
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small Rh nanoparticles supported on WO3−x nanowires as efficient catalysts for visible-light-enhanced hydrogen evolution from ammonia borane†
Xiao
Li
,
Yucong
Yan
,
Yi
Jiang
 ,
Xingqiao
Wu
,
Shi
Li
,
Jingbo
Huang
,
Junjie
Li
,
Yangfan
Lin
,
Deren
Yang
and
Hui
Zhang
*
,
Xingqiao
Wu
,
Shi
Li
,
Jingbo
Huang
,
Junjie
Li
,
Yangfan
Lin
,
Deren
Yang
and
Hui
Zhang
*
State Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, P. R. China. E-mail: msezhanghui@zju.edu.cn
First published on 21st August 2019
Abstract
Hydrolysis of ammonia borane (AB) is a safe and convenient means of H2 production when efficient catalysts are used. Here we report a facile one-pot solvothermal method to synthesize Rh/WO3−x hybrid nanowires. Ultra-small Rh nanoparticles with an average size of ∼1.7 nm were tightly anchored on WO3−x nanowires. Rh/WO3−x catalysts exhibited substantially enhanced activity for hydrolytic dehydrogenation of AB under both dark and visible light irradiation conditions relative to mixed Rh nanoparticles and WO3−x nanowires (Rh + WO3−x), and Rh/C and WO3−x nanowires. X-ray photoelectron spectroscopy (XPS) analysis indicated that the synergistic effect between Rh nanoparticles and WO3−x nanowires was responsible for such an enhancement in activity. Specifically, Rh/WO3−x achieved the highest turnover frequency (TOF) with a value of 805.0 molH2 molRh−1 min−1 at room temperature under visible light irradiation. The H2 release rate as a function of reaction time exhibited a volcano plot under visible light irradiation, indicating that a self-activation process occurred in the hydrolytic dehydrogenation of AB due to additional oxygen vacancies arising from in situ reduction of WO3−x nanowires by AB, and thus an enhanced localized surface plasmon resonance (LSPR). Such a self-activation process was responsible for the enhanced catalytic activity under visible light irradiation relative to that under dark conditions, which was supported by the lower activation energy (45.2 vs. 50.5 kJ mol−1). In addition, Rh/WO3−x catalysts were relatively stable with only little loss in activity after five cycles due to the tight attachment between two components.
Introduction
Hydrogen (H2) has been considered as one of the best alternative energy carriers with several promising advantages including high-energy density, environmental friendliness, and rich abundance compared with traditional fossil fuels.1–3 Based on the fuel-cell technique, it is easy to convert chemical energy of H2 to electrical energy through an electrochemical process for driving electronic devices and vehicles. However, the low density of H2 makes it hard to be liquefied and compressed, and thus brings about the difficulty of storage. To address this issue, it is essential to develop suitable hydrogen storage materials with secure and efficient features.4,5 Ammonia borane (AB), as the simplest B–N compound, possesses the advantages of high hydrogen capacity (19.6 wt%), low molecular weight (30.7 g mol−1), and high stability both in solid state and solution, thereby making it a highly attractive candidate for the storage of hydrogen.6–9 Generally, the hydrogen stored in AB can be released through two distinct ways: thermal dehydrogenation and hydrolysis. Compared to thermal dehydrogenation, the hydrolysis pathway is obviously more desirable due to its low energy consumption and high H2 selectivity and productivity.10–12 However, self-hydrolysis of AB is negligible at room temperature in the absence of proper catalysts. So far, lots of heterogeneous nanocatalysts consisting of noble13–15 and non-noble metals10,16,17 as well as their alloys18–21 and compounds22–24 have been developed to promote the hydrolysis of AB. Among these nanocatalysts, Rh-based catalysts have shown almost the highest activities for AB hydrolysis.13,25–28 In view of the high cost and scarce abundance associated with Rh, massive efforts have been devoted to enhance the catalytic properties of Rh-based catalysts and maximize the utilization efficiency of Rh.In heterogeneous catalysis, supports in a nanocatalyst might play a key role throughout the process.27,29–32 On one hand, supports could be utilized to disperse and downsize metal nanoparticles during the synthesis, and thus expose more active sites and lead to the enhancement in catalytic performances.15,25,26 On the other hand, coupling metal nanoparticles with supports could introduce additional synergistic effects, which intrinsically change the physical and chemical nature of the interface between them.13,19,33–35 Therefore, the catalytic activity and selectivity could be tuned by varying different supports. As a kind of typical transition metal oxide, tungsten oxide-based materials display fascinating performances, which result from the unique feature of the outer d valence electrons.24 Sub-stoichiometric tungsten oxides (WO3−x) with rich oxygen vacancies have been widely investigated in various fields, such as gas sensing,36 electrocatalysis,37 photocatalysis,38 electrochromism,39 lithium-ion batteries,40etc. In addition, some studies show that the catalytic performances of the metal nanoparticles could be enhanced when coupling them with WO3−x supports.41–43 Although there are some interesting reports about photocatalytic hydrogen evolution from AB, where other photoactive semiconductors are used to support metal nanoparticles,44,45 there are few reports on the preparation of noble metal/WO3−x hybrid nanocomposites and their application in this field.
Herein, we present a simple one-pot solvothermal method to synthesize Rh/WO3−x hybrid nanowires. The as-obtained Rh/WO3−x hybrid nanocatalysts display remarkable catalytic activity for AB hydrolytic dehydrogenation in the dark with further enhancement in catalytic activity under visible light irradiation. In addition, we find an interesting self-activation process during the catalytic reaction, which might be due to additional oxygen vacancies arising from the reduction of WO3−x nanowires by AB.
