
Anchoring anions with metal–organic framework-functionalized separators for advanced lithium batteries†
Li
Shen
a,
Hao Bin
Wu
 *b,
Fang
Liu
a,
Chen
Zhang
a,
Shengxiang
Ma
a,
Zaiyuan
Le
a and
Yunfeng
Lu
*a
*b,
Fang
Liu
a,
Chen
Zhang
a,
Shengxiang
Ma
a,
Zaiyuan
Le
a and
Yunfeng
Lu
*a
aDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, 420 Westwood Plaza, Los Angeles, 90095, California, USA. E-mail: luucla@ucla.edu
bSchool of Materials Science and Engineering, Zhejiang University, Hangzhou, China. E-mail: hbwu@zju.edu.cn
First published on 17th January 2019
Abstract
The low Li+ transference number (tLi+), or the relatively small proportion of Li+ conductivity with respect to total ionic conductivity, has been identified as a key drawback of binary electrolytes. The lack of approaches to restrain anion mobility results in the poor cyclability of energy-dense electrodes as the high rate of anion movement induces concentration polarization. Herein, we propose regulating the ion conduction behavior using a nanocomposite separator fabricated via the functionalization of a glass fiber (GF) separator with a metal–organic framework (MOF). The open metal sites in the MOF serve as the anchoring sites for anions, and the resulting tLi+ is increased by 100%. The MOF-functionalized nanocomposite separator with high tLi+ substantially improves the electrochemical performance of a Li metal anode, with an areal capacity exceeding 2 mA h cm−2, and intercalation-type electrodes (LiFePO4 and Li4Ti5O12), with high active material loadings of 45 mg cm−2.
Conceptual insightsProgress toward next-generation batteries with high energy densities and power capabilities faces significant challenges due to the limitations of Li metal anodes and thick electrodes, especially under high-current operating conditions. The challenges largely originate from the inefficient transport of Li+ in conventional liquid electrolytes, in which effective Li+ conductivity only accounts for a small fraction of total conductivity, namely, a low Li+ transference number. Inefficient Li+ transport leads to concentration polarization and the proliferation of dendritic Li, deteriorating the interfacial stability of Li metal anodes and the capacity retention of thick electrodes. To address these issues, we developed nanocomposite functional separators (MOG) by modifying glass fiber (GF) membranes with a metal–organic framework (MOF), which effectively regulates the intrinsic ionic transport behavior of the liquid electrolyte. The open metal sites in the MOF anchor anions in the liquid electrolyte and promote effective Li+ transport, doubling the Li+ transference number. The tuning of the Li+ transport efficiency by MOG enables remarkably prolonged cycling of Li anodes and thick electrodes compared to cells with GF. This work opens a new route for regulating liquid electrolytes and battery performance with nanocomposite functional separators. |
Introduction
