
An enhanced enzymatic reaction using a triphase system based on superhydrophobic mesoporous nanowire arrays†
Fengying
Guan‡
,
Jun
Zhang‡
,
Heming
Tang
,
Liping
Chen
and
Xinjian
Feng
 *
*
College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: xjfeng@suda.edu.cn
First published on 27th September 2018
Abstract
Gaseous reactants play a key role in a wide range of biocatalytic reactions, however reaction kinetics are generally limited by the slow mass transport of gases (typically oxygen) in or through aqueous solutions. Inspired by the morphologies of natural non-wetting surfaces, herein we address this limitation by developing a triphase reaction system possessing a triphase gas–solid–liquid interface. As a proof of concept, we study the kinetics of glucose oxidase (GOx) catalyzed reactions using a triphase system fabricated by layering GOx upon superhydrophobic mesoporous ZnO nanowire arrays through which oxygen, needed for the enzymatic reaction, is supplied directly from the atmosphere to the liquid–solid interface. We find that the enzymatic reaction rate is enhanced by a factor of 30 over that obtained from a conventional diphase system where oxygen is supplied through and from the liquid. The triphase system offers the opportunity to develop high performance bioassay systems, serving as an enabling platform for addressing challenges posed by gas-deficit kinetics.
Conceptual insightsSuperhydrophobic substrates have been known for decades and are used in many areas. A unique property of superhydrophobic surfaces is that they can trap gas at the liquid–solid interface, enabling the direct transport of gas through the gas phase to the solid–liquid interface. This offers us an opportunity to enhance the kinetics of multiphase catalytic reactions, which used to be restricted by the low concentration and slow diffusion rate of gas in the liquid phase. Herein, we fabricated a superhydrophobic substrate by using mesoporous ZnO nanowire arrays that possess both rapid charge transport pathway (along the nanowires) and gas transport pathway (through the free spaces between nanowires). Taking an oxidase catalytic reaction as an example, where O2 is used as a natural electron acceptor, we demonstrate that the kinetics of the oxidase catalytic reaction was significantly enhanced. We further developed high performance triphase bioassay systems; as illustrative of this, we show that oxidase-based bioassays using such an enzymatic system exhibit a 40× higher linear detection upper limit than that obtained using a conventional solid–liquid diphase system, with excellent detection sensitivity and selectivity. The triphase system appears to be an enabling platform for understanding and boosting the efficiency of gas-involved multiphase catalytic reactions. |
Introduction
Gaseous reactants inherent to aqueous solution (bio)catalytic reactions are of great importance in determining the efficiency of a wide range of biochemical and industrial processes.1–6 For instance, almost all oxidases will produce H2O2 upon substrate oxidation when oxygen is involved. While oxidase-based electrodes for bioelectronics and bioassay applications are achieved by utilizing the measurement of enzymatic product H2O2,7–13 with conventional solid–liquid diphase electrode systems, see Fig. S1 (ESI†), the availability of oxygen at the reaction interface depends upon its mass transfer through the liquid phase. The low solubility and slow mass transport rate of oxygen in aqueous solutions suppress oxidase kinetics, in turn suppressing the amount of H2O2 production, and consequently the performance of the bioelectronics.In this work, this limitation was addressed by developing a triphase enzymatic reaction system. An oxidase layer is immobilized atop a superhydrophobic mesoporous nanowire (NW) arrays surface after being modified with H2O2 electrocatalysts, as shown in Fig. 1. Inspired from the natural non-wetting surfaces,14,15 scientists have fabricated various artificial superhydrophobic substrates16–21 that have been used in a wide variety of research areas,22–30 such as heat transfer,23 photocatalysis,24 drug release25,26 and self-cleaning.27 When immersed in an aqueous solution, these superhydrophobic surfaces can trap a number of gas pockets at the liquid–solid interface, leading to the formation of a gas–solid–liquid joint interface,31,32 where gaseous reactants can be rapidly supplied from the gas phase. Fig. 1b is a magnified view of the triphase enzymatic reaction zone. The enzyme layer is immobilized on top of the superhydrophobic NW arrays, and so can be readily wetted by the aqueous solution. The analyte can thus diffuse freely from a liquid phase into the enzyme layer, resulting in oxidization and thus formation of enzymatic product H2O2 that is, in turn, electrochemically reduced at the electrode surface. For oxygen, its diffusion coefficient in air is approximately 104 times higher than that in liquid,33 hence, the oxygen level at the triphase interface is sufficient to sustain reaction kinetics and constant, offering us an opportunity to enhance bioelectronics device performance.