Experimental section
Chemicals and materials
Rhodium(II) acetate dimer (Rh2(OAc)4, 99.99%), polyvinylpyrrolidone (PVP, Mw ≈ 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), tungsten hexacarbonyl (W(CO)6, 97%), sodium hexachlororhodate(III) (Na3RhCl6) and sodium borohydride (NaBH4, ≥98.0%) were purchased from Sigma-Aldrich. Tungsten chloride (WCl6, 99%) and ammonia borane complex (AB, 97%) were purchased from Aladdin. Benzyl alcohol (BA, AR), ethanol (AR), and acetone (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Deionized water (18.2 MΩ cm−1) used in all experiments was prepared by passing through an ultra-pure purification system. All the chemicals were used as received without further purification.
000), tungsten hexacarbonyl (W(CO)6, 97%), sodium hexachlororhodate(III) (Na3RhCl6) and sodium borohydride (NaBH4, ≥98.0%) were purchased from Sigma-Aldrich. Tungsten chloride (WCl6, 99%) and ammonia borane complex (AB, 97%) were purchased from Aladdin. Benzyl alcohol (BA, AR), ethanol (AR), and acetone (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Deionized water (18.2 MΩ cm−1) used in all experiments was prepared by passing through an ultra-pure purification system. All the chemicals were used as received without further purification.
Synthesis of Rh/WO3−x hybrid nanowires
Rh/WO3−x hybrid nanowires were synthesized by a simple one-pot solvothermal method. In a standard procedure, 6 mg of Rh2(OAc)4 and 120 mg of PVP were dissolved in 10 mL of BA and stirred at room temperature for 1 h. The obtained homogeneous green solution and 100 mg of W(CO)6 were transferred and sealed in a 15 mL Teflon-lined stainless steel autoclave in an Ar atmosphere. After that, the autoclave was heated at 200 °C for 12 h and cooled at room temperature after the reaction. The final product was collected by centrifugation and washed three times with ethanol and deionized water. The catalysts (denoted as Rh/WO3−x-2) were re-dispersed in 5 mL of 10% ethanol solution and dried by freeze-drying. The loading of Rh in the catalysts was tuned by varying the amount of Rh2(OAc)4 to 3 mg (denoted as Rh/WO3−x-1) and 12 mg (denoted as Rh/WO3−x-3), respectively, under otherwise identical experimental conditions.Synthesis of Rh nanoparticles
19 mg of Na3RhCl6 and 555 mg of PVP were dissolved in 8.75 mL of deionized water. The mixture was further stirred for 30 min at 0 °C. After that, an aqueous solution of NaBH4 (4.75 mg of NaBH4, 1.25 mL) was rapidly injected into the mixture under vigorous stirring. Then the mixture was kept stirring for 5 min. The final product was collected by centrifugation, washed two times with acetone and ethanol, and then re-dispersed in 1 mL deionized water.Synthesis of WO3−x nanowires
WO3−x nanowires were synthesized according to a previous report with slight modification.46 150 mg of WCl6 was dissolved in 10 mL of ethanol to form a transparent solution, which was then mixed with 30 mL of aqueous solution containing 250 mg of PVP in a beaker (50 mL) under vigorous stirring. After 3 min, the solution was transferred into a 50 mL Teflon-lined stainless steel autoclave. After that, the autoclave was heated at 200 °C for 4 h and cooled at room temperature after the reaction. The final product was collected by centrifugation and washed three times with ethanol.Characterization
Transmission electron microscopy (TEM) images were obtained with a Hitachi HT-7700 microscope operated at 100 kV. High-resolution transmission electron microscopy (HRTEM) was performed using a FEI Tecnai G2 F20 microscope operated at 200 kV. High-angle annular dark-field scanning TEM (HAADF-STEM) was conducted on an FEI Titan ChemiSTEM operated at 200 kV. X-ray powder diffraction (XRD) patterns were recorded on a Rigaku D/max-ga X-ray diffractometer with graphite monochromatic Cu Kα radiation (λ = 1.54178 Å). X-ray photoelectron spectroscopy (XPS) analysis was performed on a scanning X-ray microprobe (Axis Supra, Kratos Inc.) with Al Kα radiation. The corresponding binding energies were calibrated with a C–C 1s peak of 284.5 eV. The absorption spectra of the samples were recorded on an ultraviolet-visible-near infrared (UV-vis-NIR) spectrophotometer (Agilent, Cary 5000). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was performed using an IRIS Intrepid II XSP (TJA Co.).Hydrolytic dehydrogenation of AB
The catalytic reaction was carried out in a three-necked flask (25 mL) containing a stir bar connected to a gas buret. The flask was placed in a water bath at 25 °C on a magnetic stirrer under visible light irradiation (λ > 400 nm, Xe lamp equipped with a 400 nm cutoff filter, 300 W, CEL-HXUV300, Beijing China Education Au-light Co., Ltd). In a typical procedure, 4 mL of the catalyst suspension with 6.86 × 10−4 mmol of Rh atoms was added to the flask under magnetic stirring. The reaction was started when 1 mL of aqueous solution containing 26 mg of AB was added to the flask. In all the catalytic reactions, the atomic ratio of Rh to AB was fixed at 0.084%. The volume of H2 was measured through the water-displacement method by calculating the volume of drained water. The catalytic reaction was completed when no bubble was observed. After that, another 1 mL of AB aqueous solution was further added to the flask to investigate the durability of the catalysts. This step was repeated five times. The above-mentioned reaction was also performed in the absence of the Xe lamp (defined as dark condition).Calculation methods
Since the Rh nanoparticles were well dispersed on supports, all the metal species in the Rh nanoparticles could be considered as active sites in the catalytic reaction. Therefore, the turnover frequency (TOF) was calculated from the equation below:where TOF is the turnover frequency, Patm is the atmospheric pressure (101.325 kPa), Vgas is the volume of the generated H2, R is the universal gas constant (8.314 m3 Pa mol−1 K−1), T is the temperature (298 K), nRh is the moles of metal species in the catalyst and t is the total reaction time in minutes.