The electrification of automobiles using lithium-ion batteries (LIBs) represents a mainstream trend in sustainable transportation.1 The widespread deployment of electric vehicles (EVs) relies heavily on developing low-cost and energy-dense batteries with fast charge/discharge capability. Since the commercial debut of LIBs in the early 1990s, the energy density has been increased by almost two-fold, and the cost has been reduced by more than 90% based on advancements in battery chemistry and cell engineering.2 However, the short driving range and slow charging time significantly retard the further market penetration of EVs.3Two feasible approaches have been proposed to boost the energy densities of Li-based rechargeable batteries. Firstly, advanced electrode materials that enable high cell voltage and high capacity have been actively explored. For example, replacing commercial graphite anodes (372 mA h g−1, ∼0.2 V vs. Li/Li+) with Li (3860 mA h g−1, 0 V vs. Li/Li+) could potentially double the energy densities of full cells.4 However, the growth of Li dendrites, low Coulombic efficiency and the unstable deposition/stripping process of the Li anode pose formidable challenges for commercialization.5 Second, increasing the areal loadings of electrode materials enhances the energy density and reduces pack costs at the cell level from an engineering prospective.6 However, thick electrodes substantially compromise the cycle lifespan under high power output, which originates from poor ionic transport and polarization.7 The limitations of the above two strategies can be largely attributed to the paradox that poor Li+ transport efficiency does not support the passage of large numbers of electrons. Namely, effective Li+ conduction only accounts for a small portion of total ionic conductivity (σ) in binary electrolytes due to the bulkier solvation sheath of Li+ compared to anions.8 The Li+ transference number (tLi+), which is defined as the fraction of σ that is carried by Li+, of commercial liquid electrolytes is typically around 0.3.9 The low tLi+ of liquid electrolytes has been identified as the major cause of concentration polarization and proliferation of dendritic Li, which limits charge/discharge rates, degrades cycle life and hampers the use of thick electrodes.7,10,11
Separators, which are an indispensable component in batteries, prevent contact between the electrodes and accommodate liquid electrolyte. The properties of separators, such as porosity, tortuosity and wettability, affect the apparent battery performance.12 While separators are mostly considered as inert containers for liquid electrolytes, a few studies indicate that the ionic transport properties of liquid electrolytes could be affected by the separators.13 For example, parallel separators with single-ion conducting ionomers enhance tLi+, leading to negligible concentration polarization and suppressed deposition of dendritic Li.14,15 The modification of commercial polyolefin separators with ceramic nanoparticles and/or polymers has been widely practiced to fabricate functional separators.16–18 Incorporating ceramics such as ZrO2, SiO2 and Al2O3 has been found to improve the thermal stability and wettability of the separators while also promoting Li+ transport. However, the only slight improvement in tLi+ to around 0.45 renders this approach inefficient. Thus, effectively modulating the intrinsic ionic transport behavior and boosting the performance of liquid electrolytes using functional separators have not yet been achieved.
