
Superaerophilic copper nanowires for efficient and switchable CO2 electroreduction†
Yusheng
Zhang‡
a,
Zhao
Cai‡
 a,
Yuxin
Zhao‡
b,
Xuemei
Wen
a,
Wenwen
Xu
a,
Yang
Zhong
ac,
Lu
Bai
d,
Wen
Liu
a,
Ying
Zhang
e,
Ying
Zhang
*f,
Yun
Kuang
*a and
Xiaoming
Sun
*ac
a,
Yuxin
Zhao‡
b,
Xuemei
Wen
a,
Wenwen
Xu
a,
Yang
Zhong
ac,
Lu
Bai
d,
Wen
Liu
a,
Ying
Zhang
e,
Ying
Zhang
*f,
Yun
Kuang
*a and
Xiaoming
Sun
*ac
aState Key Laboratory of Chemical Resource Engineering, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: sunxm@mail.buct.edu.cn; kuangyun@mail.buct.edu.cn
bSchool of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, P. R. China
cCollege of Energy, Beijing University of Chemical Technology, Beijing 100029, P. R. China
dNational Center for Nanoscience & Technology, Beijing 100049, P. R. China
eState Key Laboratory for Heavy Oil Processing, China University of Petroleum, Qingdao 266580, P. R. China
fSchool of Chemistry, Monash University, Clayton 3800, Australia. E-mail: sherry.zhang@monash.edu
First published on 9th November 2018
Abstract
Copper is one of the most efficient electrocatalysts for switchable carbon dioxide conversion, but the design of an advanced Cu-based catalyst with high selectivity while suppressing hydrogen evolution remains a great challenge. Herein, we use Cu nanowires (Cu NWs) as the starting materials and polytetrafluoroethylene (PTFE) as the surface modifier to make a superaerophilic electrode using a wettability control strategy. This strategy allows tuning of the selectivity of the CO2 reduction reaction (CO2RR) and a decrease of the hydrogen evolution rate simultaneously by facilitating the supply of CO2 reactants and inhibiting the adsorption of water (protons). The transferring point from a pinning to bursting state turned out to be the optimized condition leading to the highest CO2RR faradaic efficiency without significant interference of current density. The optimized superaerophilic Cu NW catalyst showed CO-selectivity with a Faraday efficiency of 71% at −0.4 V vs. RHE and HCOOH-selectivity with a Faraday efficiency of 68% at −0.6 V vs. RHE. Moreover, the accelerated gas and ion diffusion and homogenized reactions also avoided accumulative damage on the surface of the Cu NWs and enhanced the stability of the Cu catalyst. This wettability tuning strategy provides a facile and efficient way to optimize the gas and ion diffusion layers, therefore promoting the performance of the CO2RR. This strategy potentially can be extended to the design of other gas consumption electrocatalysts.
Conceptual insightsA key challenge for the electrocatalytic CO2 reduction reaction (CO2RR) is the lack of nanocatalysts with both high efficiency and durability toward liquid fuels or valuable chemicals (L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069). Different from numerous research studies focusing on exploration of new catalysts for better intrinsic activity/selectivity, here, using Cu nanowires as the starting materials and polytetrafluoroethylene as the surface modifier, we designed superaerophilic Cu nanowires, which exhibited both excellent activity and durability towards CO2RR. Our wettability tuning strategy presents a facile and effective approach to improve CO2RR via optimizing the gas and ion diffusion layers. This method should be inspiring for the design of other gas consumption electrolyzer materials. |