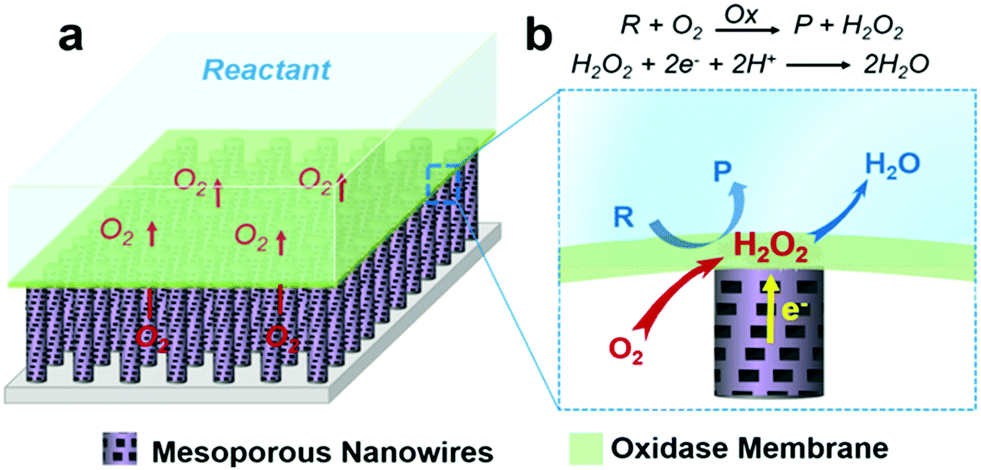 | ||
| Fig. 1 Schematic illustration of the gas–solid–liquid triphase enzymatic reaction system. (a) The oxidase film is immobilized on superhydrophobic mesoporous nanowire (NW) arrays modified with a H2O2 electrocatalyst. When immersed in the aqueous solution, the gas–solid–liquid triphase will be formed due to the air pockets being trapped. (b) The triphase bio-electrochemical reaction interface with an enlarged view, where adequate oxygen needed at the oxidase reaction zone can be supplied directly from the air phase via the free spaces between NWs. The oxidase kinetics are monitored by in situ cathodic measurement of the formation of enzymatic product H2O2. R = reactant, P = product. | ||
Results and discussion
To demonstrate the triphase enzymatic reaction system, glucose oxidase (GOx) and mesoporous ZnO NW arrays34 were selected as the model enzyme and electrode substrate, respectively. The ZnO NWs have a wurtzite structure (JCPDS card No. 65-3411, Fig. S2, ESI†), an average length of 12 μm, and nanopores (∼20 nm) on the surface, see Fig. 2a and b. As shown in Fig. 2c, from a typical transmission electron microscopy (TEM) image, we can see that the nanopores are distributed across the entire width of the NWs. The NWs were then sensitized with Pt nanoparticles with a diameter of approximately 3 nm (Fig. S3, ESI†). After being treated with perfluorooctane sulfonates (PFOS), the contact angle (CA) is 150 ± 2° (Fig. S4a, ESI†), which means a superhydrophobic surface was obtained. Onto the nanowire substrate, a mixture of GOx and Nafion solution was then cast, with an electrode area of 0.25 cm2. As shown in Fig. 2d, a field-emission scanning electron microscopy (FE-SEM) image of the