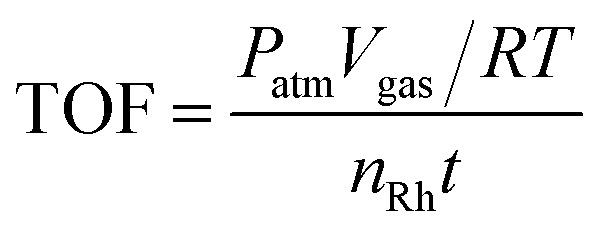
Results and discussion
The Rh/WO3−x hybrid nanowires were synthesized by a simple one-pot solvothermal approach in BA containing Rh2(OAc)4, W(CO)6, and PVP. Fig. 1 shows the morphological and structural characterization of the sample (Rh/WO3−x-2) prepared using the standard procedure by adding 6 mg of Rh2(OAc)4. From the TEM image in Fig. 1a, the sample exhibited a wire-like shape with average lengths of hundreds of nanometers and diameters of several nanometers. Careful observation shows that a rich variety of small Rh nanoparticles were anchored on the surface of the WO3−x nanowires to form the hybrid Rh/WO3−x nanostructures. Such hybrid nanowires were further confirmed by HAADF-STEM characterization, as shown in Fig. 1b. From size statistics (Fig. S1†), the average diameter of Rh nanoparticles was calculated to be ca. 1.7 nm. In addition, the WO3−x nanowires tended to aggregate and form bundles, leading to an uneven diameter distribution. The coalescence of the nanowires could be attributed to their intrinsic oxygen vacancies and strong interactions among individual ones.46,47Fig. 1c shows the atomic-resolution HAADF-STEM image of an individual hybrid nanowire, indicating a highly crystalline nature. The fringes with a lattice spacing of 0.38 nm in the nanowire can be indexed to the {010} planes of monoclinic WO2.72 (JCPDS no. 05-0392). Such lattice fringes were perpendicular to the longitudinal axis of the nanowire, indicating that it grew along the 〈010〉 direction.48 The fringes with a lattice spacing of 0.22 nm in the nanoparticle correspond to the {111} planes of face-centered cubic (fcc) Rh (JCPDS no. 05-0685). Obviously, the Rh nanoparticles were tightly anchored on the WO3−x nanowires, leading to a possible synergistic effect between them in the catalytic reaction. The crystal structure of these hybrid nanowires was further characterized using the XRD technique, as shown in Fig. 1d. As observed, two obvious diffraction peaks can be assigned to the (010) and (020) planes of monoclinic WO2.72, which is consistent with the HAADF-STEM result (Fig. 1c). However, no obvious diffraction peaks associated with Rh were detected probably due to the small size of the nanoparticles and low loading of Rh. ICP-AES measurements indicated that the Rh loading in the hybrid nanowires reached approximately 1.22 wt%. | ||
| Fig. 1 (a) TEM image, (b) HAADF-STEM image, (c) atomic-resolution HAADF-STEM image, and (d) XRD pattern of Rh/WO3−x-2 hybrid nanowires prepared using the standard procedure by adding 6 mg of the Rh precursor. | ||
The loading of Rh in the hybrid nanowires was tuned by varying the amount of Rh2(OAc)4 to 3 and 12 mg. From the TEM images in Fig. S2a and c,† both samples (Rh/WO3−x-1 and Rh/WO3−x-3) were observed to exhibit a wire-like shape with well-dispersed nanoparticles being anchored on the surface. Obviously, the density of Rh nanoparticles on the hybrid nanowires increased with the amount of Rh precursors fed in the synthesis. The size statistics (Fig. S2b and d†) indicates that the average diameters of Rh nanoparticles in these two samples (Rh/WO3−x-1 and Rh/WO3−x-3) are close to 1.7 nm, which are similar to that of Rh nanoparticles in Rh/WO3−x-2. The size of WO3−x nanowires was also similar in the above-mentioned three samples. In addition, ICP-AES analysis showed that the Rh loading of Rh/WO3−x-1 and Rh/WO3−x-3 was 0.58 wt% and 2.55 wt%, respectively.
XPS measurements were carried out to investigate the electronic interaction between Rh nanoparticles and WO3−x nanowires. For comparison, Rh nanoparticles (Fig. S3†) and WO3−x nanowires (Fig. S4†) were synthesized (see the Experimental section for details). From the TEM image in Fig. S3,† the size of Rh nanoparticles reached 1.7 nm, which was similar to that of Rh nanoparticles in Rh/WO3−x. As shown in Fig. S4,† the as-synthesized WO3−x nanowires were the same as those in Rh/WO3−x both in morphology (Fig. S4a†) and crystal structure (Fig. S4b†). After that, we simply mixed the as-prepared Rh nanoparticles with a carbon support and WO3−x