Due to the energy crisis caused by depletion of fossil fuels and accumulation of greenhouse gases, converting carbon dioxide (CO2) to renewable fuels or value-added chemicals represents a highly-promising way to meet these challenges from both sides.1–3 In past decades, numerous efforts have been devoted to increasing the commercial viability of the electrocatalytic CO2 reduction reaction (CO2RR) by reducing operating overpotential.4–8 However, more efficient electrocatalysts with high activity, selectivity, and durability for CO2RR have yet to be developed. Though a range of materials including metals/alloys,9 oxides,10,11 carbons,12,13 chalcogenides,14,15 and molecular complexes16 are known to produce mostly low order hydrocarbon products, only Cu-based catalysts have shown distinct advantages for easy accessibility and appreciable activity towards various reaction pathways with higher order hydrocarbons or oxygen-containing compounds.17–20
Recent investigations have shown that nanostructured Cu catalysts with high-index facets or grain boundaries could vastly improve activity or selectivity over bulk counterparts at lower overpotentials.21,22 This is mainly because their surface atoms are in a low-coordination environment and thus provide abundant active sites that can be used as a “knob” to modulate surface adsorption/desorption energies. Besides, a local pH gradient layer might form in situ surrounding these highly active sites during a CO2RR process, leading to an altering of the reaction pathway.23 Cu nanowires (Cu NWs) with five-fold twinned crystalline are rich in grain boundaries in their cross sections and also in step atoms on their surfaces.24 These structural features make Cu NWs a promising electrocatalyst platform for further improving CO2RR performance. However, three main issues need to be addressed: (1) competition of the CO2RR and hydrogen evolution reaction in the highly active sites due to the strong philicity of these active sites to water or electrolytes;25,26 (2) CO2RR selectivity among value-added products (e.g., CO vs. HCOOH) is difficult to be tailored;27 and (3) durability of the Cu NW catalysts still lacks an efficient approach to improve it.28 Strategies including alloying28 or oxidative pulses29 have been explored, but they still require complex processes and so are far from being used practically.
Recent progress with superwetting electrodes provides an alternative sight to solve the above issues by tailoring the adsorption/desorption affinity/behavior of reagents or products.30–32 Biomimic micro-/nano-structures have shown specific superwetting behaviors with numerous materials with desirable properties and intriguing functionality.33,34 In the past three years, superwetting electrodes exhibited remarkable performance in gas-involved electrocatalytic reactions. For instance, a superaerophobic MoS2 electrode and a superaerophilic carbon nanotube electrode have shown ultrahigh activity and stability for hydrogen evolution and the oxygen reduction reaction, respectively.30–32 Due to CO2RR being a gas consumption reaction, we had the idea that the formation of an aerophilic (hydrophobic) interface would facilitate gas diffusion to the surface of the catalyst and thus enhance the CO2 reduction rate. To the best of our knowledge, related works have never been reported.
Herein, we demonstrate a facile polytetrafluoroethylene (PTFE) treated strategy to promote the CO2RR activity of Cu NWs (Fig. 1). The as-modified Cu NW electrode displays both superaerophilic and superhydrophobic properties. The superaerophilic surface enables a fast diffusion and affinity of CO2 onto the Cu surface under aqueous electrolyte media, and the superhydrophobic property inhibited excessive protons absorbed to the Cu surface, thus simultaneously boosting CO2RR and suppressing the side-hydrogen evolution reaction (HER). The catalytic results show that the CO faradaic efficiency (FE) of the optimized PTFE–Cu NW electrode at −0.4 V vs. RHE increased by 2.73-fold (from 19% to 71%), meanwhile the HCOOH efficiency at −0.6 V vs. RHE increased by 1.42-fold (from 28% to 68%). Besides, high selectivity and significantly improved durability were observed using this modified electrode due to stabilized active sites.