Herein, we demonstrate a new type of functional separator that can effectively enhance Li+ transport efficiency in liquid electrolytes, enabling the development of high-energy-density lithium batteries using a lithium metal anode and thick electrode. These functional separators with high thermal stability and mechanical robustness are fabricated by modifying glass fiber (GF) with a metal–organic framework (MOF). The tLi+ of the liquid electrolyte is doubled by anchoring the anions with MOF moieties. As a result, the Li+ transport limitation and concentration polarization in lithium batteries are effectively alleviated, enhancing the long-term cycling stability. This work demonstrates a general and practically feasible approach for improving the performance of lithium batteries using functional separators that regulate the ionic transport behavior of liquid electrolytes.
Results and discussion
As illustrated in Fig. 1, commercially available GF membranes were modified with MOF nanoparticles to obtain the MOF-GF composite separator (Fig. 1b, denoted as MOG). As a rapidly expanding family of crystalline porous solids, MOFs are constructed by periodically bridging inorganic metal clusters with organic ligands.19,20 The functionality of MOF herein is exemplified by a zirconium-based MOF, UiO-66-NH2, which consists of Zr6O4(OH)4 octahedra interconnected by 2-aminoterephthalic acid (NH2–BDC).21 The capped μ3-OH on Zr6O4(OH)4 undergoes dehydration upon thermal activation, yielding open metal sites (OMSs) on Zr6O6 (coordination number reduced from 8 to 7 for Zr4+, Fig. S1, ESI†).22 The resulting OMSs with Lewis acidity throughout the microporous scaffolds serve as anchoring points for anionic species in liquid electrolytes, thereby increasing Li+ transport efficiency (Fig. 1c).23 This concept is different from a recent study on a MOF-coated polyolefin separator, in which the MOF contains no OMSs, and the functional groups on the MOF work as ion regulators.24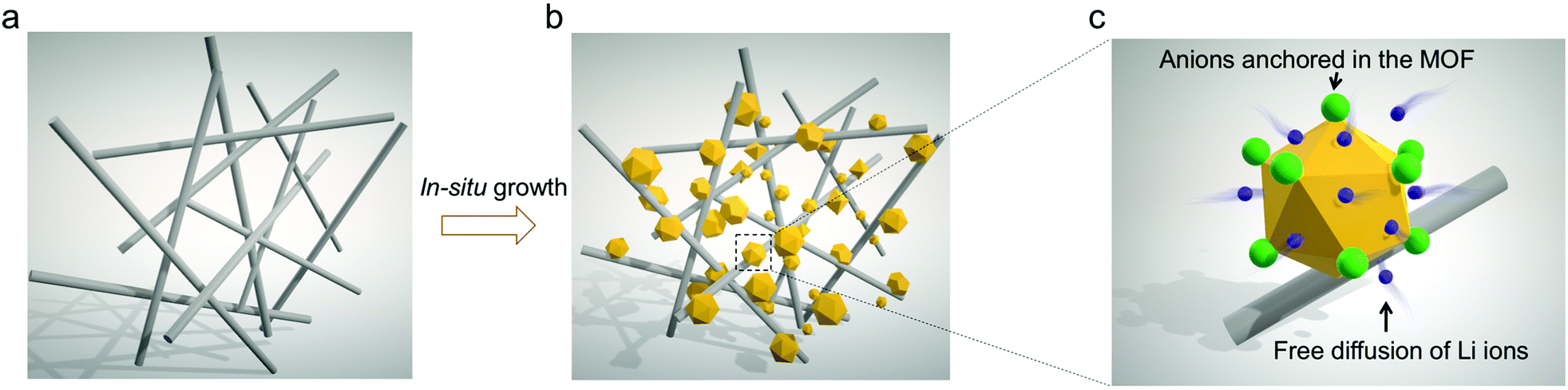 | ||
| Fig. 1 Schematic illustrations of (a) glass fiber (GF) and (b) the MOF-GF composite separator (MOG) and (c) an enlarged view showing ion transport behaviours in MOG. | ||
GF was selected as the porous matrix in view of its excellent thermal stability (>500 °C), high chemical stability and low cost.25,26 Moreover, the abundant hydroxyl groups on the borosilicate backbones interact with the dangling groups (e.g., –COOH and –NH2) on the MOF nanoparticles,27 facilitating the uniform deposition of MOF on GF. The facile in situ growth of MOF nanoparticles on GF was achieved by the infiltration of MOF precursors into GF porous membranes and subsequent heat treatment.28 As shown in Fig. 2a and c, the pristine GF membrane is composed of entangled fibers with micro-/submicrometer-sized diameters. After the in situ growth of the MOF, the fibrous structure was retained, while the GF surfaces were decorated with numerous nanoparticles (Fig. 2b and d). The successful growth of MOF on GF is also suggested by the change in the white color of the pristine GF to the yellow color of MOG after the in situ growth of MOF, as shown in the inset photograph of Fig. 2a. The crystal structure of the composite membrane was determined by powder X-ray diffraction (XRD), as shown in Fig. 2e. The pristine GF shows no diffraction peak due to its amorphous nature. The XRD pattern of MOG is similar to that of MOF synthesized without GF. Both patterns can be well indexed to the simulated pattern of parent UiO-66 (vertical lines at the bottom of Fig. 2e) with characteristic peaks at 2θ ≈ 7.3° and 8.5°. The slight deviation in the XRD pattern of MOG from those of pure and simulated MOF indicates possible defects (e.g., missing linkers) in the MOF moieties of MOG.29 The content of MOF in MOG was estimated to be around 35 wt% according to the weight gain after the MOF deposition process.