enzyme electrode showed that a thin GOx/Nafion film is layered upon the NW arrays. After the immobilization of GOx, a water CA of 57 ± 2° (Fig. S4b, ESI†) is attained, which shows that the surface became hydrophilic. Based on such a unique enzyme electrode system, aqueous reactants can readily diffuse from the liquid phase into the hydrophilic enzyme layer, while electrons and oxygen can, respectively, transport rapidly along the NWs and through the free-spaces between the NWs to the enzymatic reaction zone. A schematic diagram of the experimental setup is shown in Fig. S5 in the ESI.†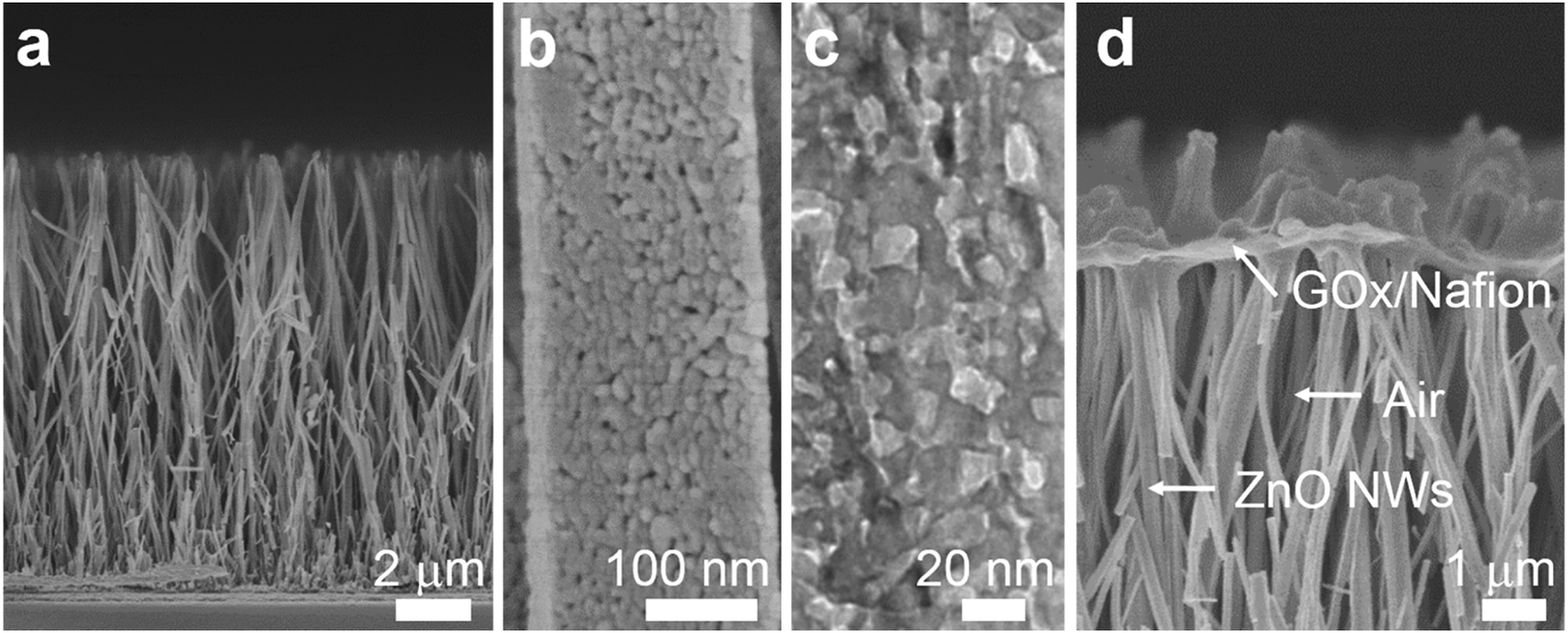 | ||
| Fig. 2 (a and b) Cross-section FE-SEM images of mesoporous ZnO NW arrays at different magnifications. (c) TEM image of part of a NW, indicating a mesoporous microstructure. (d) FE-SEM side-view of the oxidase enzyme electrode architecture: a thin GOx/Nafion composite film is immobilized on the top surface of the NW arrays. | ||
Under an air atmosphere (as illustrated in Fig. 3a), the background subtracted current increases linearly with glucose concentration up to about 40 mM (R2 = 0.99) (Fig. 3c) at an optimized potential of 0 V (see Fig. S6, ESI†). In contrast, when the same electrode is applied under a nitrogen atmosphere, see Fig. 3b, the background subtracted current increases with glucose concentration up to only ≈1 mM (Fig. 3d), a value 40-times lower than that obtained under an air atmosphere. Since the currents shown in Fig. 3c and d are all background subtracted the increase in current is solely attributed to reduction of enzymatic product H2O2, while the enhanced enzyme activity observed under air should be attributed to the sustained oxygen transportation from the air phase to the interface.
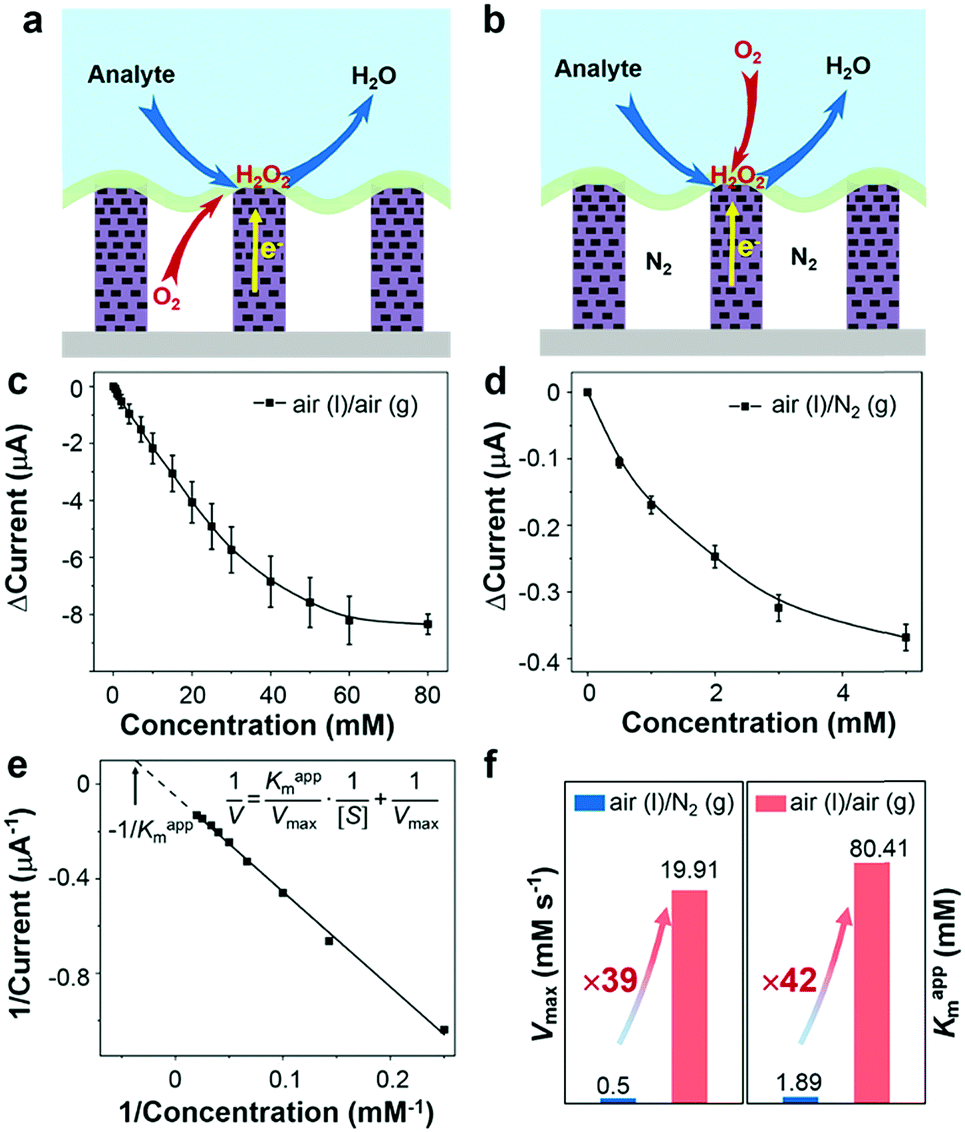 | ||
| Fig. 3 Performance of the triphase enzymatic system. (a and b) are, respectively, illustrations of the triphase enzyme electrode under air and nitrogen atmospheres. (c and d) Background subtracted current vs. glucose concentration at 0 V vs. Ag/AgCl, under air and N2 atmospheres, respectively. The g and l represent the gas phase and liquid phase, respectively. ΔCurrent = Currents − Current0, Current0 is the background current and Currents is the current measured in the solution with different glucose concentrations. (e) The corresponding Lineweaver–Burk plot of the triphase electrode in an air atmosphere over an extended concentration range. Vmax is the maximum reaction rate obtained from the Lineweaver–Burk plot, [S] is the reactant concentration, and Kappm is the apparent Michaelis–Menten constant. (f) Kappm and Vmax comparisons between air and nitrogen atmospheres. In our study, when the triphase electrode was placed under a nitrogen atmosphere (air(l)/N2(g)) during the electrochemical measurements, the oxygen required for the enzymatic reaction is derived from the dissolved oxygen in the aqueous solution, which is equivalent to the traditional solid–liquid diphase electrode system. | ||
In order to further explore the enzyme electrode process, cyclic voltammograms (CVs) of the triphase system were recorded in a 10 mM glucose solution with variable scan rates (10–300 mV s−1) under air (Fig. S7a, ESI†). The cathodic current linearly correlates with the square root of the scan rate ν1/2 (Fig. S7b, ESI†), indicating that the triphase electrode process is not surface reaction controlled, but instead glucose diffusion controlled. In distinct contrast, under a nitrogen atmosphere the cathodic current linearly correlates to scan rate ν (Fig. S7c and d, ESI†), which indicates that the oxidase kinetics are limited when the oxygen concentration at the electrode surface is restricted. The kinetic parameters of an enzymatic reaction were further evaluated. As shown in Fig. 3c and d, the dose-dependent plot is relatively flat at high glucose concentration, which is consistent with the unique property of the Michaelis–Menten kinetic mechanism.35 The parameters of the enzymatic reaction kinetics, such as the maximum reaction rate (Vmax) and the apparent Michaelis–Menten constant (Kappm) can be obtained using the Lineweaver–Burk plot equation (insert of Fig. 3e).36Kappm can be used to characterize the performance of the triphase system since the value of Kappm represents the linear dynamic range (1/2Kappm),37 and Vmax can be used to characterize the enzymatic reaction rate. As shown in Fig. 3f, we found that the Kappm and Vmax of GOx at the triphase interface in the air phase are 42 and 39 times higher than that in a nitrogen atmosphere, respectively.