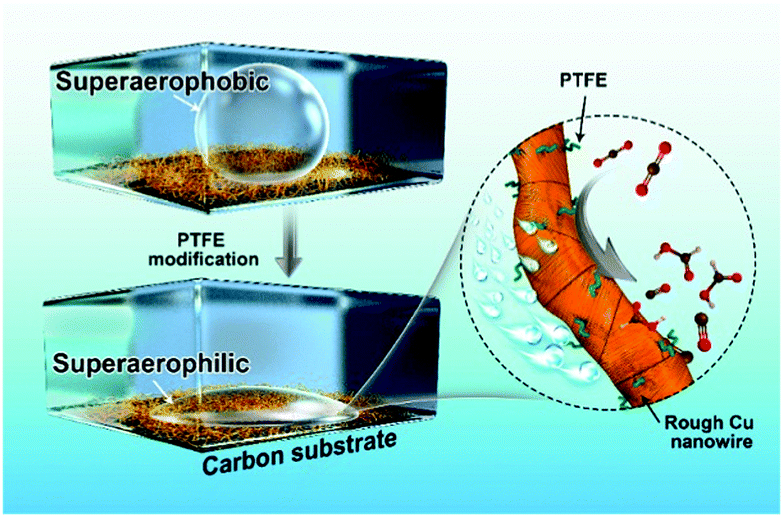 | ||
| Fig. 1 Schematic illustration of the superaerophilic Cu nanowire catalyst for CO2 reduction. After PTFE modification, the grain boundary-enriched Cu NWs shows a superaerophilic surface to accelerate the carbon dioxide diffusion process. | ||
Pristine Cu NWs were prepared by an aqueous reduction route at room temperature according to a previous report.35 After modification by PTFE (see Experimental section for details), the PTFE-treated Cu NWs (PTFE–Cu NWs) still maintained a well-defined ultralong nanowire structure. Fig. 2a shows the scanning electron microscopy (SEM) image of the as-prepared PTFE–Cu NW electrode. The ultralong nanowire network is composed of PTFE–Cu NWs with uniform diameters of ∼120 nm and a length of hundreds of micrometers. It was noted that the copper oxide nanowires were reduced to metallic Cu after electrochemical reduction. Specifically, only the metallic Cu pattern was observed in the XRD pattern (Fig. S1, ESI†), and the Cu 2p XPS spectrum indicated the presence of Cu0/1+ and the absence of Cu2+ after electrochemical reduction (Fig. S2 and S3, ESI†).22 The transmission electron microscopic (TEM) image suggested a rough nanowire morphology of these PTFE–Cu NWs (Fig. 2b). The high-resolution TEM image clearly revealed the polycrystalline nature of Cu NWs with many grain boundaries (Fig. 2c). A selected area electron diffraction (SEAD) pattern of a single Cu NW showed concentric rings composed of polycrystalline diffraction spots (Fig. S4, ESI†), which is consistent with the HRTEM results.
 | ||
| Fig. 2 (a) SEM image and (b) TEM image of PTFE–Cu NWs; (c) HRTEM image of the PTFE–Cu NWs, suggesting a grain boundary-rich nature, scale bars in (a) 1 μm, (b) 50 nm, and (c) 2 nm. Carbon dioxide bubble adhesion behavior of (d) pristine Cu NW and (f) PTFE–Cu NW electrodes in 0.1 M KHCO3 electrolyte. Digital images of (e) pristine Cu NWs and (g) PTFE–Cu NWs electrodes in 0.1 M KHCO3 electrolyte. The mirror-like red color of PTFE–Cu NW electrode demonstrates its superaerophilic nature. | ||
PTFE–Cu NWs were prepared by mixing a pristine Cu NW dispersed solution with a PTFE solution in a proper proportion, then annealed at 350 °C for 30 min and electrochemically reduced in 0.1 M KHCO3. As revealed in Fig. 2d and e, the CO2 gas bubble (with a volume of ∼2 μL) was pinned on the pristine Cu NW electrode under aqueous electrolyte and the contact angle was determined to be ∼150.4 ± 3.2°, indicating a superaerophobic nature. Notably, the CO2 gas bubble in 0.1 M KHCO3 liquid showed a pinning behavior even after 60 ms. However, the CO2 gas contact behavior was distinctly different after PTFE modification. The PTFE coating is conformal on the Cu surface (Fig. S5, ESI†) to minimize the surface energy, therefore causing the superaerophilicity. As shown in Fig. 2f and g, the CO2 gas bubble burst once it touched the PTFE–Cu NW electrode surface (within 1 ms), demonstrating a superaerophilic nature. This “bursting” behavior indicates a much stronger affinity of the CO2 gas bubble to the PTFE–Cu NW electrode, which facilitates the CO2 reactant supply near the surfaces of catalysts for CO2RR.