nanowires to form Rh/C and Rh + WO3−x catalysts, respectively. The morphological (Fig. S5a and S6a†) and structural (Fig. S5b and S6b†) characterization indicates that Rh nanoparticles are uniformly dispersed on the supports with lattice fringes of the Rh {111} planes being clearly observed. Due to the similar structure of Rh/WO3−x-2 to Rh/WO3−x-1 and Rh/WO3−x-3, only the XPS spectra of Rh/WO3−x-2 are shown here for simplicity. Compared to the Rh 3d XPS spectrum of Rh/C, the Rh 3d peak for Rh/WO3−x-2 shifts to a lower binding energy by 0.2 eV, as shown in Fig. 2a. However, there is almost no shift of the Rh 3d peak for Rh + WO3−x catalysts. This result indicates the increase in the surface electron density on Rh nanoparticles once they are coupled with WO3−x nanowires by direct synthesis instead of a simple mixture.46 On the other hand, the binding energy of W 4f in Rh/WO3−x-2 is positively shifted (Fig. 2b) compared with that of the WO3−x nanowires and Rh + WO3−x, further confirming the strong electronic interactions between Rh nanoparticles and WO3−x nanowires in Rh/WO3−x-2. In addition, the W 4f XPS spectra demonstrated that W species showed two oxidation states and the W 4f peaks could be fitted into two doublets: W5+ and W6+ species. WO3−x (WO2.72) and Rh have different work functions, which are 4.50 and 4.98 eV, respectively.49 So when Rh nanoparticles were coupled with WO3−x nanowires, electrons would transfer from WO3−x to Rh, leading to a negative charge state on Rh nanoparticles. Therefore, such an electron-rich state will benefit the electron transfer from Rh to the antibonding orbital of H2O molecules, and thus accelerate hydrolytic dehydrogenation of AB by forming activated transition state complexes.50
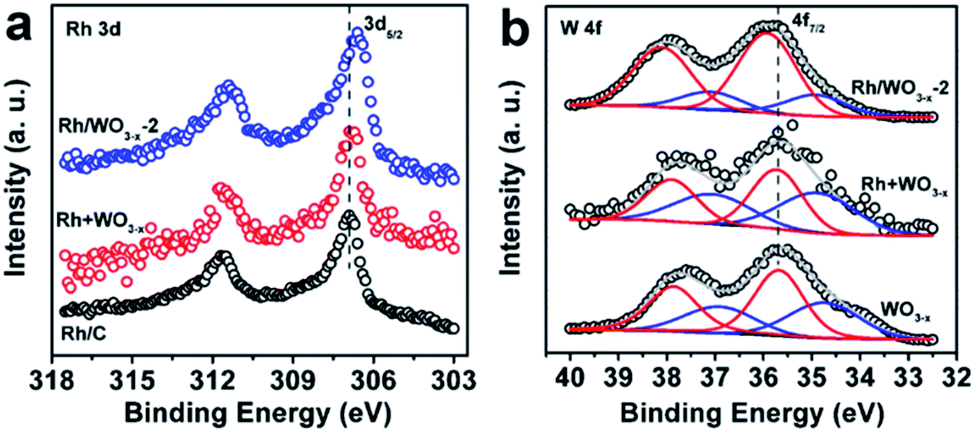 | ||
| Fig. 2 XPS spectra of (a) Rh 3d orbitals for Rh/C, Rh + WO3−x, and Rh/WO3−x-2 catalysts and (b) W 4f orbitals for WO3−x nanowires, Rh + WO3−x, and Rh/WO3−x-2 catalysts. | ||
Rh/WO3−x hybrid nanowires with different Rh loadings were then evaluated as catalysts for hydrolytic dehydrogenation of AB with WO3−x nanowires, Rh + WO3−x and Rh/C as the references. Rh/WO3−x-1, Rh/WO3−x-2 and Rh/WO3−x-3 were initially tested for the hydrolysis of AB under dark conditions. As shown in Fig. S7,† these three catalysts exhibited high catalytic activities for the dehydrogenation of AB with a similar TOF approaching 500 molH2 molRh−1 min−1. This result could be attributed to the same amount of Rh being involved in the catalytic reaction. So we chose Rh/WO3−x-2 with an appropriate Rh loading as the representative example for the following research on the catalytic properties and then we called it Rh/WO3−x for simplification. Fig. 3 shows a comparison of the catalytic properties of Rh/WO3−x, Rh + WO3−x, Rh/C, and WO3−x nanowires in the dark and under visible light irradiation (λ > 400 nm), respectively. Obviously, WO3−x nanowires were inactive for the dehydrogenation of AB under both conditions. As can be seen from Fig. 3a and c, Rh/WO3−x exhibited a much rapider H2 release rate and higher TOF value compared to those of Rh + WO3−x and Rh/C in the dark, implying higher activity for hydrolytic dehydrogenation of AB. Simultaneously, the total amount of produced H2 for Rh/WO3−x was ∼3.0 equivalent per mole of AB added, suggesting complete H2 conversion in the catalytic reaction. However, the dehydrogenation of AB catalyzed by Rh + WO3−x or Rh/C was incomplete since the amount of produced H2 was less than ∼3.0 equivalent per mole of AB. The incomplete dehydrogenation may arise from B-containing species poisoning during the catalytic process. According to XPS analysis (Fig. 2), Rh nanoparticles would be negatively charged in Rh/WO3−x compared with those in Rh/C and Rh + WO3−x. Such an electron-rich state not only accelerates the formation of activated transition state complexes, but also may promote the desorption of BO2− anions that were often regarded as poisoning active sites.51 However, Rh nanoparticles in Rh/C and Rh + WO3−x are vulnerable to BO2− anions, which may cause incomplete dehydrogenation.
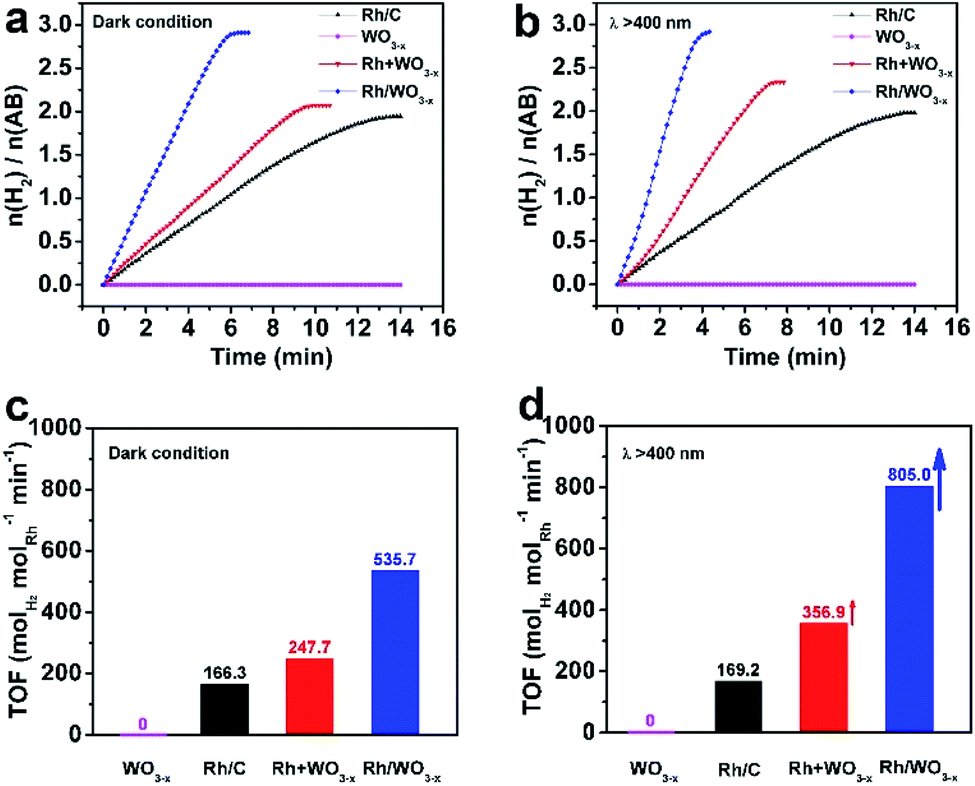 | ||
| Fig. 3 Plots of time versus volume of hydrogen generated from the catalytic hydrolysis of AB over different catalysts including Rh/WO3−x, Rh + WO3−x, Rh/C, and WO3−x nanowires (a) in the dark and (b) under visible light irradiation at a reaction temperature of 298 K. Their corresponding TOF values achieved (c) in the dark and (d) under visible light irradiation. | ||
It is well-known that the photocatalytic efficiency is highly related to light absorption of catalysts. As such, the UV-vis-NIR absorption spectra of Rh/WO3−x and WO3−x nanowires were measured and are shown in Fig. S8.† In addition to strong valence-to-conduction band transition absorption in the UV region, WO3−x nanowires also showed broad absorption in the visible and near infrared regions due to the presence of oxygen vacancies in the crystal structure.52,53 After deposition of Rh nanoparticles, Rh/WO3−x still exhibited strong adsorption in the detected regions, especially in the UV and visible regions due to localized surface plasmon resonance (LSPR) associated with Rh nanoparticles and WO3−x nanowires as well as strong coupling between them. This distinct adsorption feature endowed Rh/WO3−x with possibly enhanced catalytic properties for hydrolytic dehydrogenation of AB with the help of visible light irradiation. Fig. 3b shows H2 generation from the hydrolysis of AB catalyzed by such catalysts as a function of reaction time under visible light irradiation. It can be seen that Rh/WO3−x still showed a much higher H2 release rate compared to Rh + WO3−x and Rh/C. Interestingly, both Rh/WO3−x and Rh + WO3−x displayed improved catalytic activities for hydrolytic dehydrogenation of AB under visible light irradiation relative to that under dark conditions. However, there was no obvious enhancement by using Rh/C as catalysts in the absence of WO3−x, indicating the important role of WO3−x in photo-assisted hydrolytic dehydrogenation of AB. Specifically, the TOF value of Rh/WO3−x under visible light irradiation reached 805.0 molH2 molRh−1 min−1, which is remarkably higher than that of Rh + WO3−x (356.9 molH2 molRh−1 min−1) and Rh/C (169.2 molH2 molRh−1 min−1).
The temperature-dependent reaction kinetics for hydrolytic dehydrogenation of AB catalyzed by Rh/WO3−x was further investigated in the range of 298–328 K under dark conditions and visible light irradiation, as shown in Fig. S9a and c,† respectively. Clearly, the hydrogen generation rate increased with the reaction temperature under both conditions. Fig. S9b and d† display the corresponding Arrhenius plots of lnTOF vs. 1/T, from which the activation energy Ea could be calculated to be 50.5 kJ mol−1 in the dark and 45.2 kJ mol−1 under visible light irradiation, respectively. This decrease in Ea under visible light irradiation suggested that visible light irradiation might provide a favorable transition state with a lower energy barrier. To further understand the enhancement in the photocatalytic activities of the catalysts, the dependence of the light intensity on the catalytic H2 evolution was investigated. We adjusted the power of the Xe lamp from 33.3% to 66.7% and 100%. The results showed that increasing the light intensity led to an increase in the H2 evolution rate of AB and the corresponding TOF values (Fig. S10a and b†). These results indicated that more photogenerated electrons were excited from WO3−x nanowires under visible-light irradiation with a higher intensity and then injected into Rh nanoparticles, which resulted in the enhanced electron density of the supported Rh nanoparticles and the corresponding enhanced catalytic activities. Scheme 1 shows the possible reaction mechanism for the enhancement in catalytic activity towards AB hydrolytic dehydrogenation under visible light irradiation. When visible light is used to irradiate Rh/WO3−x catalysts, the WO3−x nanowires would generate hot electrons through LSPR. Thanks to the tight attachment of Rh nanoparticles to WO3−x nanowires, the hot electrons would rapidly transfer to the Rh nanoparticles through the interface between them. Lots of new discrete energy levels below the conduction band of WO3−x semiconductor nanowires were generated by the introduction of oxygen vacancies,54 and metallic Rh nanoparticles were the electron trapping centers, which effectively decreased the recombination of hot electrons. As such, visible light irradiation could bring more electrons to the surface of Rh nanoparticles, and thus facilitate hydrolytic dehydrogenation of AB.