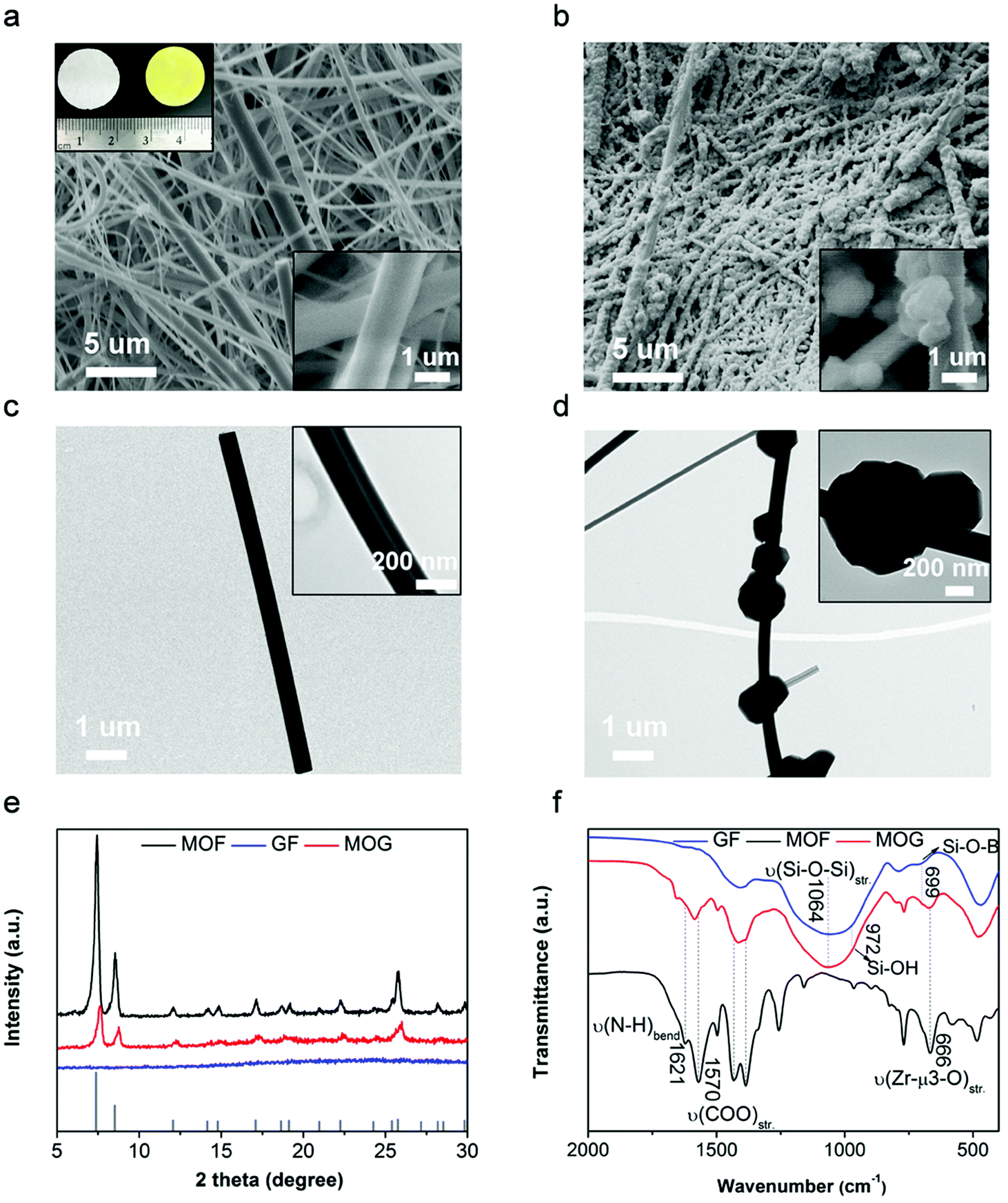 | ||
| Fig. 2 SEM images of (a) GF (inset in the top left shows images of white GF and yellowish MOG) and (b) MOG. TEM images of (c) GF and (d) MOG. (e) XRD patterns of MOF, GF and MOG (bottom: simulated pattern of MOF). (f) FTIR spectra of MOF, GF and MOG. | ||
The introduction of MOF moieties in MOG notably altered the pore structure and surface properties of the separators. N2 adsorption/desorption isotherms were collected to evaluate the pore structure (Fig. S2, ESI†). The commercial GF membrane exhibits a small Brunauer–Emmett–Teller (BET) surface area of 9 m2 g−1 and irregular macrospores, which are expected to result in very limited interaction with the liquid electrolyte. As a reference, pure MOF exhibits a high BET surface area of 795 m2 g−1 and dominant micropores with sizes of 9 Å. MOG shows a reasonable surface area of 453 m2 g−1 due to the limited loading of microporous MOF on GF. The slightly expanded pore size of 13 Å possibly originates from defective MOF growth on GF,30 which is consistent with the slight shift in the diffraction peaks of MOG compared to MOF. Fourier-transform infrared spectroscopy (FTIR) was performed to shed light on the evolution of chemical bonding (Fig. 2f). In the spectrum of GF, the broad peak centered at 1064 cm−1 accompanied by a pronounced shoulder at 972 cm−1 are attributed to the characteristic vibrations of Si–O–Si and Si–OH, respectively.31 In the spectrum of pure MOF, Zr-μ3-O (metal clusters) and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O/N–H (NH2–BDC linkers) are indicated by sharp signals at 666 cm−1 and 1570/1621 cm−1, respectively.32 In contrast, the spectrum of the MOG composite retains the major characteristic peaks of both GF and MOF; the decreased peak intensity of Si–OH and the redshifts in the peaks of –COO/N–H are indicative of interactions between GF and MOF.