In order to show the impact of the superaerophilic property on electrocatalytic performance, the pristine Cu NWs and PTFE–Cu NWs were examined as CO2RR catalysts with a 0.1 M KHCO3 electrolyte. As shown in Fig. 3a and b, the pristine Cu NWs exhibited an ultralow onset potential at −0.3 V vs. RHE with a small current density of 0.4 mA cm−2, but both the CO and HCOOH selectivity are very low (total faradaic efficiencies (FE) of CO2 reduction products were lower than 40%) under a wide range working potentials (−0.3 to −0.8 vs. RHE) and the FE of H2 was higher than 50%, which is in good accordance with previous reports.22 Upon PTFE modification, the PTFE–Cu NW electrode showed no obvious change in current density (remained 92% at −0.8 V vs. RHE compared to pristine Cu NWs), but with a significant difference in product distribution. As shown in Fig. 3c and d, a high selectivity of CO (FE of 71%) was achieved at −0.4 V vs. RHE. FE of H2 which was obviously lower than that observed with the pristine Cu NW electrode, demonstrating that the HER process was effectively suppressed after PTFE modification. More importantly, the main product could be switched from CO to HCOOH (i.e., 68%) by facilely switching the potential to −0.6 V vs. RHE. An efficient and switchable CO2RR performance with the superaerophilic Cu electrode was realized and attributed to the fine-tuned superaerophilic and superhydrophobic properties of PTFE–Cu NWs, which facilitated CO2 gas diffusion and depressed excessive proton supply from the electrolyte. To find an optimized PTEE concentration for the modification, PTFE–Cu NW catalysts with different percentages of PTFE (20–80% mass ratios, see Experimental section for details, Fig. S7–S9, ESI†) were tested. It was found that the CO2RR performance of the PTFE–Cu NWs was highly dependent on the PTFE concentration, as shown in Fig. 3e and f. With an increasing amount of PTFE used, both the current density and electrochemical surface area (ECSA) decreased, suggesting the PTFE modification would cover the Cu active surface (Fig. S6 and Table S1, ESI†), thus decreasing the total current density. In addition, surface PTFE introduces electric resistance and charge transfer resistance to the electrode which is another factor causing the decrease of total current density, as demonstrated by Fig. S10 (ESI†). To better understand the impact of the aerophilic/hydrophobic property on CO2RR selectivity, a correlation of partial current densities of CO2RR and HER to the corresponding PTFE percentage is displayed in Fig. 3f.
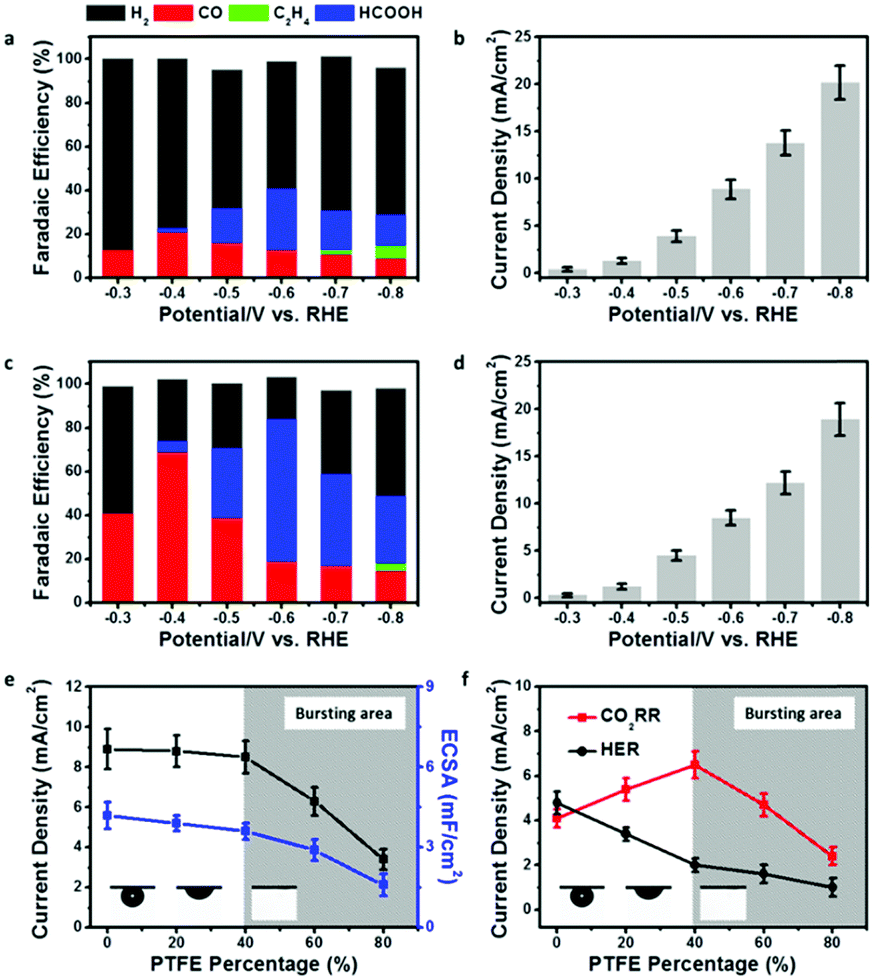 | ||
| Fig. 3 (a) Product distribution and (b) total current density using pristine Cu NW electrode for CO2RR. (c) Product distribution and (d) total current density using PTFE–Cu NW electrode for CO2RR. (e) Comparison of total current density and ECSA using Cu NW catalyst modified with different amounts of PTFE. (f) Comparison of CO2RR/HER current density using Cu NW catalyst modified with different amounts of PTFE. | ||