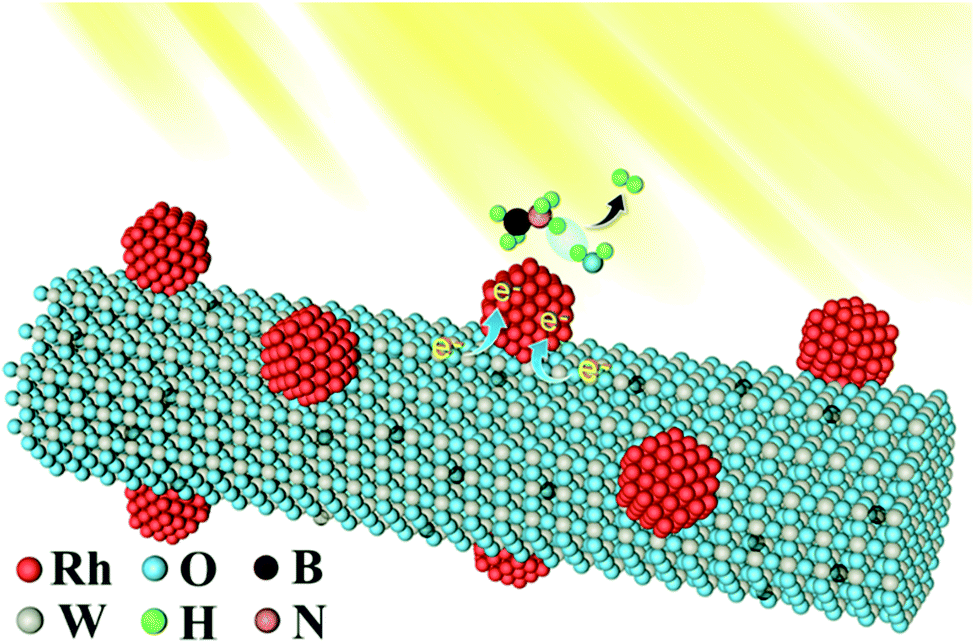 | ||
| Scheme 1 Schematic illustration of the Rh/WO3−x hybrid nanowire catalysts for visible-light-enhanced hydrogen evolution. | ||
Interestingly, we noticed that the color of the Rh/WO3−x suspension changed from yellow-green to dark blue during the hydrolytic dehydrogenation of AB (Fig. S11a and b†). After the catalytic reaction finished, the color of the Rh/WO3−x suspension would remain dark blue if the flask was kept sealed. Once the Rh/WO3−x suspension was exposed to the air for a little while, its color would turn back to yellow-green quickly, as shown in Fig. S11c.† According to a previous report,55 the color of substoichiometric WO3−x gradually turned more blue with the increase of oxygen vacancies. As such, we can speculate that WO3−x nanowires might be in situ reduced by AB during the hydrolytic dehydrogenation, leading to the increase in oxygen vacancies. In addition, the differential of the H2 production volume V to H2 generation time t (dV/dt) was calculated and the corresponding plot of dV/dt vs. t is shown in Fig. S12.† In this plot, dV/dt can be considered as the H2 release rate. As observed, the H2 release rate remains stable for the first few minutes and then gradually decreases under dark conditions, which can be attributed to the decrease in the concentration of AB over reaction time. Upon visible light irradiation, however, the H2 release rate rises first and then falls, showing a volcano plot. This result indicates that a self-activation process occurred during hydrolytic dehydrogenation of AB only under visible light irradiation. The color change of the Rh/WO3−x suspension demonstrated the generation of additional oxygen vacancies during the catalytic reaction, leading to the formation of more discrete energy levels below the conduction band of WO3−x and thus the enhanced LSPR intensity.56,57 As such, much more hot electrons were generated from LSPR under visible light irradiation, and then injected into tightly coupled Rh nanoparticles. Such additional hot electrons further facilitated the hydrolytic dehydrogenation of AB, causing the self-activation process.
The stability of the Rh/WO3−x catalyst was also evaluated since it is another critical parameter for practical application. Reusability tests were conducted under visible light irradiation at room temperature by continuous addition of a new proportion of AB aqueous solution when the previous run was completed. As shown in Fig. 4, the catalyst can be reused at least five times and the total time for completing the hydrolytic dehydrogenation of AB gradually increases slightly. Then we further prolonged the reusability tests to ten times, and the corresponding time vs. volume plots and TOF values of each cycle are shown in Fig. S13a and b.† After five cycles, the Rh/WO3−x catalysts retain 29.7% of the initial TOF value, which is similar to a previous report,13 but only 19.1% of the initial TOF value was retained after ten cycles. There are some possible reasons for such a decrease in the activity during cycling: (1) reactants were diluted in solution with the increase of the cycling number, (2) the viscidity of the solution also increased with the number of cycles,58 and (3) the catalyst surface was covered with the hydrolysis product metaborate that had the deactivation effect.29,51 In addition, the catalyst was characterized after the fifth run by TEM (Fig. S14a†) and XRD (Fig. S15a†), showing unchanged morphology and phase of the catalyst. The corresponding size distribution of Rh nanoparticles (Fig. S14b†) indicated the slight increase of their size with the number of cycles. XPS analysis (Fig. S15b†) showed that the Rh 3d peak shifts to a higher binding energy by 0.1 eV after the fifth run, which indicated the decrease in the surface electrons on Rh nanoparticles and weakness of the interaction between Rh nanoparticles and WO3−x. It might be part of the reasons for activity degradation.
