
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near-infrared excited luminescence and in vitro imaging of HeLa cells by using Mn2+ enhanced Tb3+ and Yb3+ cooperative upconversion in NaYF4 nanocrystals
Katarzyna
Prorok
 *a,
Michał
Olk
b,
Michał
Skowicki
c,
Agnieszka
Kowalczyk
c,
Agata
Kotulska
a,
Tomasz
Lipiński
c and
Artur
Bednarkiewicz
a
*a,
Michał
Olk
b,
Michał
Skowicki
c,
Agnieszka
Kowalczyk
c,
Agata
Kotulska
a,
Tomasz
Lipiński
c and
Artur
Bednarkiewicz
a
aInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, Okolna 2, 50-422 Wroclaw, Poland. E-mail: k.prorok@intibs.pl
bInstitute of Immunology and Experimental Therapy, Polish Academy of Sciences, R. Weigla 12, 53-114 Wroclaw, Poland
cLukasiewicz Research Network – PORT Polish Center for Technology Development, Stablowicka 147, 54-066 Wroclaw, Poland
First published on 12th July 2019
Abstract
Advanced biodetection and bioimaging require fluorescent labels which exhibit many, easily distinguishable colors to identify or study numerous biotargets in a single sample. Although numerous different colors have been demonstrated with lanthanide doped nanoparticles, these colors usually originate from various ratios of overlapping multiple emission bands from activators, which severely limits the number of available labels. As a consequence, different lanthanide doped labels cannot be easily distinguished from each other (e.g. Er3+ from Ho3+) in a quantitative way, when such labels are co-localized during microscopy wide-field imaging. It is therefore reasonable to expand the available choice of spectral signatures and not rely on just different colors. Other ions, such as Tb3+ or Eu3+, can offer new possibilities and unique spectral features in upconversion mode in this respect. For example, despite partial overlap with Er3+ or Ho3+ emission spectra, Tb3+ ions display also unique and easily distinguishable spectral features at 580 nm. Unfortunately, in terms of brightness, Tb3+ emission in upconversion mode is typically too weak to be useful. To improve the Tb3+ upconversion emission intensity, a new approach, i.e. Mn2+ co-doping, has been proposed and verified in this work. A versatile optimization of Tb3+, Yb3+ and Mn2+ ion concentrations has been performed based on luminescence spectra and lifetime studies. The most intense emission was achieved for nanoparticles doped with 10% Mn2+ ions, with over 30 times brighter intensity of Tb3+ ions compared to the emission of nanocrystals without the addition of Mn2+ ions. Additionally, as a proof of the concept, the surface of nanoparticles was coated with proteins and conjugated with folic acid, and such biofunctionalized nanoparticles were subsequently used for bioimaging of HeLa cells.
1 Introduction
Upconverting nanoparticles (UCNPs) have gained considerable interest as nano-colloidal biolabels in biodetection and bioimaging.1–5 However, some high-throughput detection/imaging schemes require the use of labels exhibiting many, distinguishable spectral fingerprints to identify or study many biotargets co-localized in a single sample. It is therefore reasonable to expand the available choice of color signatures. Many colors have been obtained with UCNPs based on ratiometric designs, where the content of one emitter changes with respect to the other one (e.g. Er3+ with respect to Tm3+).6 While sufficient to provide many colors, such probes are indistinguishable in homogeneous solutions because overlapping spectral bands are present in all labels (e.g. green 540 nm emission is present in Er3+ and Ho3+, and red 650 nm emission is present in Er3+, Ho3+, and Tm3+). In practical applications many nanometric labels co-localize spatially (i.e. within the point spread function volume) and spectrally (i.e. in optical band-pass filter based wide-field imaging), therefore spectral purity of luminescent labels is critical for multi-target biolabeling.7 While Er3+ and Tm3+ emission spectra are clearly different, the Er3+ and Ho3+ emission is spectrally similar. Although spectrally more unique,8 other lanthanides' emission intensity (i.e. Pr3+, Tb3+, Eu3+, and Sm3+) is typically too weak in upconversion mode to be applicable. Weak emission can be explained by high concentration quenching (e.g. Pr3+ and Sm3+) which limits the amount of activators and consequently limits emission intensity, or could be due to the energy upconversion mechanism other than energy transfer upconversion (ETU). The latter case is typical for Yb3+ sensitized Eu3+ and Tb3+ emission, which involves cooperative energy transfer (CET).9–13 These ions display unique and easily distinguishable spectral features, but CET is around 2–3 orders of magnitude less efficient in comparison to energy transfer upconversion found in the Yb3+–Tm3+/Er3+/Ho3+ pairs.14 For these reasons, terbium upconversion emission should be enhanced in order to provide labels with brightness comparable to that of Yb3+–Er3+ or Yb3+–Tm3+ in UCNPs and thus offer sufficient sensitivity for bioassays and bioimaging.In our previous studies, we have proposed and demonstrated approaches to optimize the Yb3+–Tb3+ upconversion efficiency.8,11,15 Our results showed that up to 40-fold upconversion intensity enhancement may be obtained for the NaYF4:Yb3+/Tb3+ nanoparticles8 by NP surface passivation with an undoped shell compared to bare core NPs of the same composition. The enhancement of Tb3+ upconversion emission was also observed when thulium ions acting as a secondary sensitizer were admixed, since a more efficient energy transfer upconversion mechanism becomes involved (i.e. ETU instead of CET).11,16,17 In triply doped samples such as Tm3+–Yb3+–Tb3+, the emitting level of the Tb3+ level is populated by the cooperative process from ytterbium pairs and simultaneously through much more efficient energy transfer upconversion from excited thulium ions.14,18 Thulium ion co-doping allowed five-fold enhancement of the upconversion emission intensity from the 5D4 terbium level,11 but contaminated the emission spectrum of such labels with Tm3+ emission.
Recently, a new approach to the energy transfer between lanthanide ions and transition metal ions has also been investigated. Zhao's group demonstrated a new strategy for the rational manipulation of erbium green and red upconversion emission.19 They obtained pure red emission of NaYF4:Yb3+/Er3+ nanoparticles by manganese ion co-doping. The transition possibilities between green and red emissions of Er3+ were disturbed by the existence of Mn2+ ions leading to “concentrated” pure red emission.19 Because the energy transfer between Er3+ and Mn2+ is extremely efficient, as a result, single-band red upconversion emission was observed.19–21 Doping with Mn2+ ions has also been used to control the color of emission of NaYF4:Yb3+/Ho3+ nanocrystals to achieve enhanced red emission,22,23 and Zhang's group proposed an energy transfer mechanism from the Yb3+–Mn2+ dimer to Eu3+. The 4T1 energy level of Mn2+ matches well with the 5D0 energy level of Eu3+.24–26 Pure and intense orange upconversion luminescence of Eu3+ in NaY(Lu)F4 nanoparticles was obtained through the upconversion sensitization by the Yb3+–Mn2+ dimer. This is in stark contrast to Eu3+ upconversion achieved through Yb3+–Tm3+ upconversion, which concomitantly shows emission from both Tm3+ and Eu3+ ions.27 Such a new approach would conceptually be much more efficient than the Yb3+ → Tm3+ → Gd3+ → Eu3+ cascade and migration assisted ET in core–shell nanoparticles, although Liu et al. demonstrated it to be comparable or even higher in intensity than that in Yb3+/Er3+ co-doped nanoparticles.6
While energy transfer between Mn2+ and Tb3+ ions was investigated in various host matrixes,28–31 the UC emission intensity enhancement and Mn2+ sensitized ET processes between Tb3+ and Yb3+ co-doped NaYF4 nanocrystals have never been reported up to now. Tb3+ emission bands are located in the range from about 450 to 700 nm, whereas the Mn2+ broad emission band is located in the region from about 460 to 700 nm and strongly depends on the crystal environment of the host materials.28,32 Partial overlap of the emission bands confirms that energy transfer processes are likely to occur, and thus the major motivation for this research was to provide an additional possibility to enhance upconversion emission intensity of the Yb3+–Tb3+ co-doped β-NaYF4 nanocrystals through the sensitization by Yb3+ and Mn2+. To reach this goal, concentration dependent structural as well as spectroscopic studies have been performed which revealed the possibility of improving the UC efficiency. Additionally, the surface of the obtained nanoparticles was subsequently coated with proteins and conjugated with folic acid. Such prepared nanoparticles were used as an in vitro bioimaging agent. This is one of the very rare reports of cellular imaging experiments using Tb3+ pure upconversion emission,33 and the only one so far which demonstrates the applicability of core only β-NaYF4 colloidal nanoparticles doped with Tb3+ and Yb3+ ions in optical wide-field UC imaging.
