
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of supramolecular self-assembly of an antimicrobial designer peptide by single amino acid substitution: implications on peptide activity†
Zhou
Ye
 and
Conrado
Aparicio
*
and
Conrado
Aparicio
*
MDRCBB, Minnesota Dental Research Center for Biomaterials and Biomechanics, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: apari003@umn.edu
First published on 31st October 2019
Abstract
Hydrophobicity and charge are key properties of antimicrobial peptides (AMPs). We compared the self-assembly performance and its correlation with antimicrobial activity of a designer AMP and analogues with substitution of hydrophobic or cationic residues by alanine. Peptides that formed supramolecular self-assemblies under the studied conditions were those that have higher antimicrobial potency.
Antimicrobial peptides (AMPs) have evolved greatly in the past two decades due to the increasing bacterial resistance to conventional antibiotics.1 Hydrophobicity and charge are important variables in natural and synthetic AMPs. Natural AMPs have a wide range of hydrophobicity and charge, but most frequently contain 40–60% hydrophobic residues and +4 to +6 charges.2 A good balance between hydrophobic and cationic residues in AMPs has been correlated to their strong antimicrobial activity.3–7 Despite evidence for the relevance of hydrophobicity and charge in AMPs activity, the mechanisms are still not well understood. Studies have suggested relations between AMPs permeability into bacterial membrane, prevention of peptide aggregation and interaction with hydrophobic cores of bacterial membranes.3–5,7
One main reason for the difficulty of studying AMPs antimicrobial mechanisms is the discovery of thousands of AMPs with high diversity in distribution of hydrophobic and charged residues along the amino acid sequence. Unlike the complexity inherent to this diversity, some short amphipathic peptides provide simpler models of AMPs to study the role of inter- and intramolecular hydrophobic interactions, electrostatic interactions and hydrogen bonding in peptide structure–activity correlations. These peptides typically contain a hydrophobic end and a charged end or repeated blocks of hydrophobic and charged residues. As a result, they readily self-assemble to supramolecular nanostructures in aqueous solutions. The size and shape of the self-assembled structures are highly affected by the peptide's hydrophobic and electrostatic interactions.8,9 Due to the more complex amino acid sequences, AMPs usually do not self-assemble to supramolecular structures in physiological saline or buffer solutions at neutral pH like it is the case for short surfactant-like peptides. However, previous work showed that AMPs can form supramolecular structures and change secondary structures triggered by increasing the solution pH or in proximity to negatively charged phospholipid vesicles or bacteria membranes.6,10,11 Moreover, similar to a number of AMPs, surfactant-like peptides are also toxic to bacteria by rupturing the bacterial membrane.12
With similarities in amphipathicity, cationicity, supramolecular self-assembly and antimicrobial activity; approaches used to study the self-assembly of short surfactant-like peptides can be applied to the study of AMPs. Thus, a better understanding of the role of hydrophobic and electrostatic interactions can be achieved by studying the structural (i.e. molecular secondary structure and self-assembly) and functional (i.e. antimicrobial activity) relationship in AMPs. However, previous studies with AMPs often focused on the antimicrobial activity without addressing AMPs structural properties in relation to their supramolecular self-assembly. We aimed to understand how single amino acid substitution with alanine of charged or hydrophobic residues modulated supramolecular self-assembly of a designer AMP and its correlation with peptides antimicrobial activity.