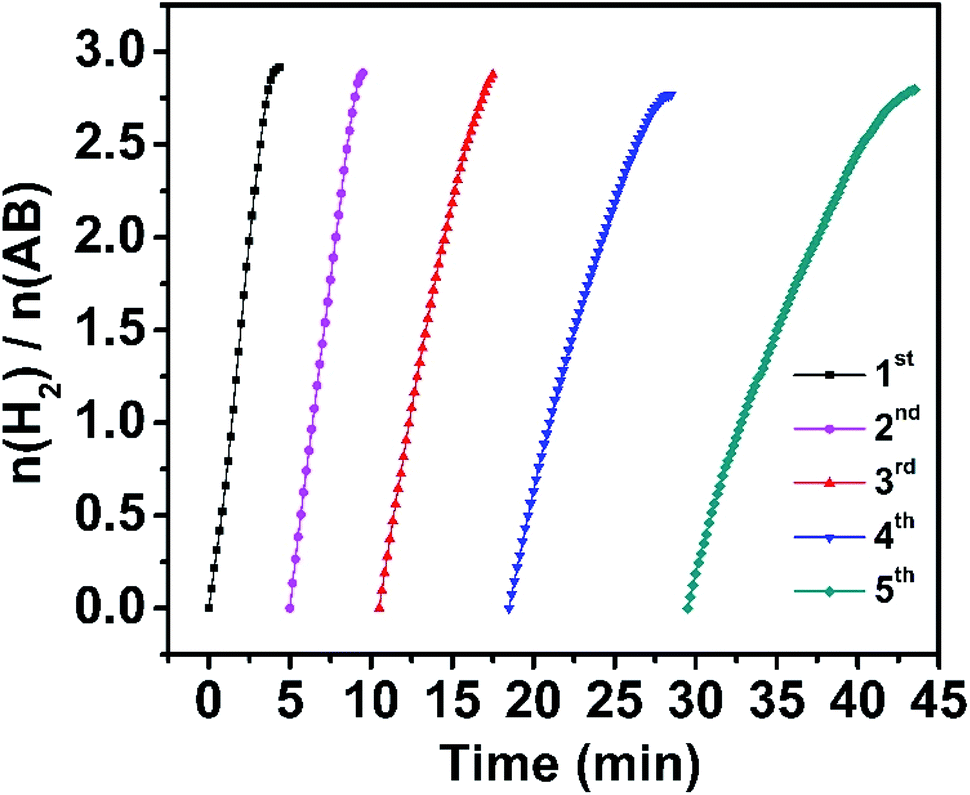 | ||
| Fig. 4 Plots of time versus volume of hydrogen generated from the hydrolysis of AB catalyzed by Rh/WO3−x for five cycles. | ||
Conclusions
In summary, we have successfully synthesized Rh/WO3−x hybrid nanowires by a one-pot solvothermal method, in which ultra-small Rh nanoparticles were tightly anchored on WO3−x nanowires. Thanks to the synergistic effect between Rh nanoparticles and WO3−x nanowires, Rh/WO3−x catalysts exhibited substantially enhanced activity for hydrolytic dehydrogenation of AB under dark conditions compared to Rh + WO3−x, Rh/C and WO3−x nanowires. Benefiting from the excellent visible-light harvesting ability of the WO3−x nanowires, the activity of Rh/WO3−x catalysts was further improved with a TOF as high as 805.0 molH2 molRh−1 min−1 and an apparent activation energy of 45.2 kJ mol−1. In addition, an interesting self-activation process was observed during the hydrolytic dehydrogenation of AB under visible light irradiation, which can be attributed to additional oxygen vacancies arising from in situ reduction of WO3−x nanowires by AB. Rh/WO3−x catalysts were also relatively stable with only little loss in activity after five cycles due to the tight hybrid nanostructures. This work offers a promising strategy to design highly efficient heterogeneous metal-oxide photocatalysts for AB hydrolytic dehydrogenation.Conflicts of interest
There are no conflict to declare.Acknowledgements
The work on electron microscopy was carried out in the Center for Electron Microscopy of Zhejiang University. This work was supported by the National Science Foundation of China (51522103, 51871200, and 61721005) and the National Program for Support of Top-notch Young Professionals.Notes and references
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS.
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS.
- J. Turner, Science, 1999, 285, 687–689 CrossRef CAS.
- L. Schlapbach, Nature, 2009, 460, 809–811 CrossRef CAS.
- Q. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 RSC.
- A. Gutowska, L. Li, Y. Shin, C. Wang, X. Li, J. Linehan, R. Smith, B. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS.
- Z. Xiong, C. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. Jones, S. Johnson, P. Edwards and W. David, Nat. Mater., 2008, 7, 138–141 CrossRef CAS.
- C. Hamilton, R. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- W. Zhan, Q. Zhu and Q. Xu, ACS Catal., 2016, 6, 6892–6905 CrossRef CAS.
- Ö. Metin, V. Mazumder, S. Özkar and S. Sun, J. Am. Chem. Soc., 2010, 132, 1468–1469 CrossRef.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC.
- U. Demirci and P. Miele, Energy Environ. Sci., 2011, 4, 3334–3341 RSC.
- S. Akbayrak, Y. Tonbul and S. Özkar, Appl. Catal., B, 2016, 198, 162–170 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2007, 168, 135–142 CrossRef CAS.
- L. Zhang, Y. Wang, J. Li, X. Ren, H. Lv, X. Su, Y. Hu, D. Xu and B. Liu, ChemCatChem, 2018, 10, 1–8 CrossRef.
- J. Yan, X. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287–2289 CrossRef CAS.
- M. Shen, H. Liu, C. Yu, Z. Yin, M. Muzzio, J. Li, Z. Xi, Y. Yu and S. Sun, J. Am. Chem. Soc., 2018, 140, 16460–16463 CrossRef CAS.
- F. Fu, C. Wang, Q. Wang, A. Martinez-Villacorta, A. Escobar, H. Chong, X. Wang, S. Moya, L. Salmon, E. Fouquet, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2018, 140, 10034–10042 CrossRef CAS.
- K. Mori, K. Miyawaki and H. Yamashita, ACS Catal., 2016, 6, 3128–3135 CrossRef CAS.
- Q. Wang, F. Fu, S. Yang, M. Moro, M. Ramirez, S. Moya, L. Salmon, J. Ruiz and D. Astruc, ACS Catal., 2019, 9, 1110–1119 CrossRef CAS.
- H. Zhang, X. Gu, P. Liu, J. Song, J. Cheng and H. Su, J. Mater. Chem. A, 2017, 5, 2288–2296 RSC.
- C. Peng, L. Kang, S. Cao, Y. Chen, Z. Lin and W. Fu, Angew. Chem., Int. Ed., 2015, 54, 15725–15729 CrossRef CAS.
- C. Hou, Q. Li, C. Wang, C. Peng, Q. Chen, H. Ye, W. Fu, C. Che, N. López and Y. Chen, Energy Environ. Sci., 2017, 10, 1770–1776 RSC.
- Y. Lou, J. He, G. Liu, S. Qi, L. Cheng, J. Chen, Y. Zhao and J. Zhu, Chem. Commun., 2018, 54, 6188–6191 RSC.
- Q. Yao, Z. Lu, Y. Jia, X. Chen and X. Liu, Int. J. Hydrogen Energy, 2015, 40, 2207–2215 CrossRef CAS.
- R. Lu, M. Hu, C. Xu, Y. Wang, Y. Zhang, B. Xu, D. Gao, J. Bi and G. Fan, Int. J. Hydrogen Energy, 2018, 43, 7038–7045 CrossRef CAS.
- L. Wang, H. Li, W. Zhang, X. Zhao, J. Qiu, A. Li, X. Zheng, Z. Hu, R. Si and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 4712–4718 CrossRef CAS.
- H. Zou, B. Jin, R. Wang, Y. Wu, H. Yang and S. Qiu, J. Mater. Chem. A, 2018, 6, 24166–24174 RSC.
- Q. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS.
- J. S. Du, T. Bian, J. Yu, Y. Jiang, X. Wang, Y. Yan, Y. Jiang, C. Jin, H. Zhang and D. Yang, Adv. Sci., 2017, 4, 1700056 CrossRef.
- Y. Jiang, X. Wu, Y. Yan, S. Luo, X. Li, J. Huang, H. Zhang and D. Yang, Small, 2019, 15, 1805474 CrossRef.
- Y. Jiang, Y. Yan, Y. Han, H. Zhang and D. Yang, RSC Adv., 2017, 7, 43373–43379 RSC.
- Q. Yao, Z. Lu, Y. Yang, Y. Chen, X. Chen and H. Jiang, Nano Res., 2018, 11, 4412–4422 CrossRef CAS.
- M. Khalily, H. Eren, S. Akbayrak, H. Susapto, N. Biyikli, S. Özkar and M. Guler, Angew. Chem., Int. Ed., 2016, 55, 12257–12261 CrossRef CAS.
- Y. Ge, W. Ye, Z. H. Shah, X. Lin, R. Lu and S. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 3749–3756 CrossRef CAS.
- A. Ponzoni, E. Comini, G. Sberveglieri, J. Zhou, S. Deng, N. Xu, Y. Ding and Z. Wang, Appl. Phys. Lett., 2006, 88, 203101 CrossRef.
- T. Zheng, W. Sang, Z. He, Q. Wei, B. Chen, H. Li, C. Cao, R. Huang, X. Yan, B. Pan, S. Zhou and J. Zeng, Nano Lett., 2017, 17, 7968–7973 CrossRef CAS.
- J. Liu, O. Margeat, W. Dachraoui, X. Liu, M. Fahlman and J. Ackermann, Adv. Funct. Mater., 2014, 24, 6029–6037 CrossRef CAS.
- J. Liu, J. Zheng, J. Wang, J. Xu, H. Li and S. Yu, Nano Lett., 2013, 13, 3589–3593 CrossRef.
- K. Bao, W. Mao, G. Liu, L. Ye, H. Xie, S. Ji, D. Wang, C. Chen and Y. Li, Nano Res., 2017, 10, 1903–1911 CrossRef CAS.
- Z. Xi, D. Erdosy, A. Mendoza-Garcia, P. Duchesne, J. Li, M. Muzzio, Q. Li, P. Zhang and S. Sun, Nano Lett., 2017, 17, 2727–2731 CrossRef CAS.
- Y. Lu, Y. Jiang, X. Gao, X. Wang and W. Chen, J. Am. Chem. Soc., 2014, 136, 11687–11697 CrossRef CAS.
- F. Li, H. Gong, Y. Wang, H. Zhang, Y. Wang, S. Liu, S. Wang and C. Sun, J. Mater. Chem. A, 2014, 2, 20154–20163 RSC.
- J. Song, X. Gu, J. Cheng, N. Fan, H. Zhang and H. Su, Appl. Catal., B, 2018, 225, 424–432 CrossRef CAS.
- J. Song, X. Gu, Y. Cao and H. Zhang, J. Mater. Chem. A, 2019, 7, 10543–10551 RSC.
- H. Zhang, C. Huang, R. Tao, Y. Zhao, S. Chen, Z. Sun and Z. Liu, J. Mater. Chem., 2012, 22, 3354–3359 RSC.
- X. Lou and H. Zeng, Inorg. Chem., 2003, 42, 6169–6171 CrossRef CAS.
- G. Xi, S. Ouyang, P. Li, J. Ye, Q. Ma, N. Su, H. Bai and C. Wang, Angew. Chem., Int. Ed., 2012, 51, 2395–2399 CrossRef CAS.
- F. Liu, F. Mo, S. Jin, L. Li, Z. Chen, R. Sun, J. Chen, S. Deng and N. Xu, Nanoscale, 2011, 3, 1850–1854 RSC.
- Z. Li, T. He, L. Liu, W. Chen, M. Zhang, G. Wu and P. Chen, Chem. Sci., 2017, 8, 781–788 RSC.
- W. Chen, J. Ji, X. Feng, X. Duan, G. Qian, P. Li, X. Zhou, D. Chen and W. Yuan, J. Am. Chem. Soc., 2014, 136, 16736–16739 CrossRef CAS.
- Y. Zhang, Y. Shi, R. Chen, L. Tao, C. Xie, D. Liu, D. Yan and S. Wang, J. Mater. Chem. A, 2018, 6, 23028–23033 RSC.
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef CAS.
- J. Yan, T. Wang, G. Wu, W. Dai, N. Guan, L. Li and J. Gong, Adv. Mater., 2015, 27, 1580–1586 CrossRef CAS.
- G. Wang, Y. Ling, H. Wang, X. Yang, C. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2012, 5, 6180–6187 RSC.
- H. Bai, W. Yi, J. Liu, Q. Lv, Q. Zhang, Q. Ma, H. Yang and G. Xi, Nanoscale, 2016, 8, 13545–13551 RSC.
- K. Manthiram and A. Alivisatos, J. Am. Chem. Soc., 2012, 134, 3995–3998 CrossRef CAS.
- U. Sanyal, U. Demirci, B. Jagirdar and P. Miele, ChemSusChem, 2011, 4, 1731–1739 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00424f |
| This journal is © The Royal Society of Chemistry 2019 |