When the PTFE percentage was lower than 40%, the CO2RR current density increased and the HER current density decreased, demonstrating that an enhanced CO2RR efficiency could be tuned by controlling a proper PTFE concentration. It should be noted that the FE of CO2RR was enhanced while the FE of HER was suppressed on a bare Cu NW electrode as long as the Cu NWs was modified by PTFE. A straightforward interpretation of this phenomenon would be to assume that the aerophilic/hydrophobic electrode surface facilitates CO2 diffusion and depresses excessive proton adsorption from the aqueous electrolyte. When more PTFE was used, the CO2 gas bubble turned to the bursting state, and both the CO2RR and HER current density decreased, which may be due to the over-covered Cu active surface induced by excessive PTFE modification, as evidenced by ECSA and TEM characterizations (Fig. 3e and Fig. S11, ESI†).
Stability of an electrode is equally important for an electrocatalyst.36,37 We also examined durability of the pristine and PTFE–Cu NWs under a potential of −0.6 V vs. RHE for 4 h. Pristine Cu NWs exhibited a large decay in both current density (from ∼8.9 to ∼6.5 mA cm−2 in the first 2 h) and CO2RR product efficiency (FEs of CO and HCOOH were both lower than 10% after a stability test) (Fig. 4a and b), which was a common observation in previous reports.28 In obvious contrast, the PTFE–Cu NW electrode showed almost negligible degradation towards current density (95.5% retained). Meanwhile, the CO/HCOOH FEs of PTFE Cu NWs showed little attenuation as well (FEs of CO and HCOOH retained 90.9% and 96.4%, respectively), indicating that stability of the catalysts after PTFE modification was highly enhanced. This stability enhancement could be ascribed to the existence of optimized gas and ion diffusion layers around the catalyst's surface,32 as well as the structural stability of PTFE–Cu NWs, as evident by the wettability tailoring of the electrode towards both CO2 gas (Fig. S12, ESI†) and electrolyte (Fig. S13, ESI†). Specifically, for the pristine Cu NW electrode, the superaerophobic surface leads to the occurrence of CO2RR at the outermost layer (surface layer) of the electrode, resulting in a fast attenuation, as shown in Fig. 4c. Such rapid decay during CO2 electroreduction in an aqueous electrolyte system was commonly observed on an aerophobic metallic Cu surface.24,26,28 However, after PTFE modification, a more uniform CO2 diffusion layer was formed on the surface of the superaerophilic PTFE–Cu NW electrode, as shown in Fig. 4d and Table S2 (ESI†). Previous reports confirm that polyethylenimine (PEI) modified nitrogen-doped carbon nanotubes showed enhanced CO2 electroreduction performance due to the PEI function of capturing and stabilizing CO2 reactant.38 As evidenced by the contact angle measurement and electrochemical performance test, the PTFE modification strategy shown in this work could be more effective to facilitate CO2 diffusion in a bubble (micrometer) scale thus promoting the CO2RR selectivity.
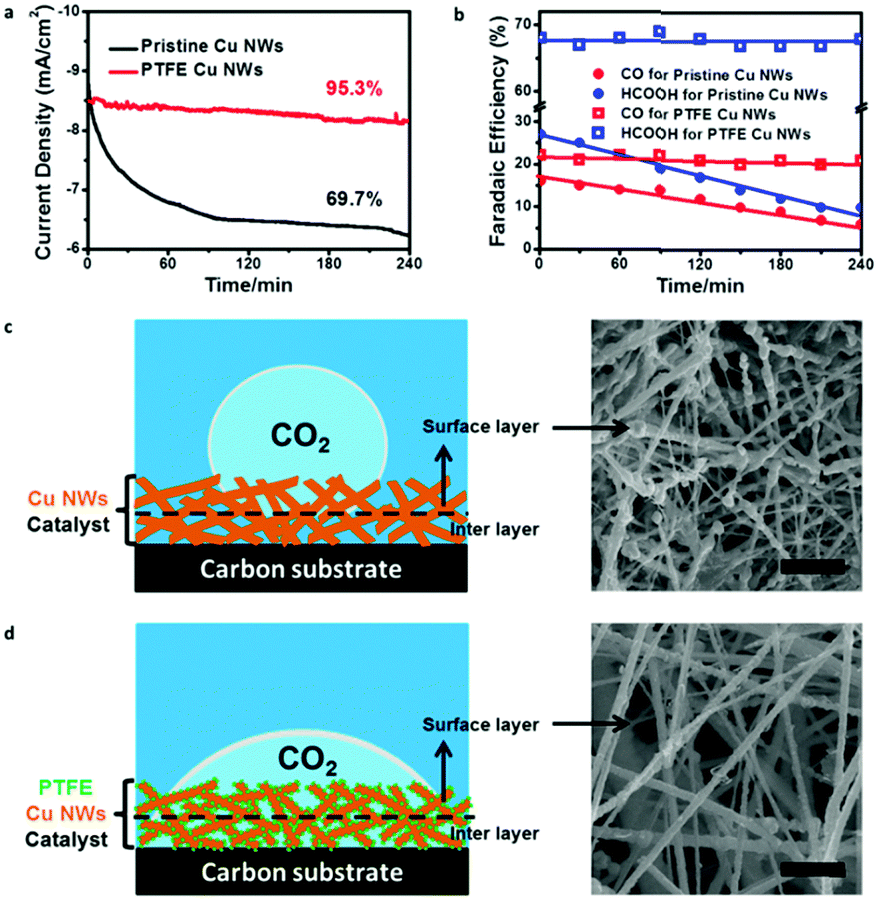 | ||
| Fig. 4 (a) Current density and (b) Faraday efficiency stability using pristine Cu NW and PTFE–Cu NW electrodes at −0.6 V vs. RHE. Schematic illustration and SEM images of (c) pristine Cu NWs and (d) PTFE–Cu NW electrode surface after stability testing, scale bars: 500 nm. | ||
Conclusions
In summary, this study demonstrates superaerophilic Cu nanowires by facile PTFE modification as an efficient and stable CO2RR electrocatalyst, featuring an efficient CO2 gas-diffusion pathway, which not only continually supplies CO2 reactants to boost the activity of switchable Cu catalysts, but also prevents excessive proton supply from the electrolyte, thus suppressing H2 production. Moreover, the product selectivity could be tuned between CO and HCOOH by facilely switching the potential. The stability of the Cu catalyst was also enhanced by the optimized gas and ion diffusion layers. Our work demonstrates the importance of optimizing electrode superwettability to promote CO2RR on a metallic catalyst and this strategy holds strong potential to be extended to other materials for different electrocatalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Lei Jiang for his helpful discussion. This work was supported by the Natural Science Foundation of China, National Key Research and Development Project (2016YFC0801302, 2016YFF0204402), the Program for Changjiang Scholars and Innovative Research Team in the University, and the Fundamental Research Funds for the Central Universities, and the long term subsidy mechanism from the Ministry of Finance and the Ministry of Education of PRC. Dr Yuxin Zhao thanks the National Science Fund of China (No. 61701543) for financial support.Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- H. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. Chen, C. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140 CrossRef CAS.
- L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed.
- F. C. Lei, W. Liu, Y. F. Sun, J. Q. Xu, K. T. Liu, L. Liang, T. Yao, B. C. Pan, S. Q. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed.
- R. Kas, K. K. Hummadi, R. Kortlever, P. de Wit, A. Milbrat, M. W. J. Luiten-Olieman, N. E. Benes, M. T. M. Koper and G. Mul, Nat. Commun., 2016, 7, 10748 CrossRef CAS PubMed.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- S. Gao, X. Jiao, Z. Sun, W. Zhang, Y. Sun, C. Wang, Q. Hu, X. Zu, F. Yang, S. Yang, L. Liang, J. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 698–702 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- Q. Zhu, J. Ma, X. Kang, X. Sun, J. Hu, G. Yang and B. Han, Sci. China: Chem., 2016, 59, 551 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS PubMed.
- C. Hahn, T. Hatsukade, Y.-G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- Y. Zhou, J. L. Silva, J. M. Woods, J. V. Pondick, Q. Feng, Z. Liang, W. Liu, L. Lin, B. Deng, B. Brena, F. Xia, H. Peng, Z. Liu, H. Wang, C. M. Araujo and J. J. Cha, Adv. Mater., 2018, 30, 1706076 CrossRef PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Z. Weng, X. Zhang, Y. Wu, S. Huo, J. Jiang, W. Liu, G. He, Y. Liang and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 13135–13139 CrossRef CAS PubMed.
- R. Shiratsuchi, Y. Aikoh and G. Nogami, J. Electrochem. Soc., 1993, 140, 3479 CrossRef CAS.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Wang, J. Luo, X. Lei and L. Jiang, Adv. Mater., 2014, 26, 2683 CrossRef CAS PubMed.
- Z. Lu, M. Sun, T. Xu, Y. Li, W. Xu, Z. Chang, Y. Ding, X. Sun and L. Jiang, Adv. Mater., 2015, 27, 2361–2366 CrossRef CAS PubMed.
- Z. Lu, W. Xu, J. Ma, Y. Li, X. Sun and L. Jiang, Adv. Mater., 2016, 28, 7155 CrossRef CAS PubMed.
- M. Liu, S. Wang and L. Jiang, Nat. Rev. Mater., 2017, 2, 17036 CrossRef CAS.
- Y. Tian, B. Su and L. Jiang, Adv. Mater., 2014, 26, 6872–6897 CrossRef CAS PubMed.
- S. Ye, A. R. Rathmell, I. E. Stewart, Y.-C. Ha, A. R. Wilson, Z. Chen and B. J. Wiley, Chem. Commun., 2014, 50, 2562–2564 RSC.
- Z. Cai, Y. Bi, E. Hu, W. Liu, N. Dwarica, Y. Tian, X. Li, Y. Kuang, Y. Li, X.-Q. Yang, H. Wang and X. Sun, Adv. Energy Mater., 2018, 8, 1701694 CrossRef.
- Z. Cai, D. Zhou, M. Wang, S. Bak, Y. Wu, Z. Wu, Y. Tian, X. Xiong, Y. Li, W. Liu, S. Siahrostami, Y. Kuang, X. Yang, H. Duan, Z. Feng, H. Wang and X. Sun, Angew. Chem., Int. Ed., 2018, 130, 9536–9540 CrossRef.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00259b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |