
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroduction of Mn(III) to regulate the electronic structure of fluorine-doped nickel hydroxide for efficient water oxidation†
Jiaqi
Lv
a,
Xiaoxuan
Yang
a,
Ke
Li
a,
Xinyu
Chen
a,
Sai
Sun
a,
Hong-Ying
Zang
 *a,
Ying-Fei
Chang
*b,
Yong-Hui
Wang
a and
Yang-Guang
Li
*a
*a,
Ying-Fei
Chang
*b,
Yong-Hui
Wang
a and
Yang-Guang
Li
*a
aKey Lab of Polyoxometalate, Science of Ministry of Education, Key Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Institute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, Jilin, P. R. China. E-mail: zanghy100@nenu.edu.cn; liyg658@nenu.edu.cn
bInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, Jilin, P. R. China. E-mail: changyf299@nenu.edu.cn
First published on 2nd September 2019
Abstract
OER is the key step to increase the rate of water-splitting reaction. Design and construction of appropriate defects is an effective strategy to enhance catalytic activity. Mn has stronger e−–e− repulsion by the local influence of its 3d orbital electrons. When Mn(III) was successfully introduced into two dimensional F-doped Ni(OH)2, it can tune the surface electronic structure of the F-doped Ni(OH)2 to increase its oxygen deficiency content. In this work, the as-synthesized Mn and F co-doped Ni(OH)2–NF on Ni foam (Mn–F/Ni(OH)2–NF) shows remarkable oxygen evolution performance, exhibiting 233 mV overpotential at 20 mA cm−2, and the Tafel slope is 56.9 mV dec−1 in 1 M KOH. The performance is better than that of the same loading of IrO2 on Ni foam. Density functional theory (DFT) calculations further show that the introduction of oxygen defects can significantly improve the OER catalytic performance of Mn–F/Ni(OH)2–NF.
Introduction
Since the first industrial revolution, the consumption of traditional fossil fuels has exacerbated, leading to the greenhouse effect and melting of glaciers.1–5 It is urgent to find new energy sources to alleviate the existing crisis.6,7 Hydrogen energy is environment-friendly and efficient.8–10 Therefore, it is important to study the water-splitting reaction which includes two parts: hydrogen evolution reaction (HER) and oxygen evolution reaction (OER). The multi-step electron transfer process of the oxygen evolution reaction is the rate-determining step which controls the overall rate of the reaction.11–14 Catalysts currently used as benchmarks for water oxidation are traditional noble metal oxides such as IrO2, RuO2, etc.15–18 which show excellent OER catalytic activity. However, they are expensive, scarce and not stable in alkaline electrolyte solution,19,20 which limits their use to some extent. Therefore, it is necessary to find a novel catalyst with abundant yield, good stability and low price.Previous investigations have shown that transition metal hydroxides have good OER activity in alkaline solution, especially nickel hydroxide, whose structure is similar to layered hydrotalcite.21,22 The structure can provide electronic channels and facilitate electron transport,23 which makes it suitable as an electrocatalytic material. But its structure is unstable in acid electrolyte solution during long-term operation and conductivity is not very good.24 Some researchers suggest that the introduction of anions (–N, –B, –S and –P) or defects can improve the OER activity of hydroxides and stabilize their structure.25–30
Recently, Ishizaki31et al. reported that F can combine with ionic and semi-ionic C to change the charge transfer rate between F and C atoms, thereby increasing the conductivity and tuning the electronic properties of the original carbon. Then the effect further improved the ORR catalytic activity. Zhang et al. found that the introduction of F− into NiFe hydroxide can improve its catalytic activity for hydrogen evolution reaction,32 and then studied the effect of F− on the OER catalytic activity of NiFe hydroxide containing F− (NiFe–OH–F), discovering that NiFe–OH–F only needs an overpotential of 243 mV to reach 10 mA cm−2 in alkaline media.33 Meanwhile, 3D Co(OH)F reported by Cao's group34 and recent work of our group35 confirmed that the F− anion can increase the OER activity of hydroxide to a certain extent. So far, there are few studies about the impact of F− on OER, because of the limited choice of F-source materials, lower amounts of F-doping, and the lack of safe and rapid doping methods. All the above factors limit research on F. Therefore, it is very important to find a reasonable way to introduce F.
In general, the introduction of trace amounts of foreign atoms into a composite material can increase the catalytic activity of the material.36,37 The catalytic activity of transition metal manganese has been receiving much attention. In 1977, Morita38et al. began to study the oxygen evolution activity of manganese oxides in alkaline solution. Song39et al. confirmed that the introduction of Mn into a material has a significant effect on the improvement of the OER activity. Zhao40et al. studied NiMn-LDH/CNT and found that its catalytic activity is higher than that of nickel hydroxide. At the same time, the material NiMn-LDH/CNT has a relatively high energy density, which proves that Mn can be a promising OER catalytic element.
The influence of oxygen vacancies on the catalytic activity of materials has attracted attention. Yang41et al. reported that the amorphous and defect-rich CoMoS4 material was a good OER catalytic material. Gao42et al. proposed that holes in CoO can improve the catalytic activity of OER. Xu43et al. reported that oxygen holes in Co3O4 nanosheets can modulate the OER catalytic activity of metal oxides. We know that chemical reactions occurring on the surface of a catalyst are related to the surface active sites and defect sites of the material. This surface effect has always attracted research interest.44,45
Herein, we designed one type of nickel hydroxide/nickel foam composite material with rich oxygen vacancies, as an OER electrocatalyst (Scheme 1). First, we used a classical hydrothermal method to grow F-doped nickel hydroxide on nickel foam (F/Ni(OH)2–NF), and then introduced Mn(III) into the system to synthesize Mn and F co-doped nickel hydroxide/Ni foam composites (Mn–F/Ni(OH)2–NF). With the introduction of Mn(III), we found that the internal electronic structure of the F-doped Ni(OH)2 composite changed. This caused an increase of surface oxygen defect content, thus improving the intrinsic activity and current density of the nickel hydroxide material.46–49 Especially, the hierarchical morphology of Mn–F/Ni(OH)2–NF forming on the Ni foam is favorable for electron transport in the reaction process. In 1 M KOH solution, its OER activity is better than that of commercial IrO2–NF. The method of doping foreign atoms to generate oxygen defects on the surface of the hydroxide material by a simple hydrothermal reaction might be a new promising strategy that can be widely used for synthesizing other defect-rich hydroxide materials.
 | ||
| Scheme 1 Schematic representation of the morphology growth process of Mn–F/Ni(OH)2–NF. | ||
Experimental section
Chemicals and materials
All chemicals were used without further treatment: Ni(NO3)2·6H2O (≥90%, Sinopharm Chemical Reagent Co., Ltd.), Mn(NO3)2·4H2O (Bailingwei Technology Co., Ltd.), CH4N2O (≥99.0%, Sinopharm Chemical Reagent Co., Ltd), NH4F (≥98.5%, Tianjin Fuchen chemical reagents factory), and Ni foam (Changchun Sanbang Pharmaceutical Technology Co., Ltd.).Treatment of Ni foam
Nickel foam with a 2 cm × 2 cm size was ultrasonically washed with deionized water and ethanol for 10 min, and then the nickel foam was soaked in 5% HCl solution for 15 min with ultrasonication. Finally, the nickel foam was washed several times with deionized water and dried at 40 °C in a vacuum.Synthesis of Ni(OH)2–NF composites
0.262 g Ni(NO3)2·6H2O and 0.144 g urea were put into 80 mL deionized water and stirred well for 30 min. Then the solution was transferred into a 100 ml Teflon-lined reactor, and meanwhile the treated Ni foam was completely immersed in the solution. They were placed in a baking oven at 120 °C for 6 h and naturally cooled to room temperature.Synthesis of Mn–F/Ni(OH)2–NF composites
0.262 g Ni(NO3)2·6H2O, 0.0331 g (0.13 mmol) Mn(NO3)2·4H2O, 0.0196 g NH4F and 0.144 g urea were put into 80 mL deionized water, and then stirred at ambient temperature for 30 min. The mixture solution was transferred to a 100 ml Teflon-lined reactor, and meanwhile the treated Ni foam was completely immersed in the solution. The reactor was placed in an oven and kept at 120 °C for 6 h. Then the Teflon reactor was cooled naturally to room temperature. The Ni foam loaded with Mn–F/Ni(OH)2 was taken out and washed with deionized water, and then dried at 40 °C in a vacuum.Synthesis of F/Ni(OH)2–NF composites
The synthetic method is the same as the “Synthesis of Mn–F/Ni(OH)2–NF composites” without adding Mn(NO3)2·4H2O.Characterization
The composition and structure of the catalyst and its comparative samples were characterized using a Siemens Model D5005 X-ray diffractometer using Cu-Ka radiation. The morphology of Mn–F/Ni(OH)2–NF composites and their comparative samples were characterized by field emission scanning electron microscopy (SU-8010A SEM) and transmission electron microscopy (JEM-2100F TEM). Elemental valence changes of the catalysts were characterized by X-ray photoelectron spectroscopy (VG ESCALAB MKII).Electrochemical measurements
Electrochemical measurements of the three-electrode system were carried out in 1 M KOH solution (pH = 14) under a nitrogen-saturated atmosphere at room temperature using a CHI760E Electrochemical Workstation. Hg/HgO and a carbon rod were selected as the reference electrode and counter electrode, respectively. Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, Ni(OH)2–NF and IrO2–NF (1 cm × 1 cm, the loading was 2 mg cm−2) were used as working electrodes for electrochemical tests. The measurement results were converted to the corresponding reversible hydrogen electrode potential according to the following formula:| ERHE = EHg/HgO + 0.0592pH + EθHg/HgO |
At room temperature 25 °C, EθHg/HgO = 0.098 V, η = ERHE − 1.23, η is the overpotential. The linear sweep curves (LSV) of all the working electrodes were collected in the 0.1–1.1 V range at a sweep rate of 10 mV s−1. The LSV curve was compensated by 5% iR. All impedances (EIS) were measured at their own open circuit voltage. The frequency was 0.5–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz and the amplitude was 5 mV. The stability test was performed at a potential of 20 mA cm−2 for each catalytic material for 10 hours. The electrochemically active surface area (ECSA) was estimated by cyclic voltammetry (CV) at different sweep rates in the range of 0.94–1.14 V vs. RHE.
000 Hz and the amplitude was 5 mV. The stability test was performed at a potential of 20 mA cm−2 for each catalytic material for 10 hours. The electrochemically active surface area (ECSA) was estimated by cyclic voltammetry (CV) at different sweep rates in the range of 0.94–1.14 V vs. RHE.
As shown in Fig. 1a, we performed the XRD test on the Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, Ni(OH)2–NF and Ni foam. The black line is the XRD pattern of the base Ni foam, and the main peaks at 44.5°, 51.84° and 76.37° correspond to the nickel PDF#040850 card. The pink line is the XRD for Ni(OH)2–NF. The green line is the XRD spectrum of F-doped Ni(OH)2–NF (F/Ni(OH)2–NF). Except for the peak of Ni foam, other peaks are consistent with the PDF#380715 card of Ni(OH)2. There is no peak belonging to F, so the F in Ni(OH)2 exists in the amorphous form. The red line is the XRD spectrum of the Mn–F/Ni(OH)2–NF material. There are obvious peaks of nickel foam and Ni(OH)2 in the spectrum, but the peaks of Mn and F are not observed. This is because Mn and F are doped in the amorphous form in Ni(OH)2. We know that the radius of the Mn atom is larger than that of the nickel atom. So when Mn(III) is introduced into the F-doped Ni(OH)2, it will preferentially occupy the position of the nickel, causing a change in the crystallinity of F/Ni(OH)2. At the same time, we will find that the main peak positions of Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF and Ni(OH)2–NF are the same, but the peak intensities are different, which further demonstrates the effect of Mn and F doping on the crystallinity of nickel hydroxide. Fig. S1† shows the XRD spectra of Mn–F/Ni(OH)2–NF with different doping amounts of Mn; all the samples have similar peaks and crystallinity. Among them there are not any peaks of Mn and F, which means Mn and F occupy the Ni lattice of Ni(OH)2, but this does not affect the crystal structure of Ni(OH)2.41,50 We also notice a slightly positive shift in the XRD peak of the Mn–F/Ni(OH)2–NF material relative to F/Ni(OH)2–NF, and we believe that this is due to the exceptional doping of Mn.51
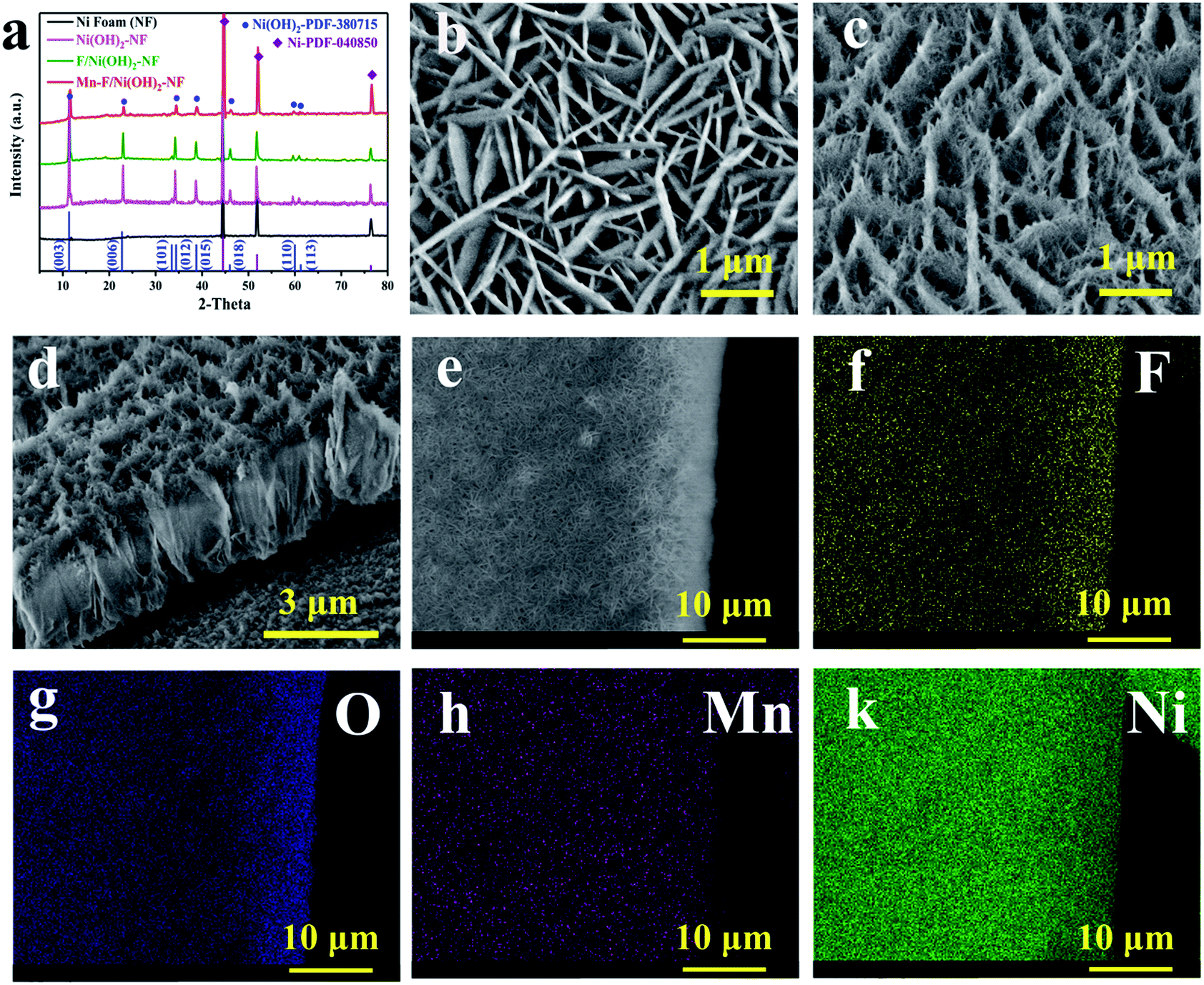 | ||
| Fig. 1 (a) XRD of Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, Ni(OH)2–NF and Ni foam. SEM images of (b) F/Ni(OH)2–NF and (c) Mn–F/Ni(OH)2–NF. (d) Side section view of Mn–F/Ni(OH)2–NF. (e) The element distribution test area. (f–i) The element distribution mapping of F, O, Mn and Ni. | ||
The scanning electron microscopy (SEM) image of F/Ni(OH)2–NF in Fig. 1b indicates that F/Ni(OH)2–NF is composed of uniform flakes which are mutually crisscrossed and grow vertically on the surface of the Ni foam. Fig. 1c shows the SEM image of Mn–F/Ni(OH)2–NF, which is a sheet-like structure that grows vertically on the surface of Ni foam, but each ‘sheet’ is stacked by filaments. Through the obvious contrast with the smooth sheet of F/Ni(OH)2–NF, it is shown that Mn is successfully doped into the F/Ni(OH)2–NF material and has an effect on the morphology of the composite material. The side cross-sectional view of Mn–F/Ni(OH)2–NF in Fig. 1d fully demonstrates the three-dimensional view of the uniform distribution of the hierarchical structure. In Fig. S2,† we show the SEM images of Mn–F/Ni(OH)2–NF with different magnifications. The results present that the overall morphology of the Mn–F/Ni(OH)2–NF is uniformly distributed.
In order to further judge the distribution of each element in Mn–F/Ni(OH)2–NF, we conducted an elemental mapping test on it. Fig. 1e shows the positions of the elements we selected. It can be seen from Fig. 1f–k that the elements F, O, Mn and Ni are uniformly distributed on the Mn–F/Ni(OH)2–NF, which further indicates that Mn(III) is uniformly doped into F/Ni(OH)2–NF.
At the same time, the Energy Dispersive X-ray spectra (EDX) of Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF are shown in Fig. S3.† Fig. S3a† shows the EDX spectrum of F/Ni(OH)2–NF. Fig. S3b† shows the EDX spectrum of Mn–F/Ni(OH)2–NF. O accounts for 34.48%, F accounts for 4.43%, Ni accounts for 59.87%, and Mn only accounts for 1.23% in the Mn–F/Ni(OH)2–NF sample. Fig. S4a† shows the TEM image of F/Ni(OH)2–NF which has a very pronounced sheet structure and clear boundaries. At the same time, since these TEM test samples are directly scraped off from the loaded nickel foam, they are relatively thin and light, and when the high-speed electrons hit the material, it will cause some deviation from the actual. Fig. S4b† shows the TEM image of Mn–F/Ni(OH)2–NF. In the angle of 20 nm view, we can only see the overlapping images due to the influence of a high number of electrons. TEM images of Mn–F/Ni(OH)2–NF with different relatively greater magnifications (Fig. S5†) show that the edges of the Mn–F/Ni(OH)2–NF are irregular and the morphology is similar to the interlacing of fibrous materials. Fig. S4c and d† show the high-resolution TEM images of F/Ni(OH)2–NF and Mn–F/Ni(OH)2–NF, and their lattice pitches are 0.2623 nm and 0.2675 nm, respectively, corresponding to the (012) (theoretical value: 0.2604 nm, PDF#38-0715) and (101) (theoretical value: 0.2676 nm, PDF#38-0715) crystal faces of Ni(OH)2. The lattice fringe spacing increases after the doping of Mn, indicating that Mn(III) is successfully doped into the lattice of Ni(OH)2. The results show that a small amount of doped Mn can significantly change the crystallinity of Mn–F/Ni(OH)2–NF and promote the lattice spacing of Mn–F/Ni(OH)2–NF growing along the (012) to (101) direction of Ni(OH)2. More precisely, the doping of Mn only leads to the crystal plane change, and the overall crystal form is still dominated by the Ni(OH)2 phase.
The structure of Ni(OH)2 is similar to layered double hydroxide (LDH) and is unstable. F atoms have higher electronegativity and can stabilize the structure of Ni(OH)2 when doped. Meanwhile, the existence of F will cause a little change in the internal structure of Ni(OH)2, leading to the loss of nearby oxygen atoms and then generating vacancies on the surface of F/Ni(OH)2. With the further introduction of Mn(III), the electron repulsive force inside the nickel hydroxide changes, and the oxygen defect content increases. We tested the oxygen defects of Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF by EPR tests as shown in Fig. 2a. A field signal of g = 2.00 indicates the presence of oxygen vacancies in Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF.52,53 It is obvious that the signal peak intensity of Mn–F/Ni(OH)2–NF is higher than that of F/Ni(OH)2–NF, which proves that Mn–F/Ni(OH)2–NF contains a higher number of oxygen defects. Thus the introduction of Mn(III) into the F/Ni(OH)2–NF can increase the content of oxygen defects, which is conducive to electrocatalytic oxygen evolution.
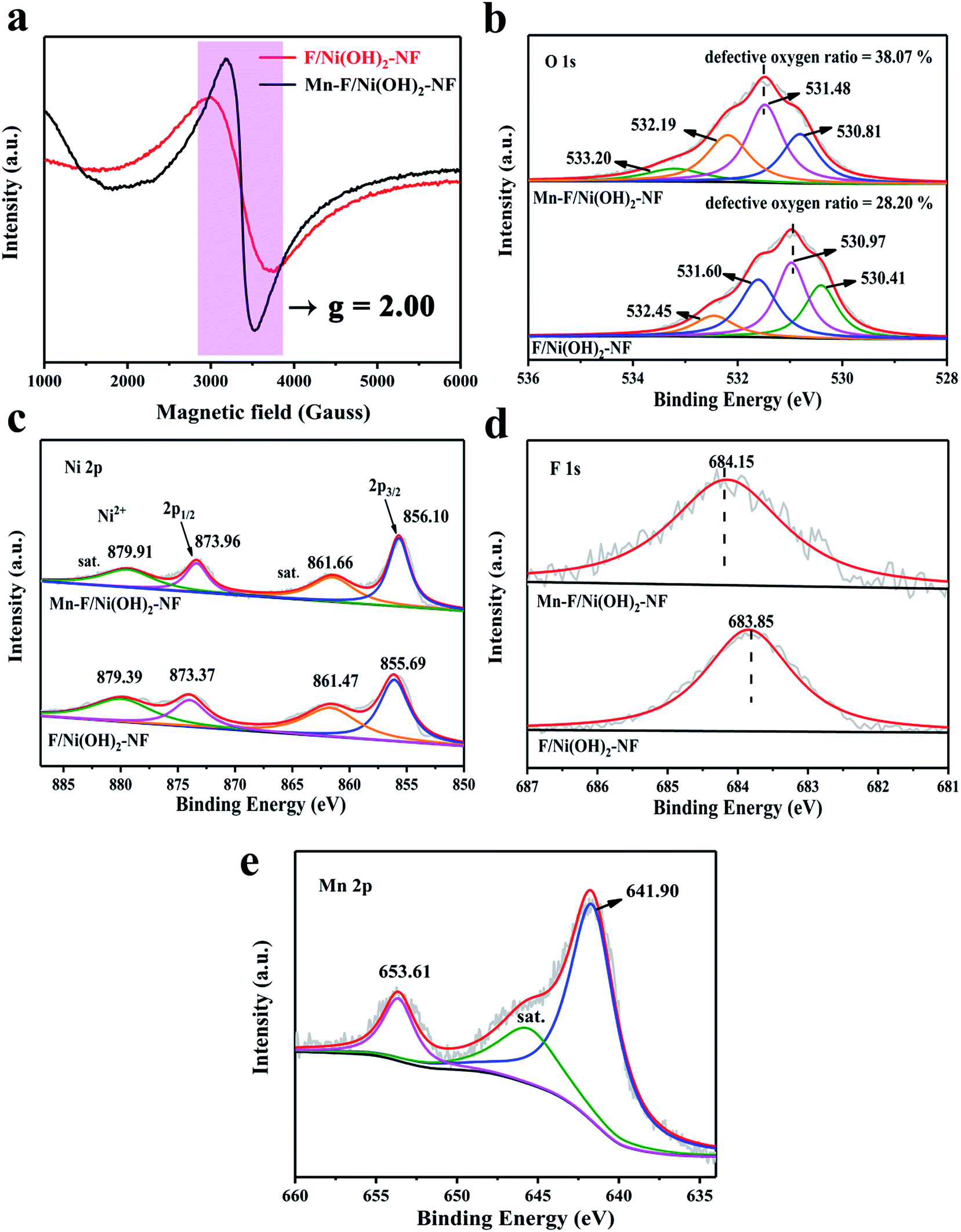 | ||
| Fig. 2 (a) Room temperature EPR spectra of Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF. (b) O 1s XPS spectra of Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF. (c–e) Ni 2p, F 1s and Mn 2p XPS peaks of Mn–F/Ni(OH)2–NF. | ||
XPS is a surface-sensitive spectroscopy technique that measures the chemical states of the elements and defects present in the catalysts. Fig. 2b shows the O XPS comparison between Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF, showing that the XPS peak of O shifts toward a high binding energy direction. Obviously, the O XPS spectrum of Mn–F/Ni(OH)2–NF is divided into four peaks. The peak of binding energy at 530.81 eV is attributed to the bond between metal and oxygen, which is recorded as O1. The peak at 531.48 eV is attributed to the defective oxygen, which is recorded as O2. The binding energy of 532.19 eV is assigned to hydroxyl oxygen or adsorbed oxygen (O3). The binding energy of 533.20 eV corresponds to the bond energy of oxygen (O4) in the adsorbed water molecules.54–56 Among them, the defective oxygen O2 accounted for 38.07%. From the O XPS spectrum of F/Ni(OH)2–NF, we can find that there is also a peak at 531.60 eV of defective oxygen, accounting for 28.20% of the total oxygen content. This indicates that F/Ni(OH)2–NF itself is a material containing oxygen defects, and with the doping of Mn, the surface oxygen defect content increases. The defect position is also called the unsaturated site. The existence of defects can increase the adsorption of water molecules and promote the OER activity. On the other hand, the defect can change the bonding energy in the composite material, which also explains why the main peaks of XPS of Ni, F and O move.
Fig. 2c shows the Ni XPS spectra of Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF. It is obvious to find that the position of Ni peaks for Mn–F/Ni(OH)2–NF has a slight positive shift compared to F/Ni(OH)2–NF. This is because the oxidizing ability of Ni2+ is stronger than that of Mn3+. When Mn(III) is doped into F/Ni(OH)2–NF, the ability to attract electrons is weakened, resulting in the loss of adjacent electrons and generation of defects. Meanwhile, the binding energy of elements becomes higher and the peaks shift. For Mn–F/Ni(OH)2–NF, the peaks at the binding energies of 856.10 and 873.96 eV correspond to Ni 2p3/2 and Ni 2p1/2, respectively. This shows that the Ni valence state here is Ni2+. The peaks at 861.66 and 879.91 eV are attributed to the satellite peak of Ni.49 Similarly, the binding energies of Ni 2p3/2 (855.69 eV) and Ni 2p1/2 (873.37 eV) are observed in the Ni XPS of F/Ni(OH)2–NF. And the peaks at binding energies of 861.47 and 879.39 eV are the satellite peaks of Ni.57Fig. 2d shows the F XPS spectra of Mn–F/Ni(OH)2–NF and F/Ni(OH)2–NF. It is clear that the binding energy of the main peak is shifted after doping with Mn. The binding energy at 684.15 eV of Mn–F/Ni(OH)2–NF is consistent with the binding energy of F to the metal hydroxyl group, indicating the formation of a bond between F and Ni(OH)2.34 So, when the peak of nickel moves, the binding energy of F also changes. The F 1s peak for F/Ni(OH)2–NF is the same as that of Mn–F/Ni(OH)2–NF. Fig. 2e shows the XPS spectrum of Mn for Mn–F/Ni(OH)2–NF. The peak at 645.59 eV is a small satellite peak. The peaks at the binding energies of 641.90 eV and 653.61 eV correspond to Mn 2p3/2 and Mn 2p1/2, respectively, indicating that Mn(III) is successfully incorporated into the F/Ni(OH)2 lattice.40
Electrochemical test performance of materials
Electrochemical tests were performed in a 1 M KOH solution using a standard three-electrode system. Carbon rods and Hg/HgO electrodes were selected as counter and reference electrodes, respectively. Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, Ni(OH)2–NF, IrO2–NF and empty nickel foam were used as the corresponding working electrodes. The size of nickel foam is 1 cm × 1 cm. The test results are shown in Fig. 3. Fig. 3a shows a linear scan curve. According to Fig. 3a, the linear scan curve shows that there is a nickel oxidation peak occurring between 1.35 and 1.65 V vs. RHE. For more obvious comparison, the simulation process for oxygen evolution during the nickel oxidation process is depicted by the dotted line. By comparison, we found that the OER catalytic activity of Mn–F/Ni(OH)2–NF was better than that of F/Ni(OH)2–NF, Ni(OH)2–NF, IrO2–NF and NF, and it could reach 1.463 V vs. RHE at 20 mA cm−2 and 1.567 V vs. RHE at 50 mA cm−2. This shows a good linear relationship between the anode current density and overpotential. Fig. 3b shows the corresponding Tafel slope. The Tafel slope of Mn–F/Ni(OH)2–NF is 56.9 mV dec−1, which is smaller than that of F/Ni(OH)2–NF (74.5 mV dec−1), Ni(OH)2–NF (78.5 mV dec−1), IrO2–NF (65.1 mV dec−1) and NF (153.0 mV dec−1). This further clearly explains the higher intrinsic catalytic activity of the Mn–F/Ni(OH)2–NF material.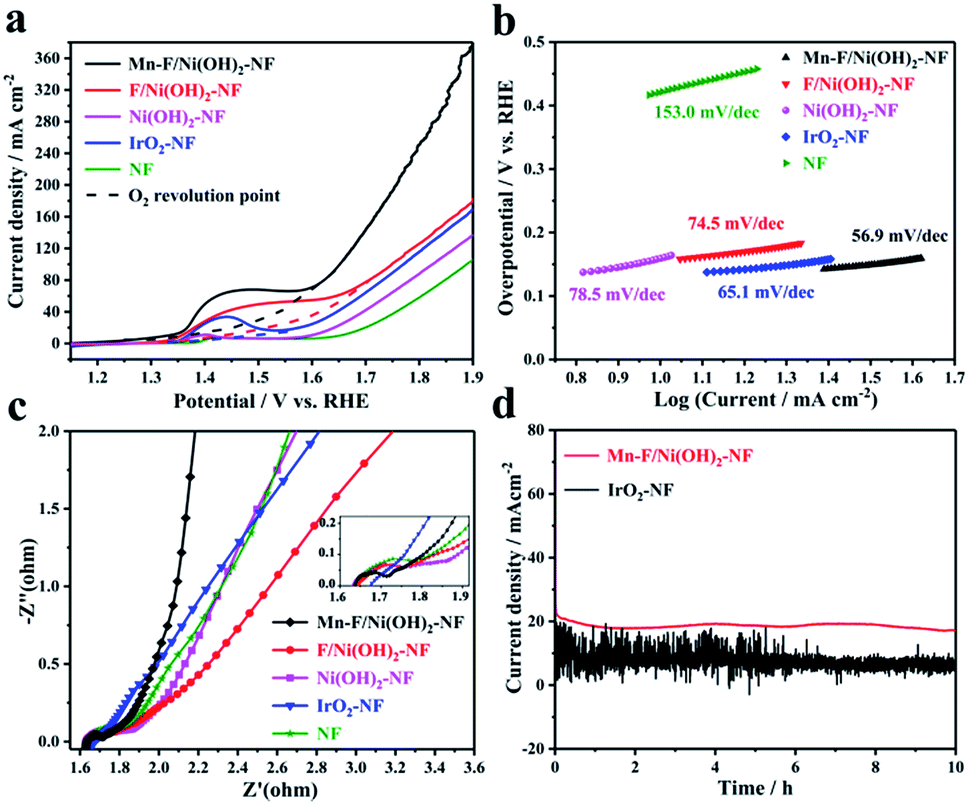 | ||
| Fig. 3 (a) LSV of Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, IrO2–NF, Ni(OH)2–NF and NF. (b) Corresponding Tafel slope of the materials. (c) Impedance spectra. (d) IT test of Mn–F/Ni(OH)2–NF and IrO2–NF at the potential corresponding to 20 mA cm−2. | ||
To further understand the reaction kinetics of OER, electrochemical impedance spectroscopy (EIS) was used to study the charge transfer process of materials in the alkaline medium at the electrode–electrolyte interface. Since IrO2 is coated on the Ni foam, its interfacial impedance is larger; as shown in Fig. 3c, the high frequency region has no semicircular shape. For Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, Ni(OH)2–NF and Ni foam, the impedance spectrum includes two parts, a line in a low frequency region and a semicircle in a high frequency region.58 The smaller the diameter of the semicircle in the high frequency region, the better the reaction rate of the material. It is found that the semicircular diameter of Mn–F/Ni(OH)2–NF is smaller than those of F/Ni(OH)2–NF, Ni(OH)2–NF and Ni foam. The charge transfer resistance Rct of Mn–F/Ni(OH)2–NF is described. The final fitting shows that the resistance values of Mn–F/Ni(OH)2–NF, F/Ni(OH)2–NF, Ni(OH)2–NF, NF and IrO2–NF are 1.70, 1.77, 1.81, 1.90, and 4.20 Ω, respectively.
The electrocatalyst's stability is a critical indicator to assess its electrochemical performance. The Mn–F/Ni(OH)2–NF and IrO2–NF shown in Fig. 3d were subjected to a stability test for 10 h at a potential corresponding to 20 mA cm−2. We can find that the current density of Mn–F/Ni(OH)2–NF is only slightly reduced and the curve is smoother. However, the line of IrO2–NF has a lot of sawtooth on the stability curve, and the current density of the curve is reduced more. This is because IrO2 is directly coated on the Ni foam, and the adhesion between IrO2 and Ni foam is limited. In the high current catalytic process of OER, IrO2 is easy to dissolve and fall off, and then the current decreases greatly. At the same time, the IrO2 catalyst coated on the Ni foam lacks a layered Ni(OH)2 structure and thus cannot transport the oxygen molecules on the surface in time, causing many bubbles on the surface. This is why the IT curve of IrO2–NF has a lot of jaggedness. These comparisons show that Mn–F/Ni(OH)2–NF has excellent stability and can be potentially applicable.
Double-layer capacitance (Cdl) was measured to roughly calculate the value of the electrochemically active surface area (ECSA), since the normalized activity of the geometric surface area of the electrode does not reflect the inherent activity of the catalyst.59 We selected the illegal range of the material and recorded the cyclic voltammogram of the capacitance current density as a function of the scan rate, thereby obtaining a Cdl that can be used to estimate ECSA. As shown in Fig. S6a,† we found that there is a linear relationship between Cdl and the active specific surface area. According to the cyclic voltammogram of Mn–F/Ni(OH)2–NF and IrO2–NF, the slope is plotted as Cdl. The Cdl of Mn–F/Ni(OH)2–NF is 4.656 mF cm−2, which is larger than the Cdl (1.507 mF cm−2) of IrO2–NF, indicating a larger active specific surface area (Fig. S6b†). The active area is consistent with the previous LSV test.
The LSV investigation of the Mn–F/Ni(OH)2–NF catalyst was conducted before and after the 10 h stability test. As shown in Fig. S7a,† we performed a simulation of oxygen evolution indicated by dotted lines at the position where nickel oxidation occurred. It is observed from the LSV that the catalytic activity of the Mn–F/Ni(OH)2–NF can be maintained up to the current density of 50 mA cm−2 before and after the reaction. Fig. S7b† shows a comparison of LSV before and after the stability test for IrO2–NF. The results show that the current drops significantly. This indicates that during this period of time, the catalyst partially detached due to the high current of the OER process, which is unavoidable. Relatively speaking, our catalyst can maintain a good current density at 50 mA cm−2. This is entirely due to the strong thermal interaction with the nickel foam substrate.
Density functional theory (DFT) calculations
The effect of Mn and F doping on the OER activity of Ni(OH)2 was studied by density functional theory (DFT). Fig. 4a shows the surface of NiOOH, which exposes four Ni5c, four O3c and four Ob atoms per unit cell built by Selloni and co-workers.60Fig. 4b shows the crystal structure of NiOOH. Fig. 4c shows the crystal structure of F/NiOOH; F replaces an Ob atom, and at the same time, the nearby Ni5c loses an O3c atom to form the oxygen vacancy. Fig. 4d shows the crystal structure of Mn–F/NiOOH. Mn occupies the position of Ni5c and creates a new oxygen vacancy at the position near Mn. Fig. 4e reveals that NiOOH is a semiconductor with a band gap of 1.21 eV by the density of states (DOS). After F and Mn doping, multiple small peaks appeared near the Fermi level in the DOS diagram, and the corresponding band gaps were 0.23 and 0.25 eV, respectively (Fig. 4f and g). The conductivity has been greatly improved, which is consistent with the experimental results.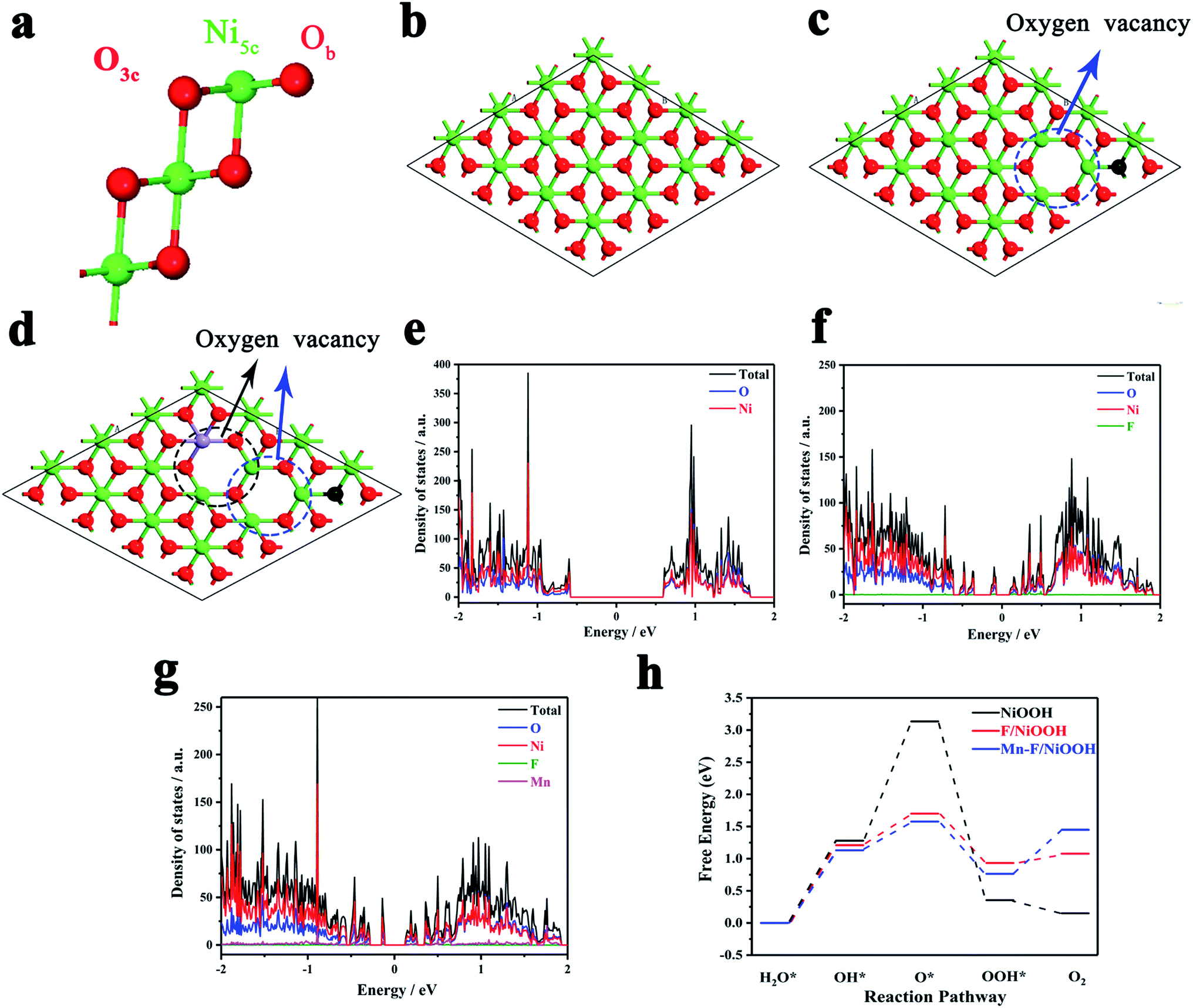 | ||
| Fig. 4 (a) Slab models for NiOOH. O and Ni atoms are colored red and green, respectively. Top views of optimized geometries of (b) pristine NiOOH nanosheets, (c) F/NiOOH, and (d) Mn–F/NiOOH. The red, green, black and purple balls represent O, Ni, F and Mn atoms, respectively. (e) Density of states of NiOOH. (f) Density of states of F/NiOOH. (g) Density of states of Mn–F/NiOOH. (h) Standard free-energy diagram of the OER process of NiOOH, F/NiOOH and Mn–F/NiOOH. | ||
Obviously, the active center is the Ni in NiOOH and F/NiOOH. However, for the Mn–F/NiOOH material, the active center may be Mn or Ni. Therefore, firstly we calculated the standard free energy of each step of the OER reaction with Mn or Ni as active sites respectively in the Mn–F/NiOOH material. The results (Fig. S8a†) show that the overpotentials centered on Mn and Ni are 0.86 eV and 0.35 eV, respectively. Obviously, when Ni is the active center, the overpotential is smaller, which is closer to our experimental result. We believe that Ni is the active center of the Mn–F/NiOOH. Then we expand the relevant calculation with Ni as the active site. The OER reaction occurs on the surface of the catalyst. Therefore, the water adsorption energy on the catalyst is a key factor affecting the OER reaction. By comparing the adsorption energy shown in Fig. S8b,† it is found that the water adsorption energy of Mn–F/NiOOH is the largest, which is more conducive to the OER reaction. This can lead to fast charge transfer from the catalyst surface to the electrolyte solution during the OER reaction. We can find the rate-determining step for the whole OER reaction is the process of OH* → O* for the NiOOH, F/NiOOH and Mn–F/NiOOH from the free energy of the reaction shown in Fig. 4h. After F and Mn doping, the barrier of the OH* → O* is greatly reduced, which is favorable for the reaction to proceed. Meanwhile, the data shown in Table S1† reveal that the theoretical overpotential is reduced from 1.90 eV (NiOOH) to 0.47 eV (F/NiOOH) and 0.35 eV (Mn–F/NiOOH), respectively. This indicates that the doping of F and Mn is critical for improving the catalytic activity of OER.
To further explain the effect of doped Mn(III) on the electronic structure of nickel hydroxide, we used crystal field theory to simply analyze the local interaction of the d electrons of Mn and Ni in the 3d orbitals of the crystal fields. The–OH present in the compound can be regarded as a weak field ligand. In Fig. S9,† Ni undergoes energy level splitting in the corresponding crystal field. From the 3d orbital distribution of Ni2+ in the Mn–F/Ni(OH)2–NF, it is found that there are two unpaired electrons in the t2g orbital. And the t2g orbital has π-symmetry, which will result in the interaction of the e−–e− repulsion between the O atom and the Ni atom in Ni(OH)2, as shown in Fig. S9c.† Since Mn occupies part of the Ni position in the molecule, its crystal field environment is the same as that of Ni. As shown in Fig. S9d,† the d orbital distributions of Mn(III) after splitting are all unfilled state occupied by a single electron except for an empty orbital. The electronic configuration of Mn itself leads to its stronger e−–e− repulsive force. There is an empty 3d orbital in Mn(III). When Ni, O and Mn exist in one system (Fig. S9a†), electrons will flow from Ni to Mn, which leads to a change in the electronic configuration of Mn and Ni. This also causes the binding energy of Ni to move towards the high direction, which is consistent with the XPS results. Considering the difference of internal e−–e− repulsive force between Mn and Ni, when the doped Mn occupies part of the Ni position, the e−–e− repulsive force becomes stronger than that of Ni (Fig. S9b†), resulting in a few of the electrons escaping from the orbital to generate oxygen vacancies. The unpaired electrons of the eg orbital exposed by oxygen vacancies easily combine with the electrons of the Pz orbital of O, resulting in better catalytic activity.61
Conclusions
In this work, we synthesized Mn and F co-doped Ni(OH)2 catalysts with oxygen defects on nickel foam by a one-step hydrothermal method. Analysis of Mn–F/Ni(OH)2–NF by XPS, EPR, SEM and TEM revealed that Mn–F/Ni(OH)2–NF has a hierarchical structure and is rich in oxygen defects. Interestingly, when Mn(III) is introduced into the F-doped Ni(OH)2–NF, the hydroxide structure of the material is unchanged and the oxygen defect content increases. We combine Mn–F/Ni(OH)2 with the conductive base nickel foam, which can improve the overall conductivity of the material and increase the contact area with the electrolyte solution to accelerate electron transport. The Mn–F/Ni(OH)2–NF has excellent OER catalytic activity, and it can be applied at a relatively high current density with good stability for about 10 hours. Finally, the DFT calculation confirmed that the doping of Mn and F effectively reduced the barrier of the OH* → O* step and effectively led to the enhancement of OER activity. The simple and tunable hydrothermal method may be suitable for the synthesis of other transition metal hydroxide-based nanomaterials, and provides a doping strategy to generate oxygen vacancies in the OER material to enhance catalytic activity and promote development of water-splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21871042, 21471028, 21673098, and 21671036), the Natural Science Foundation of Jilin Province (No. 20150101064JC), the National Key Basic Research Program of China (No. 2013CB834802), the Changbai Mountain Scholarship, the Fundamental Research Funds for the Central Universities (No. 2412015KJ012, 2412017BJ004) and the Jilin Provincial Department of Education. We would like to acknowledge the support from the Jilin Provincial Department of Education.References
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. S. Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- A. P. Wu, Y. Xie, H. Ma, C. G. Tian, Y. Gu, H. J. Yan, X. M. Zhang, G. Y. Yang and H. G. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS.
- M. J. Chen, L. Wang, H. P. Yang, S. Zhao, H. Xu and G. Wu, J. Power Sources, 2018, 375, 277–290 CrossRef CAS.
- G. Li, X. L. Wang, M. H. Seo, S. Hemmati, A. P. Yu and Z. W. Chen, J. Mater. Chem. A, 2017, 5, 10895–10901 RSC.
- W. Zhou, X. J. Wu, X. Cao, X. Huang, C. Tan, J. Tian, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 RSC.
- L. L. Huang, R. Chen, C. Xie, C. Chen, Y. Y. Wang, Y. F. Zeng, D. W. Chen and S. Y. Wang, Nanoscale, 2018, 10, 13638–13644 RSC.
- P. Z. Chen, K. Xu, T. P. Zhou, Y. Tong, J. C. Wu, H. Cheng, X. L. Lu, H. Ding, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef CAS PubMed.
- Y. F. Zeng, L. J. Chen, R. Chen, Y. Y. Wang, C. Xie, L. Tao, L. L. Huang and S. Y. Wang, J. Mater. Chem. A, 2018, 6, 24311–24316 RSC.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. Stephens and I. Chorkendorff, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- M. Vuković, J. Appl. Electrochem., 1987, 17, 737–745 CrossRef.
- H. J. Yin and Z. Y. Tang, Chem. Soc. Rev., 2016, 45, 4873–4891 RSC.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- G. Rogez, C. Massobrio, P. Rabu and M. Drillon, Chem. Soc. Rev., 2011, 40, 1031–1058 RSC.
- X. M. Zhou, Z. M. Xia, Z. Y. Zhang, Y. Y. Ma and Y. Q. Qu, J. Mater. Chem. A, 2014, 2, 11799–11806 RSC.
- N. Yang, C. Tang, K. Wang, G. Du, A. M. Asiri and X. P. Sun, Nano Res., 2016, 9, 3346–3354 CrossRef CAS.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 123241 Search PubMed.
- B. Zhang, C. H. Xiao, S. M. Xie, J. Liang, X. Chen and Y. H. Tang, Chem. Mater., 2016, 28, 6934–6941 CrossRef CAS.
- B. W. Zhang, Y. H. Lui, H. W. Ni and S. Hu, Nano Energy, 2017, 38, 553–560 CrossRef CAS.
- Z. Y. Wang, J. T. Li, X. C. Tian, X. P. Wang, Y. Yu, K. A. Owusu, L. He and L. Q. Mai, ACS Appl. Mater. Interfaces, 2016, 8, 19386–19392 CrossRef CAS PubMed.
- Y. Liang, Y. Yu, Y. Huang, Y. Shi and B. Zhang, J. Mater. Chem. A, 2017, 5, 13336–13340 RSC.
- G. Panomsuwan, N. Saito and T. Ishizaki, J. Mater. Chem. A, 2015, 3, 9972–9981 RSC.
- B. Zhang and S. Hu, Electrochem. Commun., 2018, 86, 108–112 CrossRef CAS.
- B. Zhang, K. Jiang, H. Wang and H. Shan, Nano Lett., 2019, 19, 530–537 CrossRef CAS PubMed.
- S. Wan, J. Qi, W. Zhang, W. Wang, S. Zhang, K. Liu, H. Zheng, J. Sun, S. Wang and R. Cao, Adv. Mater., 2017, 29, 1700286 CrossRef PubMed.
- J. Q. Lv, X. X. Yang, H. Y. Zang, Y. H. Wang and Y. G. Li, Mater. Chem. Front., 2018, 2, 2045–2053 RSC.
- Y. B. Li, H. M. Zhang, Y. Wang, P. Liu, H. G. Yang, X. D. Yao, D. Wang, Z. Y. Tang and H. J. Zhao, Energy Environ. Sci., 2014, 7, 3720–3726 RSC.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- M. Morita, C. Iwakura and H. Tamura, Electrochim. Acta, 1977, 22, 325–328 CrossRef CAS.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- J. W. Zhao, J. L. Chen, S. M. Xu, M. F. Shao, Q. Zhang, F. Wei, J. Ma, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2014, 24, 2938–2946 CrossRef CAS.
- Y. Sun, C. Wang, T. Ding, J. Zuo and Q. Yang, Nanoscale, 2016, 8, 18887–18892 RSC.
- R. Gao, Z. Y. Li, X. L. Zhang, J. C. Zhang, Z. B. Hu and X. F. Liu, ACS Catal., 2016, 6, 400–406 CrossRef CAS.
- L. Xu, Q. Q. Jiang, Z. H. Xiao, X. Y. Li, J. Huo, S. Y. Wang and L. M. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- D. Liu, C. Wang, Y. Yu, B. H. Zhao, W. Wang, Y. Du and B. Zhang, Chem, 2019, 5, 376–389 CAS.
- K. C. Wen, W. Q. Lv and W. D. He, J. Mater. Chem. A, 2015, 3, 20031–20050 RSC.
- K. C. Wen, Y. P. Han, M. D. Zou, W. Q. Lv and W. D. He, J. Power Sources, 2015, 291, 126–131 CrossRef CAS.
- H. W. Huang, C. Yu, H. L. Huang, C. T. Zhao, B. Qiu, X. C. Yao, S. F. Li, X. T. Han, W. Guo, L. M. Dai and J. S. Qiu, Nano Energy, 2019, 58, 778–785 CrossRef CAS.
- W. Q. Lv, Y. H. Niu, X. Jian, K. H. L. Zhang, W. Wang, J. Y. Zhao, Z. M. Wang, W. Q. Yang and W. D. He, Appl. Phys. Lett., 2016, 108, 083901 CrossRef.
- Y. Zhu, H. D. Yang, K. Lan, K. Iqbal, Y. Liu, P. Ma, Z. M. Zhao, S. Luo, Y. T. Luo and J. T. Ma, Nanoscale, 2019, 11, 2355–2365 RSC.
- T. T. Jiang, J. H. Kong, Y. Q. Wang, D. W. Meng, D. G. Wang and M. H. Yu, Cryst. Res. Technol., 2016, 51, 58–64 CrossRef CAS.
- P. Jakes and E. Erdem, Phys. Status Solidi RRL, 2011, 5, 56–58 CrossRef CAS.
- Z. Wang, K. Wang, H. Wang, X. Chen, W. Dai and X. Fu, Catal. Sci. Technol., 2018, 8, 3260–3277 RSC.
- L. Z. Zhuang, L. Ge, Y. S. Yang, M. R. Li, Y. Jia, X. D. Yao and Z. H. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- J. Kim, X. Yin, K. C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS PubMed.
- M. L. Guan, C. Xiao, J. Zhang, S. J. Fan, R. An, Q. M. Cheng, J. f. Xie, M. Zhou, B. J. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed.
- J. Zhang, H. Ren, J. Y. Wang, J. Qi, R. B. Yu, D. Wang and Y. L. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 RSC.
- X. L. Zheng, Y. F. Ji, J. Tang, J. Y. Wang, B. F. Liu, H.-G. Steinrück, K. Lim, Y. Z. Li, M. F. Toney, K. Chan and Y. Cui, Nat. Catal., 2019, 2, 55–61 CrossRef CAS.
- Y. F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- Y. Liu, S. B. Yin and P. K. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 23131–23139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00535h |
| This journal is © The Royal Society of Chemistry 2019 |