We modified the designer peptide GL13K by substituting one single cationic residue (i.e. lysine) or one very hydrophobic residue (i.e. leucine or isoleucine) to a nonpolar residue with moderate hydrophobicity (i.e. alanine). We named the peptide analogues as A3, A5, A6, A7, A10, A11 and A13 with the number in the name corresponding to the position of the alanine-substituted amino acid from the peptide N-terminus, as shown in Chart 1. As the unmodified GL13K has an alanine in position number 8, the A8 peptide here is GL13K. GL13K is a well-studied peptide derived from a human salivary protein BPIFA2 that efficiently kill multiple Gram-negative and Gram-positive bacteria.13–15 Gorr et al. have previously reported the effects of alanine substitutions on GL13K antimicrobial activity and its lipopolysaccharide binding activity.16,17
 | ||
| Chart 1 Molecular structure of GL13K. Cationic residues are marked in red and highly hydrophobic residues are marked in blue. Green rectangles mark amino acids that were substituted by alanine to obtain each of the peptide analogues. The name of each analogue peptide is under the corresponding substituted amino acid. | ||
The changes in secondary structure of GL13K and its analogues were assessed by circular dichroism (CD) for up to 5 days in a pH 9.6 borax-NaOH buffer, which is the lowest pH at which GL13K can change secondary structure at our experimental concentration (0.1 mM).6 Confirming our previous findings,6 at pH 9.6, GL13K peptides gradually rearranged over time from an unordered structure to one with large portions of β-sheet conformation as the peptides self-assembled to form twisted nanofibrils (Fig. 1a and 2a). We first substituted single lysines with one alanine to reduce the charge from +5 (GL13K has C-terminal amidation) to +4; that is, A5, A7 and A11. Similar to GL13K, all lysine → alanine analogues (A5, A7 and A11) showed secondary structure evolution that started sometime before being for 1 day in buffer solution (Fig. 1b–d). However, the changes and final secondary structures were not exactly the same among these three analogues. A7 had almost equivalent structural changes to those of GL13K with strong β-sheet CD signal and formation of strikingly similar self-assembled twisted nanofibrils (Fig. 1c and 2c). A5 also gradually changed the secondary structure to increasing content of β-sheets, but with slower dynamics and displaying lower ellipticity than A7 and GL13K (Fig. 1b). Moreover, the β-sheet characteristic peak shifted to a slightly larger wavelength (220 nm) compared to A7 and GL13K (218 nm). This suggested that the self-assembled nanostructures of A5 were moderately different from the ones from A7 and GL13K. We attributed the red-shift of the negative maximum (218–220 nm) to an increase of β-turn or strongly twisted β-sheet, which has been also observed by others.18 Accordingly, A5 not only formed nanofibrils but also showed molecular aggregation to form nanospheres as well as supramolecular aggregation of the twisted nanofibrils (Fig. 2b). The secondary structure of A11 peptides evolved differently than A5, A7 and GL13K as they changed to conformations with a high portion of α-helix from 1 day in solution (Fig. 1d). A11 formed nanofibrils that were notably shorter than those formed by GL13K and with high length variation (Fig. 2d). The A11 structures with high content in α-helix were not stable and denatured to negligible CD signal after 5 days incubation. As a result, we hardly found any supramolecular structures on the TEM grid (Fig. S1†). The different response of the three lysine → alanine analogues demonstrated the significant influence of the charged residue position on the self-assembly process for these peptides. These results highlight the notable effects that small changes in the charge distribution of the peptides had on their intermolecular interactions. Thus, a higher reduction in the number of cationic residues of these peptides will most likely inhibit their self-assembly to form supramolecular structures. For example, GL13-NH2, the natural peptide from which GL13K was derived, only has one cationic residue and maintained a highly unordered molecular structure for 5 days in buffer solution at pH 9.6 (Fig. S2†). This further suggests that a sufficient number of cationic residues is necessary for triggering self-assembly in this family of peptides.