To understand the influence of the air-retaining layer on the triphase enzymatic reaction kinetics, mesoporous ZnO NWs with different lengths were prepared and used as the substrate. As shown in Table 1 and Fig. S8 (ESI†), the Vmax and Kappm values increase with NW length. Increasing the NW length will reduce wire density and enhance the mass transport of oxygen from the air phase to the reaction interface. Hence, we concluded that the GOx catalytic reaction rate is highly dependent on the oxygen concentration at the liquid–solid interface and that our proposed triphase enzymatic system can significantly increase the oxidase kinetics, verifying its feasibility in addressing the gas-deficit challenge.
| Length (μm) | K appm (mM) | V max (mM s−1) |
|---|---|---|
| 6 | 17.85 | 4.06 |
| 9 | 56.49 | 14.28 |
| 12 | 80.41 | 19.91 |
| 15 | 90.79 | 23.54 |
The significantly enhanced oxidase kinetics offers the opportunity for developing high performance bioelectronics and bioassays, something of great scientific and technological interest. It is reported that there are numerous diabetic patients around the world, which require regular, accurate glucose level monitoring.7–9,38–40 In the presence of oxygen almost all oxidase-catalytic reactions will produce H2O2, the electrochemical measurement of which is an ideal approach for bioassay development. However, for traditional solid–liquid diphase enzyme electrode systems the slow mass transfer rate and low concentration of oxygen in aqueous solution restrict oxidase kinetics and, in turn, the linear detection dynamic range and sensitivity. Scientists have investigated lots of ways to achieve extended dynamic range, such as mass-transport-limiting membranes, analyte solution dilution and so on. However, the low concentration of analyte at the assay interface leads to restricted formation of an enzymatic product of H2O2, and consequently, limited sensitivity (nA mM−1). In our triphase system, the oxygen needed in the enzymatic reaction zone is sufficient, and a mass-diffusion-barrier is no longer required here. Consequently, as can be seen from Fig. 3c, a higher linear dynamic range upper limit (40 mM) and excellent sensitivity (0.18 μA mM−1) are achieved. We believe that the sensitivity can be further improved in the future if three-dimensional hierarchical NW arrays that offer a larger interfacial contact area with GOx can be used.
Apart from the extended linear dynamic range and good sensitivity, selectivity is also a crucial parameter for sensor performance. The interferences that are generated from many endogenous/exogenous species can be successfully eliminated via a cathodic measurement of enzymatic product H2O2. At 0 V (versus Ag/AgCl), see Fig. 4a, after the addition of 0.1 mM ascorbic acid (AA), uric acid (UA), urea (U), and acetamido phenol (AP) into 5 mM glucose electrolyte, the variation of current response is negligible indicating excellent selectivity. Repeated measurements of one triphase electrode in 10 mM glucose were recorded, and the relative standard deviation is about 4.05%, as illustrated in Fig. 4b. Besides, after adding glucose, as shown in Fig. S9 (ESI†), the triphase electrode responds rapidly within 10 s with a detection limit of 50 μM.
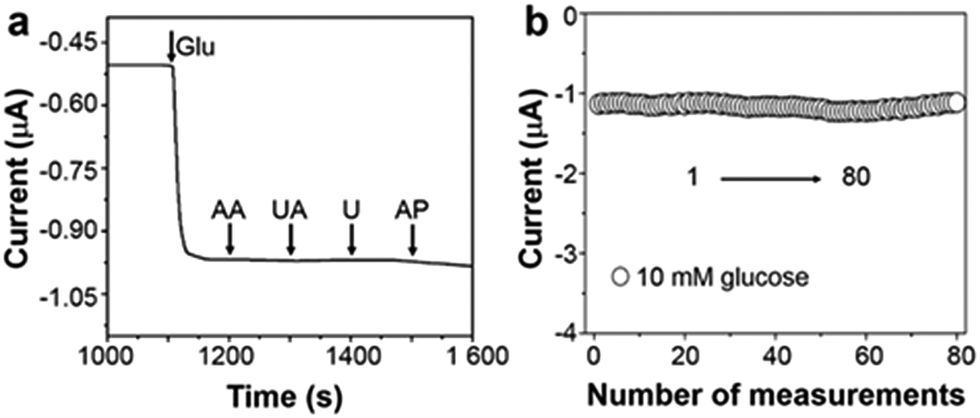 | ||
| Fig. 4 (a) At 0 V vs. Ag/AgCl, amperometric response of the triphase enzyme electrode to the successive addition of 5 mM glucose, 0.1 mM AA, UA, U and AP in 0.2 M PBS electrolyte. (b) Successive 80 measurements of 10 mM glucose using the one enzyme electrode, with a relative standard deviation of 4.05%. | ||
Conclusions
In summary, we demonstrate a triphase enzymatic reaction system possessing a triphase air–solid–liquid interface based on superhydrophobic mesoporous ZnO NW arrays. The reaction system allows sufficient oxygen to transport to the reaction zone via the gas phase, leading to over 30 times higher enzymatic reaction rate than that obtained with a conventional diphase system. Such high performance bioassay systems have great potential in clinical diagnosis applications. Our results lay the groundwork for additional exploration to understand and promote interfacial reaction kinetics and highlight the powerful interface-engineering strategy for designing high performance catalytic reaction system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (51772198), the Jiangsu Province Science Foundation for Distinguished Young Scholars (BK20150032), and the project of Scientific and Technologic Infrastructure of Suzhou (SZS201708).Notes and references
- J. Huang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2015, 137, 15015–15025 CrossRef CAS PubMed.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- S. Wang, Y. Wu, X. Kan, B. Su and L. Jiang, Adv. Funct. Mater., 2014, 24, 7007–7013 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed.
- Y. Zhao, Y. Liu, Q. Xu, M. Barahman, D. Bartusik, A. Greer and A. M. Lyons, J. Phys. Chem. A, 2014, 118, 10364–10371 CrossRef CAS PubMed.
- L. Zhang, L. Han, P. Hu, L. Wang and S. Dong, Chem. Commun., 2013, 49, 10480–10482 RSC.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS PubMed.
- G. S. Wilson and Y. B. Hu, Chem. Rev., 2000, 100, 2693–2704 CrossRef CAS PubMed.
- Y. Lei, R. Sun, X. Zhang, X. Feng and L. Jiang, Adv. Mater., 2016, 28, 1477–1481 CrossRef CAS PubMed.
- H. Lee, T. K. Choi, Y. B. Lee, H. R. Cho, R. Ghaffari, L. Wang, H. J. Choi, T. D. Chung, N. Lu, T. Hyeon, S. H. Choi and D.-H. Kim, Nat. Nanotechnol., 2016, 11, 566–572 CrossRef CAS PubMed.
- Z. Song, C. Xu, X. Sheng, X. Feng and L. Jiang, Adv. Mater., 2018, 30, 1701473 CrossRef PubMed.
- L. Li, Y. Wang, L. Pan, Y. Shi, W. Cheng, Y. Shi and G. Yu, Nano Lett., 2015, 15, 1146–1151 CrossRef CAS PubMed.
- W. Barthlott and C. Neinhuis, Planta, 1997, 202, 1–8 CrossRef CAS.
- L. Feng, S. Li, Y. Li, H. Li, L. Zhang, J. Zhai, Y. Song, B. Liu, L. Jiang and D. Zhu, Adv. Mater., 2002, 14, 1857–1860 CrossRef CAS.
- A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 0546–0550 RSC.
- S. Farsinezhad, P. R. Waghmare, B. D. Wiltshire, H. Sharma, S. Amiri, S. K. Mitra and K. Shankar, RSC Adv., 2014, 4, 33587–33598 RSC.
- L. Jiang, Y. Zhao and J. Zhai, Angew. Chem., Int. Ed., 2004, 43, 4338–4341 CrossRef CAS PubMed.
- T. Verho, J. T. Korhonen, L. Sainiemi, V. Jokinen, C. Bower, K. Franze, S. Franssila, P. Andrew, O. Ikkala and R. H. A. Ras, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10210–10213 CrossRef CAS PubMed.
- X. Feng and L. Jiang, Adv. Mater., 2006, 18, 3063–3078 CrossRef CAS.
- Y. Wu, T. Hang, Z. Yu, L. Xu and M. Li, Chem. Commun., 2014, 50, 8405–8407 RSC.
- Y. Wu, K. Liu, B. Su and L. Jiang, Adv. Mater., 2014, 26, 1124–1128 CrossRef CAS PubMed.
- J. Li, Y. Hou, Y. Liu, C. Hao, M. Li, M. K. Chaudhury, S. Yao and Z. Wang, Nat. Phys., 2016, 12, 606–612 Search PubMed.
- X. Sheng, Z. Liu, R. Zeng, L. Chen, X. Feng and L. Jiang, J. Am. Chem. Soc., 2017, 139, 12402–12405 CrossRef CAS PubMed.
- S. T. Yohe, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 2016–2019 CrossRef CAS PubMed.
- M. N. Kavalenka, F. Vuelliers, S. Lischker, C. Zeiger, A. Hopf, M. Röhrig, B. E. Rapp, M. Worgull and H. Hölscher, ACS Appl. Mater. Interfaces, 2015, 7, 10651–10655 CrossRef CAS PubMed.
- Y. Lu, S. Sathasivam, J. Song, C. R. Crick, C. J. Carmalt and I. P. Parkin, Science, 2015, 347, 1132–1135 CrossRef CAS PubMed.
- L. Mi, J. Yu, F. He, L. Jiang, Y. Wu, L. Yang, X. Han, Y. Li, A. Liu, W. Wei, Y. Zhang, Y. Tian, S. Liu and L. Jiang, J. Am. Chem. Soc., 2017, 139, 10441–10446 CrossRef CAS PubMed.
- J. Zhang, X. Sheng, J. Jin, X. Feng and L. Jiang, Nano Res., 2017, 10, 2998–3004 CrossRef CAS.
- Z. Lu, W. Xu, J. Ma, Y. Li, X. Sun and L. Jiang, Adv. Mater., 2016, 28, 7155–7161 CrossRef CAS PubMed.
- L. Wen, Y. Tian and L. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3387–3399 CrossRef CAS PubMed.
- J. Wang, Y. Zheng, F. Q. Nie, J. Zhai and L. Jiang, Langmuir, 2009, 25, 14129–14134 CrossRef CAS PubMed.
- E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge Univ. Press, New York, 2nd edn, 1997 Search PubMed.
- D. He, X. Sheng, J. Yang, L. Chen, K. Zhu and X. Feng, J. Am. Chem. Soc., 2014, 136, 16772–16775 CrossRef CAS PubMed.
- R. A. Kamin and G. S. Wilson, Anal. Chem., 1980, 52, 1198–1205 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- C. P. McMahon, G. Rocchitta, P. A. Serra, S. M. Kirwan, J. P. Lowry and R. D. O'Neill, Anal. Chem., 2006, 78, 2352–2359 CrossRef CAS PubMed.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- Q. Cai, K. Zeng, C. Ruan, T. A. Desai and C. A. Grimes, Anal. Chem., 2004, 76, 4038–4043 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00184g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |