Riccio, M. I. Davis and A. I. Faden, Metabotropic Glutamate Receptor 5 Activation Inhibits Microglial Associated Inflammation and Neurotoxicity, Glia, 2009, 57(5), 550–560 CrossRef PubMed.
- M. M. Carrasquillo, M. Allen, J. D. Burgess, X. Wang, S. L. Strickland, S. Aryal, J. Siuda, M. L. Kachadoorian, C. Medway, C. S. Younkin, A. Nair, C. Wang, P. Chanana, D. Serie, T. Nguyen, S. Lincoln, K. G. Malphrus, K. Morgan, T. E. Golde, N. D. Price, C. C. White, P. L. De Jager, D. A. Bennett, Y. W. Asmann, J. E. Crook, R. C. Petersen, N. R. Graff-Radford, D. W. Dickson, S. G. Younkin and N. Ertekin-Taner, A candidate regulatory variant at the TREM gene cluster associates with decreased Alzheimer's disease risk and increased TREML1 and TREM2 brain gene expression, Alzheimers Dement., 2017, 13(6), 663–673 CrossRef PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction, Nanomedicine, 2016, 11(6), 673–692 CrossRef CAS PubMed.
- S. Kato, K. Itoh, T. Yaoi, T. Tozawa, Y. Yoshikawa, H. Yasui, N. Kanamura, A. Hoshino, N. Manabe, K. Yamamoto and S. Fushiki, Organ distribution of quantum dots after intraperitoneal administration, with special reference to area-specific distribution in the brain, Nanotechnology, 2010, 21(33), 335103 CrossRef PubMed.
- S. Paris-Robidas, D. Brouard, V. Emond, M. Parent and F. Calon, Internalization of targeted quantum dots by brain capillary endothelial cells in vivo, J. Cereb. Blood Flow Metab., 2016, 36(4), 731–742 CrossRef CAS PubMed.
- X. Gao, J. Chen, J. Chen, B. Wu, H. Chen and X. Jiang, Quantum dots bearing lectin-functionalized nanoparticles as a platform for in vivo brain imaging, Bioconjugate Chem., 2008, 19(11), 2189–2195 CrossRef CAS PubMed.
- K. De Nolf, R. K. Capek, S. Abe, M. Sluydts, Y. Jang, J. C. Martins, S. Cottenier, E. Lifshitz and Z. Hens, Controlling the size of hot injection made nanocrystals by manipulating the diffusion coefficient of the solute, J. Am. Chem. Soc., 2015, 137(7), 2495–2505 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Experimental Determination of the Extinction Coefficient of CdTe, CdSe, and CdS Nanocrystals, Chem. Mater., 2003, 15(14), 2854–2860 CrossRef CAS.
- P. Calvo, B. Gouritin, H. Chacun, D. Desmaele, J. D'Angelo, J. P. Noel, D. Georgin, E. Fattal, J. P. Andreux and P. Couvreur, Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery, Pharm. Res., 2001, 18(8), 1157–1166 CrossRef CAS.
- C. Perez-Campana, V. Gomez-Vallejo, M. Puigivila, A. Martin, T. Calvo-Fernandez, S. E. Moya, R. F. Ziolo, T. Reese and J. Llop, Biodistribution of different sized nanoparticles assessed by positron emission tomography: a general strategy for direct activation of metal oxide particles, ACS Nano, 2013, 7(4), 3498–3505 CrossRef CAS PubMed.
- S. A. Kulkarni and S. S. Feng, Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery, Pharm. Res., 2013, 30(10), 2512–2522 CrossRef CAS PubMed.
- S. A. Bustin, V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele and C. T. Wittwer, The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments, Clin. Chem., 2009, 55(4), 611–622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Movie 1: time-lapse imaging of QD-PEG-OH (red) uptake in lysosomes (green) in the corpus callosum region in live OWH slices. The video is 10 fps and will replay 5 times. Movie 2: time-lapse imaging of QD-PEG-OMe (red) uptake in lysosomes (green) in the corpus callosum region in live OWH slices. The video is 10 fps and will replay 5 times. Movie 3: time-lapse imaging of QD-MPA (red) uptake in CD11b+ microglia (green) in corpus callosum region in live OWH slices. The video is 5 fps and will replay 5 times. Movie 4: time-lapse imaging of QD-PEG-OH (red) uptake in CD11b+ microglia (green) in corpus callosum region in live OWH slices. The video is 5 fps and will replay 5 times. Movie 5: time-lapse imaging of QD-PEG-OMe (red) uptake in CD11b+ microglia (green) in corpus callosum region in live OWH slices. The video is 5 fps and will replay 5 times. See DOI: 10.1039/c9na00334g |
| This journal is © The Royal Society of Chemistry 2019 |