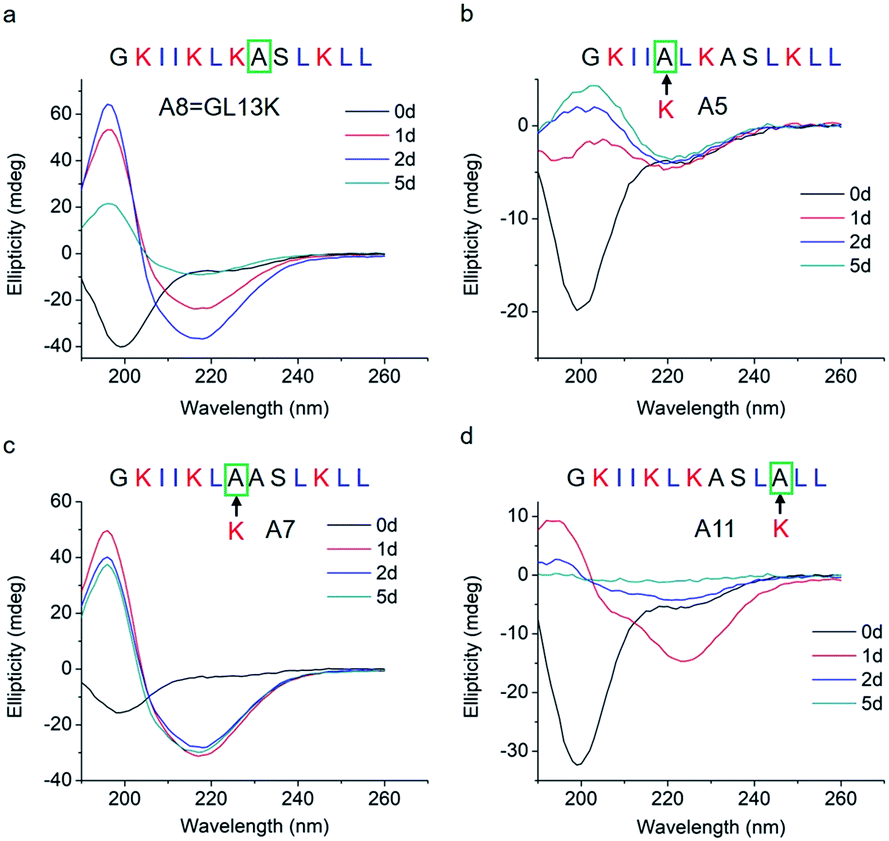 | ||
| Fig. 1 CD spectra of (a) GL13K and lysine → alanine analogue peptides (b) A5, (c) A7, and (d) A11. Arrows point to the amino acid substituted by alanine in each of the tested peptides. | ||
 | ||
| Fig. 2 TEM micrographs and corresponding CD spectra of GL13K and its lysine → alanine analogue peptides after 5 days of incubation for (a) GL13K, (b) A5 and (c) A7 or 1 day of incubation for (d) A11. Scale bars are 50 nm. | ||
To study how peptide hydrophobic interactions influence molecular self-assembly, the hydrophobicity of GL13K was slightly reduced in A3, A6, A10, and A13 peptide analogues by substituting a single leucine or isoleucine with an alanine; that is, a less hydrophobic amino acid. Notably, all four leucine/isoleucine → alanine analogues maintained unordered molecular structures for the whole experimental period (Fig. 3). Accordingly, we did not find self-assembled supramolecular structures with any of these peptides in the buffer solution (pH 9.6). All these peptides had the same number and distribution of cationic residues as GL13K; therefore, changes in hydrophobic interactions seem to have a larger effect than electrostatic interactions on the intermolecular interactions of GL13K as they all resulted in inhibition of molecular self-assembly for these analogues.
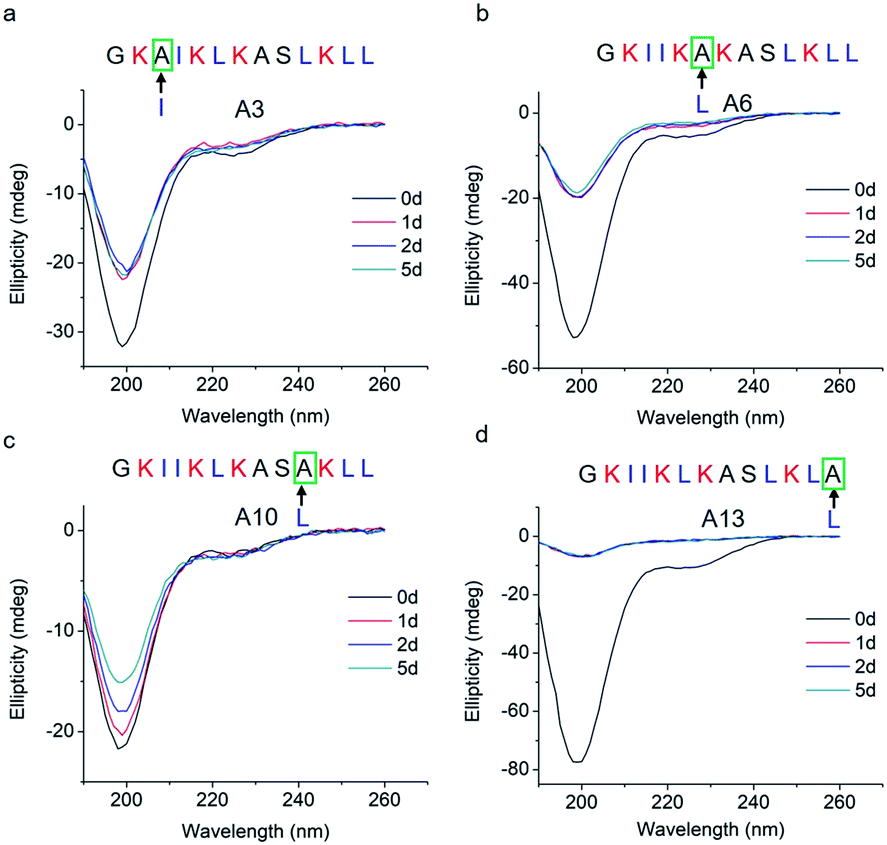 | ||
| Fig. 3 CD spectra of GL13K leucine/isoleucine → alanine analogues. Arrows point to the amino acid substituted by alanine in each of the tested peptides. | ||
Thus, a single modification of charged or hydrophobic amino acids in GL13K altered or inhibited, respectively the ability to the peptides to self-assemble into supramolecular structures. H. Hirt et al.16 assessed that the same GL13K analogues tested here had different killing potencies against Pseudomonas aeruginosa PAO1. Correlations between peptides charge, hydrophobicity and antimicrobial activity were discussed. However, potential relations between structural properties of the peptides (secondary structure, supramolecular self-assembly) and their activity were unmentioned. Here, to analyse the correlations between the ability of GL13K and analogue peptides to form self-assembled structures with their antimicrobial potency, we estimated the percentages of secondary structures (unordered, β-sheet, β-turn and α-helix) of these peptides and used the published values of minimum inhibition concentration (MIC) against P. aeruginosa PAO1.16 The percentages of secondary structures were estimated with the CD spectra after 1 day incubation using the CDPro software (https://sites.bmb.colostate.edu/sreeram/CDPro/CDPro.htm, last accessed April 7, 2019). Peptides with low percentage of unordered structure (<50%), which corresponded to those that self-assembled into supramolecular structures (GL13K = A8 and lysine → alanine analogue peptides; i.e., A5, A7 and A11), had low MICs (≤32 μg ml−1); that is, high antimicrobial potency. Contrarily, peptides with high percentage of unordered structure (>50%), which did not self-assemble (GL13K leucine/isoleucine → alanine peptide analogues with leucine or isoleucine substitution: A3, A6, A10 and A13), had higher MICs (≥64 μg ml−1, Fig. 4a); that is low antimicrobial activity. High percentage of β-sheet was related to peptides that formed self-assembled twisted nanofibrils and all had low MICs (Fig. 2a–c and 4b). The only other analogue peptide with relatively low MIC was A11, which had high α-helix instead of β-sheet percentage. Some studies also correlated the α-helical structure and antibacterial potency of AMPs, but did not discuss the role of supramolecular self-assembly.19,20 Additionally, GL13-NH2 peptides did not self-assembled and had low antimicrobial activity.13 Together, these results suggest that formation of supramolecular self-assembled structures in alkaline buffer solutions, irrespective of the dominant secondary structure in the molecular assemblies, was correlated with high antimicrobial potency.
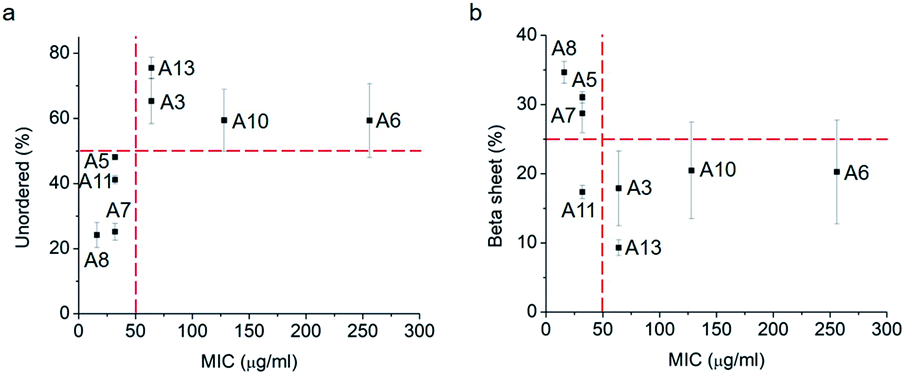 | ||
| Fig. 4 Secondary structure ((a), unordered; (b), β-sheet) percentage vs. MIC against P. aeruginosa PAO1 of GL13K (A8) and its analogues. Secondary structures were estimated from CD spectra after 1 day incubation and averaged by three methods, SELCON, CDSSTR and CONTIN/LL. | ||
The relationship between secondary structure composition and antimicrobial activity was also observed in other well studied AMPs, including 1018, DJK2 and DJK5.21 These AMPs had relatively high percentage of β-sheet in alkaline buffer solutions and presented low MICs (1–2 μg ml−1) against multispecies oral bacteria. On the contrary, other peptides that did not have antimicrobial potency (hLf1-11, L-GL13K-R and D-GL13K-R) also had lower percentage of α-helix and β-sheet. Interestingly, all these peptides have four cationic residues and similar hydrophobicity and hydrophobic moment but present different structural and functional properties with equivalent correlations between these properties to the ones studied here. The secondary structure change of AMPs was also observed in physiological solutions with phospholipid vesicles10 or bacteria,11 where part of the AMPs charge was neutralized by negatively charged phospholipids or bacterial membrane components. This indicates that peptide self-assembly might also occur when cationic AMPs are in proximity to other negatively charged substrates, such as vesicles or bacterial membranes. However, there is no direct evidence to differentiate whether the antimicrobial activity of the peptides is affected by the changes in secondary structure or the changes in the peptide sequences, irrespective of any actual change in assembled states. Further exploration of the intrinsic relationship between the AMPs self-assembly and antimicrobial activity can improve our understanding of bacteria killing mechanisms of AMPs.
The sensitivity of antimicrobial activity of AMPs analogues with minor modifications was also observed in other peptide families, such as replacement of highly hydrophobic residues (i.e. leucine) with less hydrophobic residues (i.e. alanine) in the AMP V13KL. Y. Chen et al. demonstrated an optimal window where more or less hydrophobicity beyond an specific threshold dramatically decreased the antimicrobial activity of those peptides.4 Moreover, the percentage of hydrophobic residues and the hydrophobic residue distribution had effect on the total hydrophobicity and activity of AMPs. By comparing two magainin analogues with identical amino acid composition and similar hydrophobic moments, T. Tachi et al. observed stronger pore formation on zwitterionic phospholipids and erythrocytes with the analogue of higher experimental hydrophobicity.5 Similarly, our previous work demonstrated the significant decrease of antimicrobial activity of a GL13K analogue obtained by randomization of the amino acid sequence.6 Furthermore, the number and position of cationic residues in the amino acid sequence of the peptides also determine the activity of AMPs. For example, M. Dathe et al. changed the charges of magainin II amide analogues between +3 and +7, showing the enhanced antimicrobial activity up to +5 charge.7 The established correlation between antimicrobial activity of AMPs and their ability to form self-assembled structures provides a new insight into the potential mechanism of antimicrobial activity and how this is significantly influenced by hydrophobicity and cationicity of the AMPs.
In conclusion, hydrophobicity and charge highly influenced the molecular self-assembly of the AMP GL13K and its analogue peptides. The peptides that formed supramolecular self-assemblies under studied conditions were those that had higher antimicrobial potency. This findings can lead to a better understanding of the antimicrobial mechanisms of GL13K and other similar AMPs as well as guide the design of highly efficient AMPs with optimal hydrophobic and cationic residue number and distribution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr Sven-U. Gorr, University of Minnesota for generously donating all GL13K analogues and fruitful discussions about the antimicrobial activity of GL13K peptides. We also thank Erik Skoe for language editing of the manuscript. This work was supported by the National Institute for Dental and Craniofacial Research of the National Institutes of Health [grant number R01DE026117 to C. A.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Parts of this work were carried out in the University of Minnesota I.T. Characterization Facility, which receives partial support from NSF through the MRSEC program.Notes and references
- J. L. Fox, Nat. Biotechnol., 2013, 31, 379–382 CrossRef CAS PubMed.
- A. Giangaspero, L. Sandri and A. Tossi, Eur. J. Biochem., 2001, 268, 5589–5600 CrossRef CAS PubMed.
- L. M. Yin, M. A. Edwards, J. Li, C. M. Yip and C. M. Deber, J. Biol. Chem., 2012, 287, 7738–7745 CrossRef CAS PubMed.
- Y. Chen, M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant and R. S. Hodges, Antimicrob. Agents Chemother., 2007, 51, 1398–1406 CrossRef CAS PubMed.
- T. Tachi, R. F. Epand, R. M. Epand and K. Matsuzaki, Biochemistry, 2002, 41, 10723–10731 CrossRef CAS PubMed.
- Z. Ye, X. Zhu, S. Acosta, D. Kumar, T. Sang and C. Aparicio, Nanoscale, 2019, 11, 266–275 RSC.
- M. Dathe, H. Nikolenko, J. Meyer, M. Beyermann and M. Bienert, FEBS Lett., 2001, 501, 146–150 CrossRef CAS PubMed.
- S. Han, S. Cao, Y. Wang, J. Wang, D. Xia, H. Xu, X. Zhao and J. R. Lu, Chem.–Eur. J., 2011, 17, 13095–13102 CrossRef CAS PubMed.
- H. Xu, J. Wang, S. Han, J. Wang, D. Yu, H. Zhang, D. Xia, X. Zhao, T. A. Waigh and J. R. Lu, Langmuir, 2009, 25, 4115–4123 CrossRef CAS PubMed.
- N. Harmouche, C. Aisenbrey, F. Porcelli, Y. Xia, S. E. D. Nelson, X. Chen, J. Raya, L. Vermeer, C. Aparicio, G. Veglia, S. U. Gorr and B. Bechinger, Biochemistry, 2017, 56, 4269–4278 CrossRef CAS PubMed.
- C. Avitabile, L. D. D'Andrea and A. Romanelli, Sci. Rep., 2014, 4, 4293 CrossRef PubMed.
- C. Chen, J. Hu, S. Zhang, P. Zhou, X. Zhao, H. Xu, X. Zhao, M. Yaseen and J. R. Lu, Biomaterials, 2012, 33, 592–603 CrossRef CAS PubMed.
- M. Abdolhosseini, S. R. Nandula, J. Song, H. Hirt and S. U. Gorr, Peptides, 2012, 35, 231–238 CrossRef CAS PubMed.
- K. V. Holmberg, M. Abdolhosseini, Y. Li, X. Chen, S. U. Gorr and C. Aparicio, Acta Biomater., 2013, 9, 8224–8231 CrossRef CAS PubMed.
- X. Chen, H. Hirt, Y. Li, S. U. Gorr and C. Aparicio, PLoS One, 2014, 9, 11579 Search PubMed.
- H. Hirt and S. U. Gorr, Antimicrob. Agents Chemother., 2013, 57, 4903–4910 CrossRef CAS PubMed.
- M. Abdolhosseini, S. R. Nandula, J. Song, H. Hirt and S. U. Gorr, Peptides, 2012, 35, 231–238 CrossRef CAS PubMed.
- A. Iyer, S. J. Roeters, V. Kogan, S. Woutersen, M. M. A. E. Claessens and V. Subramaniam, J. Am. Chem. Soc., 2017, 139, 15392–15400 CrossRef CAS PubMed.
- N. Wiradharma, U. Khoe, C. A. E. Hauser, S. V. Seow, S. Zhang and Y. Y. Yang, Biomaterials, 2011, 32(8), 2204–2212 CrossRef CAS PubMed.
- D. T. Yucesoy, M. Hnilova, K. Boone, P. M. Arnold, M. L. Snead and C. Tamerler, JOM, 2015, 67(4), 754–766 CrossRef CAS PubMed.
- D. G. Moussa, J. A. Kirihara, Z. Ye, N. G. Fischer, J. Khot, B. A. Witthuhn and C. Aparicio, J. Dent. Res., 2019, 98(10), 1112–1121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00498j |
| This journal is © The Royal Society of Chemistry 2019 |