2 Experimental
2.1 Materials
Yttrium oxide (99.99%), ytterbium oxide (99.99%), terbium oxide (99.99%), manganese(II) carbonate, acetic acid (99%), pure oleic acid and 1-octadecene (90%), thiazolyl blue tetrazolium bromide (MTT), and Dulbecco's Modified Eagle's Medium (DMEM) were purchased from ALDRICH Chemistry. Ethanol (96% pure p.a.), methanol, n-hexane and chloroform were purchased from POCH S.A. (Poland). Fetal bovine serum (FBS), folic acid (FA) (≥97%) and dimethyl sulfoxide (DMSO) (≥99.5%) were purchased from Sigma Aldrich. N-Hydroxysuccinimide (NHS) (≥98%) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (≥98%) were purchased from Alfa Aesar. Bovine serum albumin (BSA) (>98%) was purchased from BioShop. All of the chemical reagents were used as received without further purification.2.2 Synthesis of nanoparticles
The hexagonal β-NaYF4:Mn2+,Yb3+,Tb3+ nanoparticles were prepared using a thermal decomposition reaction of lanthanide oleates.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes and washed with hexane and ethanol. Finally, the prepared nanoparticles were dispersed in chloroform. The colloidal solution was stable without aggregation.
000 rpm for 10 minutes and washed with hexane and ethanol. Finally, the prepared nanoparticles were dispersed in chloroform. The colloidal solution was stable without aggregation.
2.3 Cell culture
The HeLa cell line was cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum and an antibiotic–antimycotic solution (Sigma-Aldrich) containing penicillin (100 units per ml), streptomycin (100 μg ml−1) and amphotericin B (250 ng ml−1). Cells were incubated in a humidified incubator at 37 °C with 5% CO2.2.4 Surface modification and functionalization of upconverting nanoparticles
200 μl of a chloroform suspension of oleic acid (OA) capped nanoparticles was added to 1 ml of 0.1 M HCl solution and vortexed for 20 minutes. The aqueous phase was placed into a new tube and centrifuged for 20 minutes (21000 × g, RT). The pellet was subsequently washed with ethanol and 0.1 M HCl and suspended in 200 μl of DMSO (dimethyl sulfoxide). The suspension of nanoparticles was mixed with 1 ml of FBS (fetal bovine serum), incubated in an ultrasonic bath for 5 minutes and vortexed for 30 minutes. To separate the nanoparticles from FBS, the sample was repeatedly centrifuged (21000 × g, 20 minutes, RT) and suspended in a solution of 0.9% NaCl and 1% BSA.
Folic acid was conjugated to the protein capped nanoparticles by the EDC/NHS chemistry. 2 mg of folic acid was activated by EDC and NHS in 140 μl of DMSO (the molar ratio of FA/EDC/NHS = 1:1:1). The solution was vortexed for 30 minutes at RT and then transferred dropwise to 1 ml of NP solution (4.25 mg ml−1) suspended in 0.1 M HBS (HEPES buffered saline), pH 8.0. The solution was vortexed for 3 hours at RT and then subsequently 100 μl of 1 M tris buffer, pH 9.0, and 200 μl of 1% BSA in saline were added to stop the reaction. Nanoparticles were purified by multiple centrifugation steps (21000 × g, 20 minutes, RT) in saline and were finally suspended in 1 ml of 0.9% NaCl and 1% BSA and stored at 4 °C.
2.5 Fluorescence microscopy assay
For in vitro studies, 5 × 104 HeLa cells were seeded on a glass-bottomed multiwell culture dish. After 8 h the cells were incubated with the FA-conjugated UCNPs for 20 h at 37 °C. Each well contained 400 μl of media with UCNPs at a concentration of 0.2 mg ml−1. To remove unbound UCNPs, the cells were washed 3 times with 0.9% NaCl and fixed with 3.7% formaldehyde solution in HBS for 10 min. Cells were washed 3 times with 0.9% NaCl and mounting media containing DAPI was added to label the nucleus.2.6 Cytotoxicity of UCNPs
The cytotoxicity of NPs was evaluated using the MTT viability assay. The cells (4000 per well) were incubated in each well of a 96-well plate with β-NaYF4:5% Yb3+,15% Tb3+ (protein coated), β-NaYF4:5% Yb3+,15% Tb3+@FA (protein coated, with folic acid), β-NaYF4:10% Mn2+,5% Yb3+,15% Tb3+ (protein coated), and β-NaYF4:10% Mn2+,5% Yb3+,15% Tb3+@FA (protein coated, with folic acid) nanoparticles in the following concentrations: 50, 100, and 200 μg ml−1 in a medium (DMEM, 10% FBS) for 72 h. After that time, the cells were washed with PBS. Then the MTT solution (0.1 mg ml−1, DMEM w/o FBS) was added and the cells were incubated for 4 h. The MTT medium was removed from each well and the cells were washed with PBS. PBS was removed from each well, DMSO was added and the plates were shaken for 10 min at room temperature to dissolve the dye. The absorption was measured at λ = 570 nm in a microplate reader. The cell viability was calculated as follows: cell viability = A/B × 100%, where A is the absorbance of the cells incubated with DMEM containing NPs and B is the absorbance of the cells incubated with DMEM w/o NPs (as a control).2.7 Characterization
Powder diffraction data were collected on an X'Pert PRO X-ray diffractometer equipped with a PIXcel ultrafast line detector, a focusing mirror and Soller slits for Cu Kα radiation. Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) were performed on an FEI Tecnai G2 20 X-TWIN microscope operating at 200 kV. TEM-EDS investigations were carried out on a double-aberration corrected FEI Titan 60-300 cubed (S)TEM, operated at 300 kV. EDS (energy dispersive X-ray spectroscopy) spectra were acquired using Bruker ChemiSTEM super-X EDS detectors, with an acquisition time of 400 s. The photo-luminescence spectra were collected with a QE65000 high-sensitivity fiber optic spectrometer (OceanOptics) under 975 nm laser diode (Pmax = 3W CW, LP975-3000 Spectra Laser, Poland) photo-excitation. Luminescence lifetimes were measured under 976 nm excitation with a CW 8.5 W laser diode modulated with a trigger from an FLS980 fluorescence spectrometer and with a PMT model R928P from Hamamatsu.HeLa cell imaging was performed with an inverted fluorescence wide-field microscope, AxioObserverZ1 (Carl Zeiss) with an EC Plan-Neofluar 40×/1.3 oil DIC M27 objective with a condenser BF (NA = 0.4) to record the white light image, and 3 W CW 975 nm laser diode excitation (Spectra-Laser) in UC mode with a custom-mounted filter cube. The filter cube was composed of FF750-SDi02 dichroic and FF01-945 emission filters to cut out the 975 nm excitation and transmit visible radiation. To collect the images EMCCD Rolera EM-C2 (QImaging) as well as ZEN2011 software (Carl Zeiss) were used. To record the fluorescence images (t = 1/5 s, f = 2.8, ISO = 100) of the samples shown in Fig. 5 and 10b, a Canon 400D digital camera with a 60 mm f = 2.8 EF macro-lens (Canon) was used.
3 Results and discussion
The hexagonal β-NaYF4:Mn2+/Yb3+/Tb3+ nanoparticles were prepared using a thermal decomposition reaction of lanthanide oleates. In order to determine the impact of manganese ion concentration on the optical properties of the Tb3+–Yb3+ upconversion emission, nanoparticles with different concentrations of ions were synthesized. The concentration of nanoparticles in all samples was the same. This enables semi-quantitative comparison and characterization of all compounds in terms of morphology, structure and spectra. Different phenomena behind the expected upconversion enhancement may be preliminary proposed, such as the energy transfer process between Mn2+, Yb3+ and Tb3+ ions as well as the distortion of the crystalline field symmetry due to the introduction of Mn2+ ions replacing yttrium ions in the β-NaYF4 lattice. These two hypotheses will be further explored and evaluated.3.1 Structural characterization of nanoparticles doped with Mn2+ ions
The influence of the Mn2+ ion concentration on the structure and the size of the nanoparticles was studied based on the powder X-ray diffraction patterns of β-NaYF4 (Fig. 1).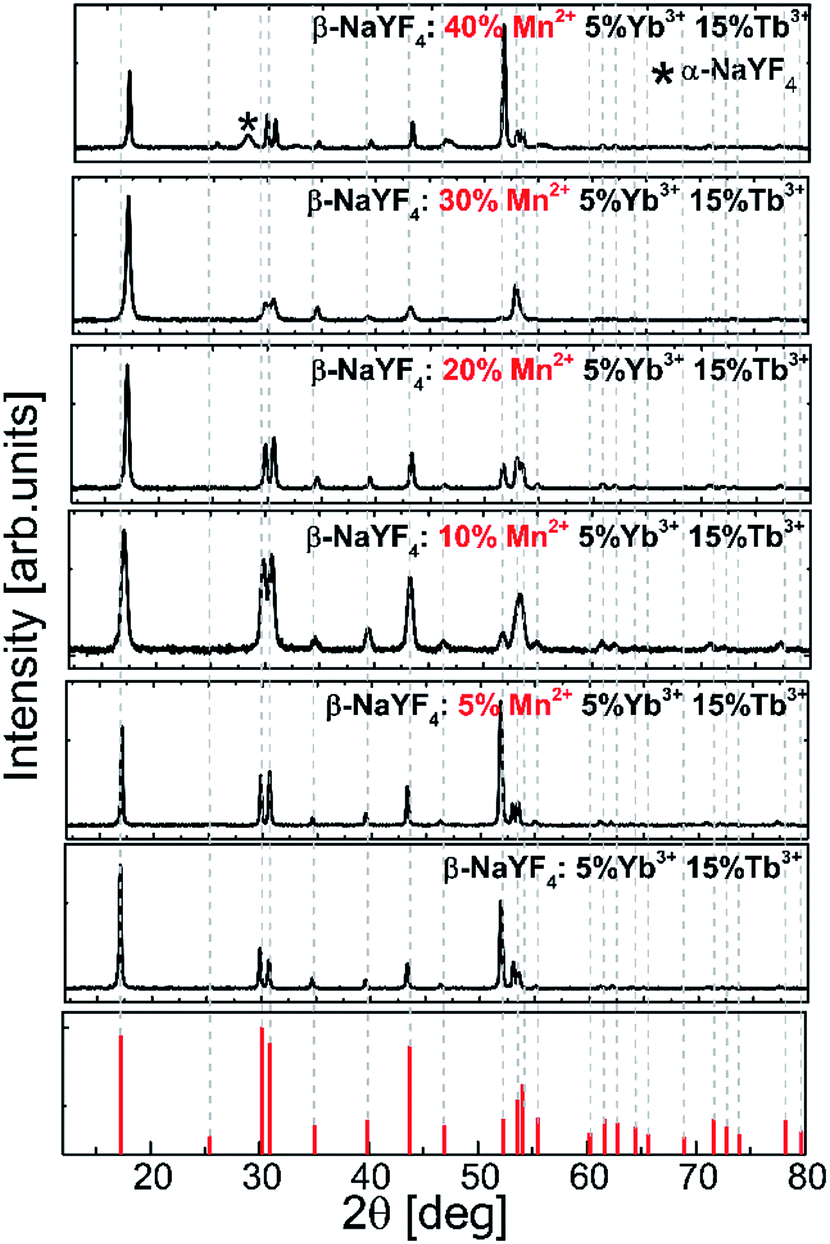 | ||
| Fig. 1 Experimental powder X-ray diffraction (XRD) patterns of the Tb3+, Yb3+, and Mn2+ codoped β-NaYF4 nanoparticles. | ||
XRD reflection peaks of the nanoparticles are in agreement with the standard pattern of JCPDS card # 00-016-0334 and indicate a pure hexagonal phase. The hexagonal phase of NaYF4 still remains stable even when smaller Mn2+ (r = 0.81 Å) ions are introduced into the host lattices to replace larger Y3+ ions (r = 0.89 Å). Nevertheless, in the nanoparticles doped with Mn2+ at amounts higher than 30%, the XRD patterns showed a mixed phase of hexagonal and cubic NaYF4 phases.
The cubic structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) contains one type of cation site, and the lanthanide and sodium ions are equally and randomly distributed in the cationic sublattice (Fig. 2a). In the hexagonal structure, there are three types of cationic sites: (I) a 9-coordinated position occupied by Y3+, (II) a 9-fold coordinated position occupied randomly by 1/2 Na+ and 1/2 Y3+, and (III) a 6-fold coordinated position occupied by 1/2 Na+. The Y3+ sites are substituted randomly with other lanthanides ions.34 Previous reports have demonstrated that the size of the substitutional dopant ions plays a key role in stabilizing a specific crystalline phase in the NaYF4 structure. The substitution ions with a smaller ionic radius favor the cubic structure, whereas the larger substitution ions tend to produce the hexagonal phase of the final nanomaterials. Doping lanthanide ions with sizes larger than that of Y3+ in NaYF4 host lattices should lead to the dominant formation of pure hexagonal-phase NaYF4 nanocrystals.35
m) contains one type of cation site, and the lanthanide and sodium ions are equally and randomly distributed in the cationic sublattice (Fig. 2a). In the hexagonal structure, there are three types of cationic sites: (I) a 9-coordinated position occupied by Y3+, (II) a 9-fold coordinated position occupied randomly by 1/2 Na+ and 1/2 Y3+, and (III) a 6-fold coordinated position occupied by 1/2 Na+. The Y3+ sites are substituted randomly with other lanthanides ions.34 Previous reports have demonstrated that the size of the substitutional dopant ions plays a key role in stabilizing a specific crystalline phase in the NaYF4 structure. The substitution ions with a smaller ionic radius favor the cubic structure, whereas the larger substitution ions tend to produce the hexagonal phase of the final nanomaterials. Doping lanthanide ions with sizes larger than that of Y3+ in NaYF4 host lattices should lead to the dominant formation of pure hexagonal-phase NaYF4 nanocrystals.35
 | ||
| Fig. 2 Schematic representations of (a) cubic- and (b) hexagonal-phase NaLnF4 structures. | ||
When the dopants have different valences than the substituted host ions, extra vacancies or interstitial ions are introduced into the host lattice during the nucleation and growth because of the charge compensation requirement. For hetero-valence ion doping, the variations of the crystal lattice are complex due to the requirement of extra vacancies or interstitial ions to compensate for the charge. When Mn2+ ions are doped into the crystal matrix, they may locate in either Ln3+ lattice sites or in interstitial positions. Doping additional ions into the crystal matrix induces a breakdown of the crystallographic site symmetry and may in consequence lead to phase transformation. The XRD pattern exhibits that the diffraction peaks of NaYF4:Tb3+,Yb3+ doped with different concentrations of Mn2+ ions shift toward higher 2θ values as the concentration of Mn2+ ions increases. It may result from the shrinkage of the unit-cell volume caused by substituting Y3+ ions with ions of a relatively smaller radius.36 This result is in good agreement with a previous report about Fe3+ and Cu2+ ion doped NaYF4 nanoparticles.37
Fig. 3 shows the representative TEM images of NaYF4:Mn2+/Yb3+/Tb3+ nanocrystals doped with different amounts of Mn2+ ions. Doping with Mn2+ ions caused a slight increase in nanoparticle size, but the NPs remained monodisperse. The average size for the Mn2+ un-doped sample can be estimated as ca. 23 nm, while for samples doped with 30% Mn2+ the nanoparticles grow to 33 nm. Further increasing the Mn2+ doping amount is detrimental for the quality of the samples, i.e. for the NaYF4:Mn2+/Yb3+/Tb3+ (40/5/15 mol%) samples, and the TEM images show two separate particle morphologies and sizes (Fig. 3d), which is consistent with the presence of two phases observed in the X-ray powder diffraction patterns (Fig. 1) exclusively for this sample. The small α-NaYF4 particles were irregular in shape and were ca. 8 nm in average size. These small NPs result from substitution of host ions by the smaller Mn2+ ions, which also tends to produce the cubic phase.10
 | ||
| Fig. 3 TEM images of β-NaYF4:Mn2+,Yb3+,Tb3+ nanocrystals doped with (a) 10%, (b) 20%, (c) 30% and (d) 40% Mn2+ ions. | ||
The presence of Mn ions (without determining the valence state) was clearly confirmed using EDX. Although many lines of Tb and Mn atoms overlap, we managed to confirm the presence of Mn ions – the K-a peak at 5.9 keV is unequivocal evidence of Mn presence within the nanocrystal structure (Fig. 4).
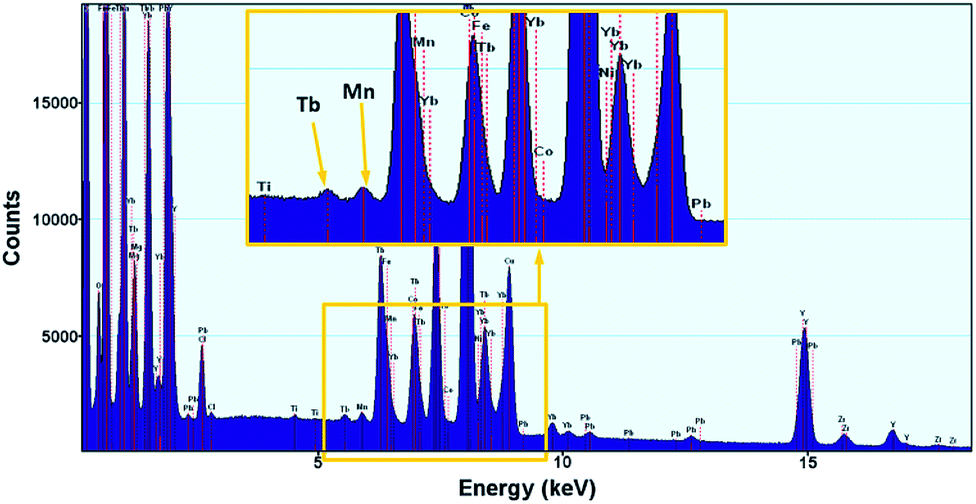 | ||
| Fig. 4 EDX spectrum of NaYF4 nanoparticles doped with Tb3+, Yb3+ and Mn2+ ions. | ||
3.2 Optical properties of β-NaYF4:Mn2+,Yb3+,Tb3+ nanoparticles
To understand the actual processes responsible for the upconversion emission from Tb3+ in the presence of intentionally varied Mn2+ ion content, the photoluminescence spectra of colloidal nanoparticles were measured under UV (370 nm, Fig. 5a) and NIR (980 nm, Fig. 5b) excitation.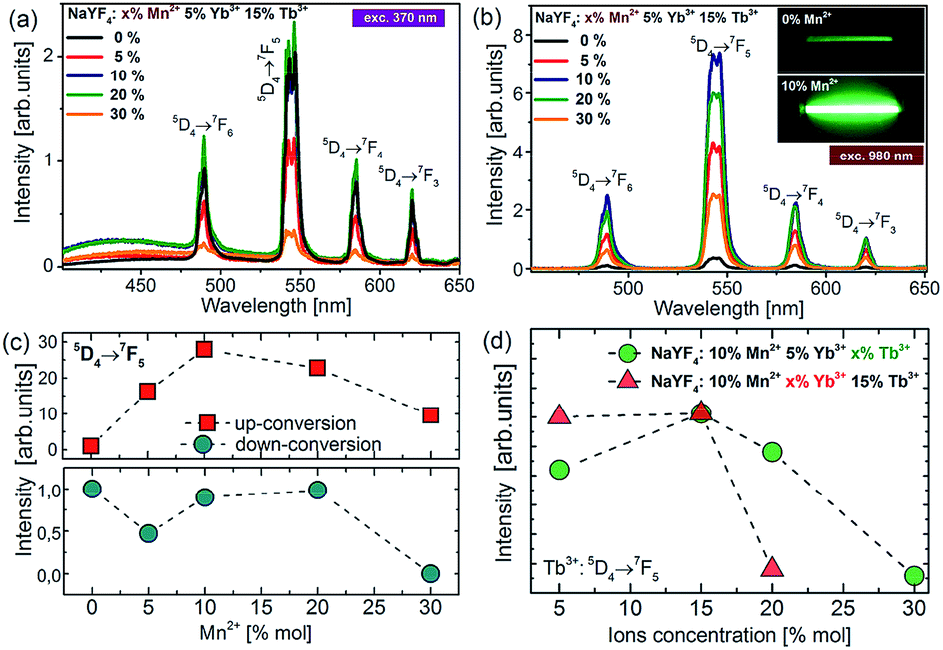 | ||
| Fig. 5 (a) The downconversion (DC) and (b) upconversion spectra of colloidal β-NaYF4:5% Yb3+,15% Tb3+ nanoparticles doped with different amounts of Mn2+ ions under 370 nm and 980 nm excitation, respectively. The inset shows the photographs of the upconversion luminescence from Mn2+ undoped (top) and co-doped (bottom) colloidal samples. (c) Terbium DC-emission and UC-emission intensity as a function of Mn2+ concentration. (d) Terbium UC emission intensity as a function of Yb3+ and Tb3+ ion concentration under 980 nm excitation. | ||
Upon the absorption of UV photons, Tb3+ ions are excited to their 4f75d1 state, and non-radiatively relax down to the emitting 5D4 level. Owing to the energy gap law and low maximum available phonons, ∼350 cm−1 in the NaYF4 matrix, this process is not very efficient. Four distinct emission bands of Tb3+ were observed and could be assigned to the 5D4 → 7FJ (J = 3, 4, 5, and 6) transitions (Fig. 5a). For the nanoparticles doped with Mn2+ ions, a small broad emission band at 450 nm was observed, which most probably is due to the 4f135d1–4f14 transition of the Yb2+ state. The broadband emission was similar to the one reported for NaYF4 nanoparticles doped with Tb3+, Yb3+ and Nd3+ ions in our previous work.15 Although Yb2+ broad band emission should typically occur at higher energy (e.g. in CaAl2O4, LiYF4, and CaF2) and at low temperatures, we observe a relationship between its intensity and the nanocrystal composition, which should not, in principle, take place, if the broad band emission arises only from the presence of organic ligands or other impurities on the surface of the nanocrystals. Nevertheless, the Yb2+ emission with the maxima at 440 nm has been observed in a few hosts, such as Sr3(PO4)3 at low temperatures, as well as the Yb2+ emission has been observed at room temperature from different matrices, showing a yellow-green-bluish colour with the maxima at ∼550 nm (in CaF2, b-SiAlON) or ∼430 nm (NaCl).
The quantitative comparison of photoluminescence properties of the as-prepared colloidal nanoparticles NaYF4:Tb3+,Yb3+ doped with Mn2+ has been presented in Fig. 5b. The nanocrystals exhibit relatively strong green upconversion emission under 980 nm NIR diode excitation. Three distinct emission peaks can be assigned to the 5D4 → 7F6 (λ = 480 nm), 5D4 → 7F5 (λ = 540 nm), 5D4 → 7F4 (λ = 585 nm) and 5D4 → 7F3 (λ = 620 nm) terbium transitions. As shown in Fig. 5, the presence of Mn2+ ions may significantly increase upconversion emission of Tb3+ ions. The most intense emission was achieved for nanoparticles doped with 10% Mn2+ ions, which was responsible for over 30 times brighter intensity of Tb3+ ions compared to the emission of nanocrystals without the addition of Mn2+ ions. A further increase of Mn2+ concentration caused a decrease in 5D4 → 7F5 transition intensity, but still the emission intensity was higher than for the samples without the addition of Mn2+ ions. Based on the result from upconversion spectra we can conclude that the concentration of Mn2+ ions should be kept similar to the amount of Yb3+ ions in nanocrystals.
To further optimize the UC intensity, besides Mn2+ optimization, additional nanocrystals with different Tb3+ and Yb3+ ion doping levels were also synthesized. The enhancement of Tb3+ emission intensity was obtained until 15 mol% Tb3+ ion doping, and then with the further increment of the doping amount, the intensity of green emission decreased, which most probably can be explained by the backward energy transfer from 5D4:Tb3+ ions to Mn2+ (Fig. 5d). A similar observation was made by Zhang's group in the case of nanoparticles doped with europium ions.26 They have studied the influence of Mn2+ ion doping on the luminescence properties of NaY(Lu)F4 nanoparticles doped with europium and ytterbium ions. They found that the optimal concentration of Eu3+ and Yb3+ ions lies in the range between 15% and 5%, respectively. Similarly in our materials, the optimal concentration of Yb3+ was estimated to be 5% because a further increase in Yb3+ concentration did not improve the UC emission intensity (Fig. 5d). We suppose that it can be linked with the high probability of occurrence of energy transfer to surface quenchers over long distances by energy migration through the Yb3+ network.38 Based on these results, the optimal concentration for effective energy upconversion transfer was estimated to be 15% Tb3+ and 5% Yb3+. In our previous work the optimum Tb3+ and Yb3+ concentration was estimated to be 40% and 20%, respectively,8,11 and the concentration ratio was equal to 2:1 previously and went up to 3:1 in this work. Obviously, this originates from Mn2+ ion co-doping; however, the exact mechanism of enhancement is not clear. Because the Tb3+ → Yb3+ energy back transfer is possible and significant energy migration to the surface quenchers at high Yb3+ doping is known,39 we may only speculate that the addition of Mn2+ ions not only supports more efficient CET, but also helps the Yb3+ network to enable more efficient energy migration and lose energy through the surface effects.
Despite the formation of Yb3+–Mn2+ dimers is well established in the literature,40–43 we have not managed to see broadband Mn2+ emission alone,28,44 which calls into question our preliminary hypothesis on the active participation of Mn2+ ions in the co-operative energy transfer in our nanoparticles (Fig. 7c). The hypothesis of cooperative sensitization of Mn2+ ions by a pair of Yb3+ ions, followed by ET to Mn2+ → Tb3+ (Fig. 7b) can also be proposed, but difficult to verify. In the view of the obtained results (i.e. mostly missing Mn2+ emission), the second, more probable explanation of the obtained enhancement of the upconversion emission can be proposed – i.e. the distortion of the crystalline field symmetry due to introduction of Mn2+ ions, replacing yttrium ions in the β-NaYF4 lattice (Fig. 7a). In the β-NaYF4 host lattice, the doped Mn2+ with a small radius can substitute cation ions or occupy interstitial sites, causing the shrinking or expansion of the host lattice and thus perturbing the symmetry of the local crystal field around Tb3+. The asymmetric surrounding environment emitting ions may lead to the faster energy transfer and in consequence increase the UC luminescence intensity.45 The maximum upconversion emission in the sample with 10% Mn2+ doping is attributed to the most asymmetric surrounding environment around Tb3+. With an increase of the doping concentration of Mn2+ from 0 to 30 mol%, the size of the nanoparticles increased monotonically, but their upconversion emission intensities increased at first and then decreased. One should also consider the fact that the size of nanoparticles increases along with the increasing Mn2+ content (Fig. 3) and by this indirect route, the Mn2+ ions could also enhance the CET emission. However, the increasing size does not correlate with the luminescence lifetimes of Yb3+ which become shorter with increasing Mn2+ levels. This observation should support the hypothesis of efficient Yb3+ → Mn2+ energy transfer, but in consequence, Mn2+ green and broadband emission should simultaneously be observed, which is not the case.
In order to study the upconversion emission mechanism and to investigate the impact of the Mn2+ ion doping, the decay profiles of Tb3+ and Yb3+ emission were measured. The examples of 5D4 luminescence decay curves and luminescence decay values for different amounts of Mn2+ ions are presented in Fig. 6.
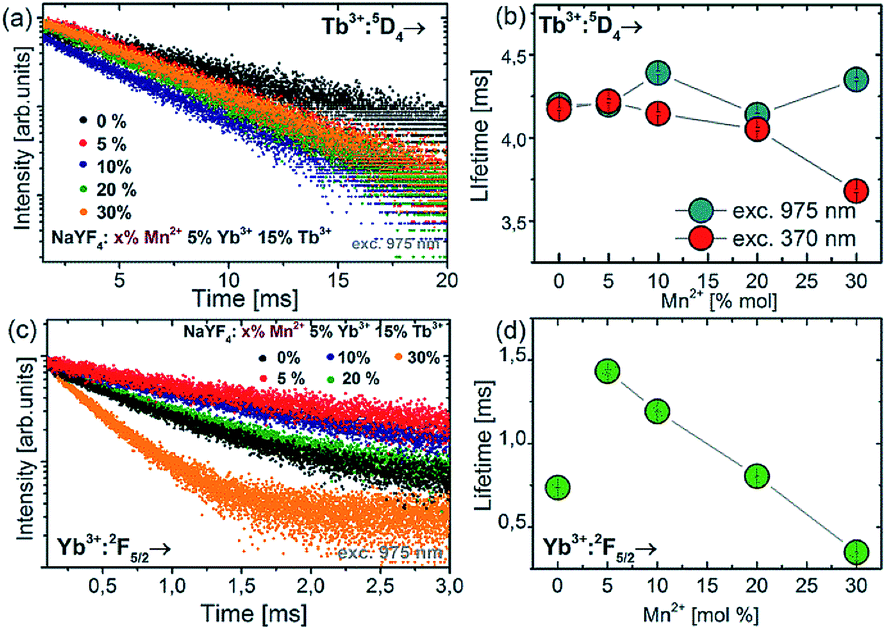 | ||
| Fig. 6 Luminescence decay curves of the 5D4 energy level of Tb3+ ions (a) and Yb3+:7F5/2 level (c) for NaYF4 colloidal nanoparticles doped with different amounts of Mn2+ ions. Concentration dependence of luminescence lifetimes for Tb3+:5D4 (b) and Yb3+:2F5/2 (d) energy levels. | ||
Doping of NaYF4:Tb3+,Yb3+ nanoparticles with Mn2+ ions did not significantly change the luminescence lifetime of the 5D4 energy level (measured at 975 nm excitation wavelength). In contrast to 975 nm excitation wavelength, increasing the Mn2+ concentration resulted in a luminescence lifetime decrease at 370 nm excitation, from ca. 4.3 ms down to the shortest value of 3.6 ms for NaYF4:5% Tb3+,15% Yb3+,30% Mn2+. The measurements of decay time were also carried out for the Yb3+ ions under 975 nm excitation. The luminescence lifetimes of the Yb3+:2F5/2 level decreased significantly as well when Mn2+ concentration was increased. Luminescence decays for Yb3+ were found to decrease monotonically with increasing Mn2+ concentration, which is evidence for efficient energy transfer from Yb3+ to Mn2+ ions.
Because we were not able to confirm unambiguously what is the reason for the observed increase in the intensity of terbium ion emission, three hypothetical upconversion mechanisms were discussed as shown in Fig. 7. Conventionally, for Tb3+ and Yb3+ ion doped nanocrystals under NIR photoexcitation at ∼980 nm, the 5D4 level population originates from cooperative energy transfer (cooperative sensitization) from two excited ytterbium ions (Yb3+–Yb3+ pair).11,46 Addition of Mn2+ ions may change the rate of energy transfer resulting from the tailored local crystal field around the active ions induced by perturbations of the symmetry of the local crystal field (Fig. 7a).47 At the same time, either the cooperative sensitization process of Mn2+ ions by two Yb3+ ions28 (Fig. 7b) or Mn2+–Yb3+ “dimer” formation40–42 (Fig. 7c) hypotheses have been postulated. This is achieved by combining the energy of two excited Yb3+ ions by a cooperative process to transfer it to Mn2+ ions. Next, the energy is transferred from the Mn2+ ions to the 5D4 state of Tb3+ ions.28,48 From the Tb3+:5D4 level, the energy can be transferred to the 7FJ levels and generate luminescence. Because we did not see concurrent Mn2+ emission (which should occur in the green spectral region), we could not confirm that Mn2+ ions contribute to upconversion energy transfer processes actively and directly, i.e. through the [Yb3+–Yb3+] → Mn2+ → Tb3+ or [Yb3+–Mn2+] → Tb3+ processes, respectively. Quenching of Tb3+ emission and Yb3+ lifetimes by increasing Mn2+ content suggests that Tb3+ → Mn2+ and Yb3+ → Mn2+ should be possible, but also here, Mn2+ emission was not found.
 | ||
| Fig. 7 Energy transfer mechanisms of NaYF4:Mn2+,Tb3+,Yb3+ nanocrystals under 975 nm excitation. The obtained enhancement of the upconversion emission intensities can potentially result from (a) the tailored local crystal field around the Tb3+ ions induced by the unit cell expansion; (b) cooperative sensitization of Mn2+ ions by a pair of Yb3+ ions, followed by ET to Mn2+ → Tb3+ and Mn2+–Yb3+ “dimer” formation.40–42 | ||
All these facts suggest that the passive role of Mn2+ co-doping dominates over detrimental quenching by Mn2+ ions, which led to the observed over 30-fold UC enhancement. Such an enhancement is, however, sufficient to make Tb3+ upconversion comparable to conventional Er3+ upconversion and can be used in biomedical imaging.
3.3 Bioimaging
Due to the hydrophobic nature of the capping ligand (oleic acid), nanoparticles’ surface is hydrophobic. Further functionalization is necessary to make the nanoparticles water dispersible and to use the synthesized nanocrystals as imaging labels.Water dispersible nanoparticles were achieved by removing the oleic acid molecules from the surface of the nanocrystals at low pH using 0.1 M hydrochloric acid.49 The hydrophilic nanoparticles were mixed with FBS (fetal bovine serum) in order to initialize the formation of a protein corona on the nanoparticles’ surface.50 To make the synthesized nanoparticles suitable for cell imaging, the NPs were further biofunctionalized with folic acid (FA) (Fig. 8a). Both Yb3+/Tb3+ and Yb3+/Tb3+/Mn2+ UCNPs were treated with exactly the same protocol.
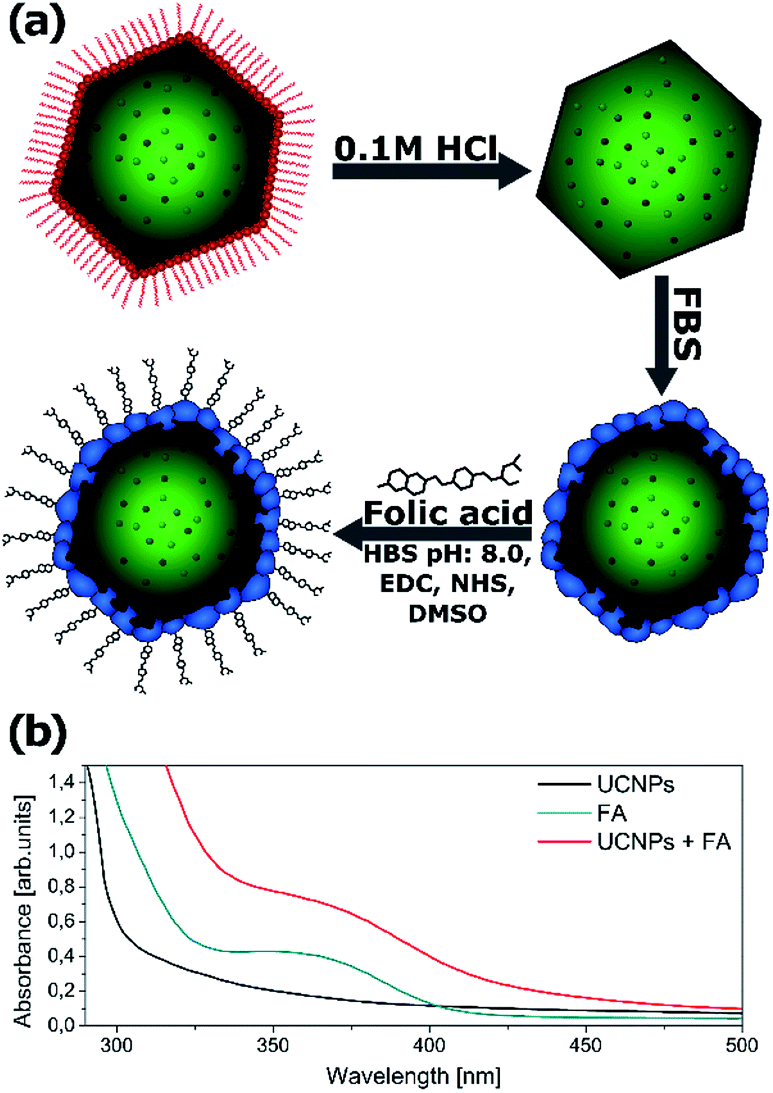 | ||
| Fig. 8 (a) Schematic illustration of the simple method used for the biofunctionalization of upconverting nanocrystals. (b) The UV-Vis absorption spectra of FBS-coated UCNPs before and after folic acid (FA) conjugation. UCNPs (0.5 mg ml−1), UCNP–FA (0.5 mg ml−1) and FA (50 μg ml−1) were dissolved in 0.1 M HEPES, pH 8. | ||
To confirm that the biofunctionalization was successfully performed and the folic acid is present on the nanoparticles’ surface, the absorption spectra of the NPs were measured after washing the free FA (Fig. 8b). The absorption spectra of the UCNPs changed after FA conjugation and exhibited an additional peak corresponding to the FA absorbance spectrum indicating successful conjugation of FA with NaYF4 nanoparticles. A similar absorbance change was reported by Hu and coworkers after folic acid conjugation to gold nanoclusters.51
To test the potential biological application of nanocrystals doped with Tb3+ ions, which offer spectrally distinguishable “fingerprints” compared to other dopants (e.g. Er3+ and Ho3+), in vitro bioimaging of the HeLa cell line was performed. HeLa is the first immortal cell line, established in 1952 by George Otto Gey. It derives from a cervical cancer patient, Henrietta Lacks, who died in 1951. Over the years, it became a model cell line in molecular cell biology, used in many publications and contributed greatly to the understanding of the biology of the human cell, cancer research and development of different vaccines.52 This particular cell line was selected due to its relatively fast growth rate with the cell count doubling in about 22 hours,53 as well as increased expression of folate receptors which is also a common feature of many other cancer cell types.54 The folate receptor effectively mediates the delivery of folate conjugated compounds and nanoparticles via receptor mediated endocytosis.55
Imaging of HeLa cells was done by incubation of the cells with FA-conjugated NaYF4:Yb3+,Tb3+,Mn2+ and NaYF4:Yb3+,Tb3+ nanocrystals. Functionalized nanoparticles were effectively internalized by HeLa cells upon 20 hours of incubation. The visible upconversion fluorescence of the NaYF4 nanocrystals doped with Mn2+, Tb3+, and Yb3+ ions loaded in HeLa cells was easily observed following excitation with 980 nm NIR radiation (Fig. 9a). The UC images were taken with 3 second exposure time. For nanocrystals without Mn2+ ions, the control luminescence imaging measurements under the same experimental conditions (the same camera setting and exposure time of 3 seconds) (Fig. 9b) showed significantly weaker emission than for the imaging of Mn2+ doped nanoparticles (Fig. 9a and b). Increasing the UC imaging acquisition time up to 30 seconds did not radically help to achieve any better signal from the sample, and only barely visible green upconversion emission was observed (not shown). Significant differences in the fluorescence intensity between both types of nanoparticles combined with the same surface bio-chemistry clearly demonstrated the advantages of nanoparticles doped with additional Mn2+ ions.
 | ||
| Fig. 9 The upconversion and DAPI microscopy images (40× magnification, UV and 975 nm excitation) of HeLa cells after incubation with folic acid-conjugated (a) NaYF4:Yb3+,Tb3+, Mn2+ – exposure time 3 seconds in UC mode, and (b) NaYF4:Yb3+,Tb3+ – exposure time 3 seconds in UC mode. Cells were incubated for 20 hours with UCNP–FA (0.2 mg ml−1). Green colour represents upconversion fluorescence, and blue colour – fluorescence from nucleic acid stained with DAPI (4′,6-diamidino-2-phenylindole dihydrochloride). | ||
While the imaging in upconversion mode is nothing extraordinary with Yb3+/Er3+ or Yb3+/Tm3+ UCNPs, the UC imaging with Yb3+/Tb3+ codoped NPs has been severely hindered so far because CET (e.g. Yb3+/Tb3+) is typically 2 orders of magnitude weaker than ETU (e.g. Yb3+/Er3+ and Yb3+/Tm3+).
Tb3+ emission is easily distinguishable from Tm3+ and Er3+ emission because of the spectrally distinct 580 nm Tb3+ emission band. Upconversion spectra of Yb3+/Er3+, Yb3+/Tm3+ and Yb3+/Tb3+ codoped NaYF4 nanoparticles and the photographs of the upconversion luminescence from colloidal NaYF4 doped with different lanthanide ions are presented in Fig. 10. The Tb3+ emission bands marked by an asterisk are spectrally distinguishable from the emission of other lanthanide ions, despite some spectral overlap exists between Tb3+ and Er3+ or Tb3+ and Tm3+ at other wavelengths (e.g. 540 or 650 nm). However, because the emission branching ratio of Tb3+ emission is fixed, i.e. the relative proportions of 5D4 → 7FJ (J = 6.0) remain constant, knowledge of this branching ratio and the intensity at 580 nm enable us to appropriately correct 540 nm and 650 nm emission intensity, aiming at quantitative analysis of multiple labels, which overlap in space. The application of other nanomaterials doped with Tb3+ ions in bioimaging has been investigated with promising results but usually under UV excitation.56–59 However, UV photoexcitation demonstrates well known drawbacks such as generation of autofluorescence from endogenous intracellular fluorophores (e.g. aromatic groups of protein amino acids, nucleic acids, and low molecular coenzymes like porphyrins, NADH, FAD, etc.),60 which results in an undesirably high background and a low signal to background ratio. Moreover, at near-UV excitation wavelengths the penetration depth of light is limited to less than 200 μm for the human skin which is due to the presence of many UV-absorbing elements and increased light scattering in the heterogeneous tissue.61 Finally, UV poses some risk of damaging biomolecules, being the result of highly reactive free radical generation by UV radiation, or disruption of DNA integrity via breaking the hydrogen bonds between DNA strands.62
 | ||
| Fig. 10 (a) Upconversion spectra of nanocrystals doped with Yb3+/Er3+, Yb3+/Tm3+ and Yb3+/Tb3+ ions. (b) Photographs of the upconversion luminescence from colloidal NaYF4 nanoparticles doped with different lanthanide ions measured at the same concentrations and with the same recording settings. | ||
In this respect the use of NIR photoexcitation and upconversion emission is advantageous because it enables signal detection without an autofluorescence background and with limited light scattering, as biomolecules are usually much less efficient non-linear emitters (e.g. in 2- or 3-photon excitation mode). Moreover, the infrared wavelength provides a much deeper penetration depth of the optical radiation into samples up to 2 cm.63
3.4 Cytotoxicity
In order to investigate the cytotoxicity of the synthesized nanoparticles, an MTT assay using HeLa cells was performed. The impact of the UCNPs on the cell proliferation after 72 hours of incubation was studied (Fig. 11). | ||
| Fig. 11 The viability of HeLa cells after 72 h of incubation with NaYF4 nanoparticles. | ||
In all bio-applications, the important potential risk of the nanoparticles is their cytotoxicity. Chemical compositions of nanoparticles as well as their surface modifications play important roles in this matter. For these reasons, the MTT assay was used to investigate the cytotoxicity of the following UCNPs: β-NaYF4:5% Yb3+/15% Tb3+ (protein coated), β-NaYF4:5% Yb3+/15% Tb3+@FA (protein coated, with folic acid), β-NaYF4:10% Mn2+/5% Yb3+/15% Tb3+ (protein coated), β-NaYF4:10% Mn2+/5% Yb3+/15% Tb3+@FA (protein coated, with folic acid). No significant difference in the cell viability was observed in the presence of 50–200 μg ml−1 UCNPs (Fig. 11). The viability of the cells was estimated to be greater than 80% for β-NaYF4:5% Yb3+/15% Tb3+ and greater than 90% for β-NaYF4:5% Yb3+/15% Tb3+@FA, β-NaYF4:10% Mn2+/5% Yb3+/15% Tb3+ and β-NaYF4:10% Mn2+/5% Yb3+/15% Tb3+@FA after 72 hours of incubation. The data show that all the examined NPs, with a modified surface, have low cytotoxicity even at a relatively high concentration (200 μg ml−1), which is in agreement with a large number of reports on the toxicity of lanthanide doped NaYF4 UCNPs.64
4 Conclusion
β-NaYF4:Yb3+,Tb3+ nanocrystals doped with different concentrations of Mn2+ ions were synthesized in response to the demand for novel UC enhancement methods of weak Yb3+–Tb3+ upconversion. The significant enhancement of the UC emission as a result of the introduction of Mn2+ ions was observed without any changes in the crystallographic phase. The upconversion emission intensity obtained from the nanoparticles co-doped with 10% Mn2+ ions increased up to 30-fold in comparison to the corresponding nanoparticles without Mn2+ ions. Three different energy transfer mechanisms of the UC process were originally proposed, but following the analysis of the experimental results, we suppose that the obtained enhancement of the upconversion emission intensities can result from the tailored local crystal field around the Tb3+ ions being induced by the unit cell expansion as well as potentially an influence of the manganese ions acting actively in the transfer of energy.The studies of the optimized Yb3+–Tb3+ UC based on Mn2+ enhanced upconversion are important for numerous reasons. First, the Yb3+ → Mn2+ → Tb3+ is a direct type of energy transfer which does not require more complicated indirect Yb3+ → Tm3+ → Tb3+ or Yb3+ → Tm3+ → Gd3+ → Tb3+ energy migration mediated upconversion, where core–(multiple)shell NPs have to be synthesized. Moreover, no spectral features originating from Tm3+ emission are present in the direct Tb3+ upconversion. In consequence, direct ETU based materials are also important when the size of the NPs is critically important, because homogeneously doped NPs can be smaller and much easier to obtain than core–shell NPs. As a proof of the performed enhancement, wide field UC imaging and visualization of folate-mediated endocytosis in FA receptor rich HeLa cells were successfully carried out, unlike with un-optimized Yb3+/Tb3+ UCNPs. Finally, a great advantage of labels doped with Tb3+ ions is the fact that their emission at 580 nm is spectrally easily distinguishable from the UC emission of conventionally used Tm3+ and Er3+ ions, which increases the selection of unique labels within the highly photostable, upconverting class of labels.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KP acknowledges the support from the Foundation for Polish Science (FNP) under the START programme. The authors acknowledge financial support from the National Sciences Centre under Grant No. DEC-2012/05/E/ST5/03901 from the NCN, Poland. The biological and bioimaging part of this work was supported by the Wroclaw Research Centre EIT+ within the project “The Application of Nanotechnology in Advanced Materials” – NanoMat (POIG.01.01.02-02-002/08) co-financed by the European Regional Development Fund (Operational Programme Innovative Economy, 1.1.2), Task 7.1 and 7.2. The authors wish to thank Mrs Anna Siudzinska (PORT) and Dr Rafal Szukiewiecz (PORT) for the EDX and XPS measurements, respectively.Notes and references
- A. Gnach and A. Bednarkiewicz, Nano Today, 2012, 7, 532–563 CrossRef CAS.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- D. Bechet, P. Couleaud, C. Frochot, M.-L. Viriot, F. Guillemin and M. Barberi-Heyob, Trends Biotechnol., 2008, 26, 612–621 CrossRef CAS PubMed.
- D. Jaque and F. Vetrone, Nanoscale, 2012, 4, 4301–4326 RSC.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu and X. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968 CrossRef CAS PubMed.
- E. M. Chan, E. S. Levy and B. E. Cohen, Adv. Mater., 2015, 27, 5753–5761 CrossRef CAS PubMed.
- K. Prorok, A. Bednarkiewicz, B. Cichy, A. Gnach, M. Misiak, M. Sobczyk and W. Strek, Nanoscale, 2014, 6, 1855–1864 RSC.
- T. Grzyb, R. J. Wiglusz, A. Gruszeczka and S. Lis, Dalton Trans., 2014, 43, 17255–17264 RSC.
- J. L. Adam, N. Duhamel-Henry and J. Y. Allain, J. Non-Cryst. Solids, 1997, 213–214, 245–250 CrossRef.
- K. Prorok, A. Gnach, A. Bednarkiewicz and W. Stręk, J. Lumin., 2013, 140, 103–109 CrossRef CAS.
- M. L. Debasu, D. Ananias, S. L. C. Pinho, C. F. G. C. Geraldes, L. D. Carlos and J. Rocha, Nanoscale, 2012, 4, 5154–5162 RSC.
- W. Strek, P. Deren and A. Bednarkiewicz, J. Lumin., 2000, 87–89, 999–1001 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139–174 CrossRef CAS PubMed.
- K. Prorok, M. Pawlyta, W. Stręk and A. Bednarkiewicz, Chem. Mater., 2016, 28, 2295–2300 CrossRef CAS.
- V. Scarnera, B. Richards, A. Jha, G. Jose and C. Stacey, Opt. Mater. (Amsterdam, Neth.), 2010, 33, 159–163 CAS.
- X. Wang, L. Liu, Q. Nie, T. Xu, X. Shen, S. Dai and X. Zhang, Spectrochim. Acta, Part A, 2007, 67, 1025–1029 CrossRef PubMed.
- W. J. Zhang, Q. J. Chen, Q. Qian, Q. Y. Zhang and Z. H. Jiang, Phys. B, 2010, 405, 1062–1066 CrossRef CAS.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- Z. Bai, H. Lin, K. Imakita, R. Montazami, M. Fujii and N. Hashemi, RSC Adv., 2014, 4, 61891–61897 RSC.
- Z. Bai, H. Lin, J. Johnson, S. C. Rong Gui, K. Imakita, R. Montazami, M. Fujii and N. Hashemi, J. Mater. Chem. C, 2014, 2, 1736–1741 RSC.
- K. L. Reddy, M. Rai, N. Prabhakar, R. Arppe, S. B. Rai, S. K. Singh, J. M. Rosenholm and V. Krishnan, RSC Adv., 2016, 6, 53698–53704 RSC.
- K. Lingeshwar Reddy, V. Srinivas, K. R. Shankar, S. Kumar, V. Sharma, A. Kumar, A. Bahuguna, K. Bhattacharyya and V. Krishnan, J. Phys. Chem. C, 2017, 121, 11783–11793 CrossRef CAS.
- P. Gerner, O. S. Wenger, R. Valiente and H. U. Güdel, Inorg. Chem., 2001, 40, 4534–4542 CrossRef CAS PubMed.
- C. Reinhard, P. Gerner, R. Valiente, O. S. Wenger and H. U. Güdel, J. Lumin., 2001, 94–95, 331–335 CrossRef.
- Z. Wang, J. Feng, S. Song, Z. Sun, S. Yao, X. Ge, M. Pang and H. Zhang, J. Mater. Chem. C, 2014, 2, 9004–9011 RSC.
- H. Du, Z. Yang and J. Sun, Guangpuxue Yu Guangpu Fenxi, 2009, 29, 2317–2320 CAS.
- H. K. Dan, D. Zhou, R. Wang, Q. Jiao, Z. Yang, Z. Song, X. Yu and J. Qiu, Mater. Lett., 2015, 150, 76–80 CrossRef CAS.
- F. Song, C. Ming, L. An, Q. Wang, Y. Yu, H. Yu, T. Sun and J. Tian, Mater. Lett., 2011, 65, 3140–3142 CrossRef CAS.
- X. Qu, L. Cao, W. Liu and G. Su, Ceram. Int., 2012, 38, 1765–1769 CrossRef CAS.
- W. Lü, Z. Hao, X. Zhang, Y. Luo, X. Wang and J. Zhang, Inorg. Chem., 2011, 50, 7846–7851 CrossRef PubMed.
- H. You, Y. Song, G. Jia and G. Hong, Opt. Mater. (Amsterdam, Neth.), 2008, 31, 342–345 CAS.
- M. Xue, X. Zhu, X. Qiu, Y. Gu, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 17894–17901 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061 CrossRef CAS PubMed.
- P. Zhang, W. Qin, D. Li and L. Wang, CrystEngComm, 2017, 19, 3215–3221 RSC.
- Y. Sui, K. Tao, Q. Tian and K. Sun, J. Phys. Chem. C, 2012, 116, 1732–1739 CrossRef CAS.
- K. Du, X. Xu, S. Yao, P. Lei, L. Dong, M. Zhang, J. Feng and H. Zhang, CrystEngComm, 2018, 20, 1945–1953 RSC.
- Z. Huang, H. Gao and Y. Mao, RSC Adv., 2016, 6, 83321–83327 RSC.
- A. Pilch, C. Würth, M. Kaiser, D. Wawrzyńczyk, M. Kurnatowska, S. Arabasz, K. Prorok, M. Samoć, W. Strek, U. Resch-Genger and A. Bednarkiewicz, Small, 2017, 13, 1701635 CrossRef PubMed.
- P. Gerner, C. Fuhrer, C. Reinhard and H. U. Güdel, J. Alloys Compd., 2004, 380, 39–44 CrossRef CAS.
- R. Martín-Rodríguez, R. Valiente, F. Rodríguez, F. Piccinelli, A. Speghini and M. Bettinelli, Phys. Rev. B, 2010, 82, 75117 CrossRef.
- C. Reinhard, P. Gerner, F. Rodrıíguez, S. García-Revilla, R. Valiente and H. U. Güdel, Chem. Phys. Lett., 2004, 386, 132–136 CrossRef CAS.
- S. Ye, J. Sun, X. Yi, Y. Wang and Q. Zhang, Sci. Rep., 2017, 7, 46219 CrossRef CAS PubMed.
- X. Liu, Y. Wang, X. Li, Z. Yi, R. Deng, L. Liang, X. Xie, D. T. B. Loong, S. Song, D. Fan, A. H. All, H. Zhang, L. Huang and X. Liu, Nat. Commun., 2017, 8, 899 CrossRef PubMed.
- S. Sinha, M. K. Mahata and K. Kumar, New J. Chem., 2019, 43, 5960–5971 RSC.
- P. Molina, V. Vasyliev, E. G. Víllora and K. Shimamura, J. Appl. Phys., 2011, 110, 123527 CrossRef.
- A. K. Singh, S. K. Singh and S. B. Rai, RSC Adv., 2014, 4, 27039–27061 RSC.
- F. Xiao, E. H. Song, S. Ye and Q. Y. Zhang, J. Alloys Compd., 2014, 587, 177–182 CrossRef CAS.
- N. Bogdan, F. Vetrone, G. A. Ozin and J. A. Capobianco, Nano Lett., 2011, 11, 835–840 CrossRef CAS PubMed.
- L. Treuel, S. Brandholt, P. Maffre, S. Wiegele, L. Shang and G. U. Nienhaus, ACS Nano, 2014, 8, 503–513 CrossRef CAS PubMed.
- D. Hu, Z. Sheng, S. Fang, Y. Wang, D. Gao, P. Zhang, P. Gong, Y. Ma and L. Cai, Theranostics, 2014, 4, 142–153 CrossRef CAS PubMed.
- J. R. Masters, Nat. Rev. Cancer, 2002, 2, 315 CrossRef CAS PubMed.
- K. A. Rafferty, Virchows Arch. B, 1986, 50, 167 CrossRef.
- D. Feng, Y. Song, W. Shi, X. Li and H. Ma, Anal. Chem., 2013, 85, 6530–6535 CrossRef CAS PubMed.
- Y. Song, W. Shi, W. Chen, X. Li and H. Ma, J. Mater. Chem., 2012, 22, 12568–12573 RSC.
- G. A. Sotiriou, D. Franco, D. Poulikakos and A. Ferrari, ACS Nano, 2012, 6, 3888–3897 CrossRef CAS.
- R. M. Petoral, F. Söderlind, A. Klasson, A. Suska, M. A. Fortin, N. Abrikossova, L. Selegård, P.-O. Käll, M. Engström and K. Uvdal, J. Phys. Chem. C, 2009, 113, 6913–6920 CrossRef CAS.
- G. K. Das, Y. Zhang, L. D'Silva, P. Padmanabhan, B. C. Heng, J. S. Chye Loo, S. T. Selvan, K. K. Bhakoo and T. T. Yang Tan, Chem. Mater., 2011, 23, 2439–2446 CrossRef CAS.
- P. Padhye, A. Alam, S. Ghorai, S. Chattopadhyay and P. Poddar, Nanoscale, 2015, 7, 19501–19518 RSC.
- M. B. T.-B. A. R. Monici, Cell and tissue autofluorescence research and diagnostic applications, Elsevier, 2005, vol. 11, pp. 227–256 Search PubMed.
- M. Meinhardt, R. Krebs, A. Anders, U. Heinrich and H. Tronnier, J. Biomed. Opt., 2008, 13, 1–5 CrossRef PubMed.
- R. M. B. T.-M. E. Tyrrell, in Singlet Oxygen, UV-A, and Ozone, Academic Press, 2000, vol. 319, pp. 290–296 Search PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC.
| This journal is © The Royal Society of Chemistry 2019 |