C–O/N–H (NH2–BDC linkers) are indicated by sharp signals at 666 cm−1 and 1570/1621 cm−1, respectively.32 In contrast, the spectrum of the MOG composite retains the major characteristic peaks of both GF and MOF; the decreased peak intensity of Si–OH and the redshifts in the peaks of –COO/N–H are indicative of interactions between GF and MOF.
MOG was thermally activated at 200 °C under dynamic vacuum to completely remove the remaining solvent and create OMSs in MOF, which is crucial to regulate the liquid electrolyte. Compared to commercial polyolefin-based separators, MOG exhibits much greater thermal stability. After thermal activation, the MOG membrane well maintained its original shape, while the color changed from light yellow to dark yellow (Fig. S3a, ESI†). In sharp contrast, commercial polypropylene (PP) separators show drastic shrinkage after the same thermal treatment due to the low melting point of PP (Fig. S3b, ESI†). The thermal stability of MOG was quantitatively evaluated by thermogravimetric analysis (TGA). The TGA curves of GF and MOG in air atmosphere are shown in Fig. S4 (ESI†). GF shows negligible weight loss at temperatures up to 600 °C, while MOG after thermal activation exhibits gradual weight loss staring from 200 °C and stabilizes at 575 °C. The corresponding weight loss of 30% is ascribed to the oxidative decomposition of UiO-66-NH2 into ZrO2. This decent thermal stability of the MOG membrane would enable the successful operation of batteries at elevated temperature.
In the nanocomposite architecture of the resulting MOG, GF serves as a mechanical support, and MOF functions as an electrolyte regulator to tune the ion transport properties. To identify the roles of MOG as a functional separator, a mediocre liquid electrolyte, 1 M LiClO4 in propylene carbonate (LPC), was used for electrochemical evaluation. The ionic conductivity was measured by electrochemical impedance spectroscopy (EIS) with electrolyte cells containing LPC-saturated separators (see ESI† for the detailed cell design). Due to the decreased macropore volume of the MOG membrane and less electrolyte being taken up, the ambient ionic conductivity of the cell with MOG (2.06 mS cm−1 at 25 °C) is ∼19% lower than that of the cell with bare GF (2.54 mS cm−1). By fitting the ionic conductivities at different temperatures to the Arrhenius equation (Fig. 3a), the derived activation energies (Ea) of the two cells were similar, indicating similar ion transport mechanisms when using MOG or GF. The tLi+ value was analyzed using Li|Li symmetric cells with the conventional Bruce–Vincent method (see ESI† for detailed calculation equations).33 As shown in Fig. 3b, a small voltage bias was applied to polarize a Li|Li cell with an MOG separator. The initial and steady-state currents, which reflect the total ion movement and Li+ movement, respectively, were recorded. The interfacial resistances derived from EIS before and after potentiostatic polarization (inset of Fig. 3b) were considered to exclude the effect of interface evolution between Li with electrolytes when determining tLi+. LPC with MOG gives a high tLi+ of 0.67, which is twice the values obtained with GF or polyolefin separators (0.32–0.34; Fig. S5, ESI†). Thus, the effective Li+ conductivity of LPC with MOG is higher than that with GF (1.38 vs. 0.86 mS cm−1). Our previous study indicated that MOFs with OMSs interact with ClO4− anions and promote the dissociation of ion pairs in the liquid electrolyte, which explains the enhanced Li+ transport in the presence of MOG.22 As shown in Fig. S6 (ESI†), the FTIR spectrum of LPC-saturated MOG reveals the emergence of an asymmetric signal (636 cm−1) corresponding to ClO4− anions, which confirms the breakdown of anion symmetry and the complexation of anions with MOF.
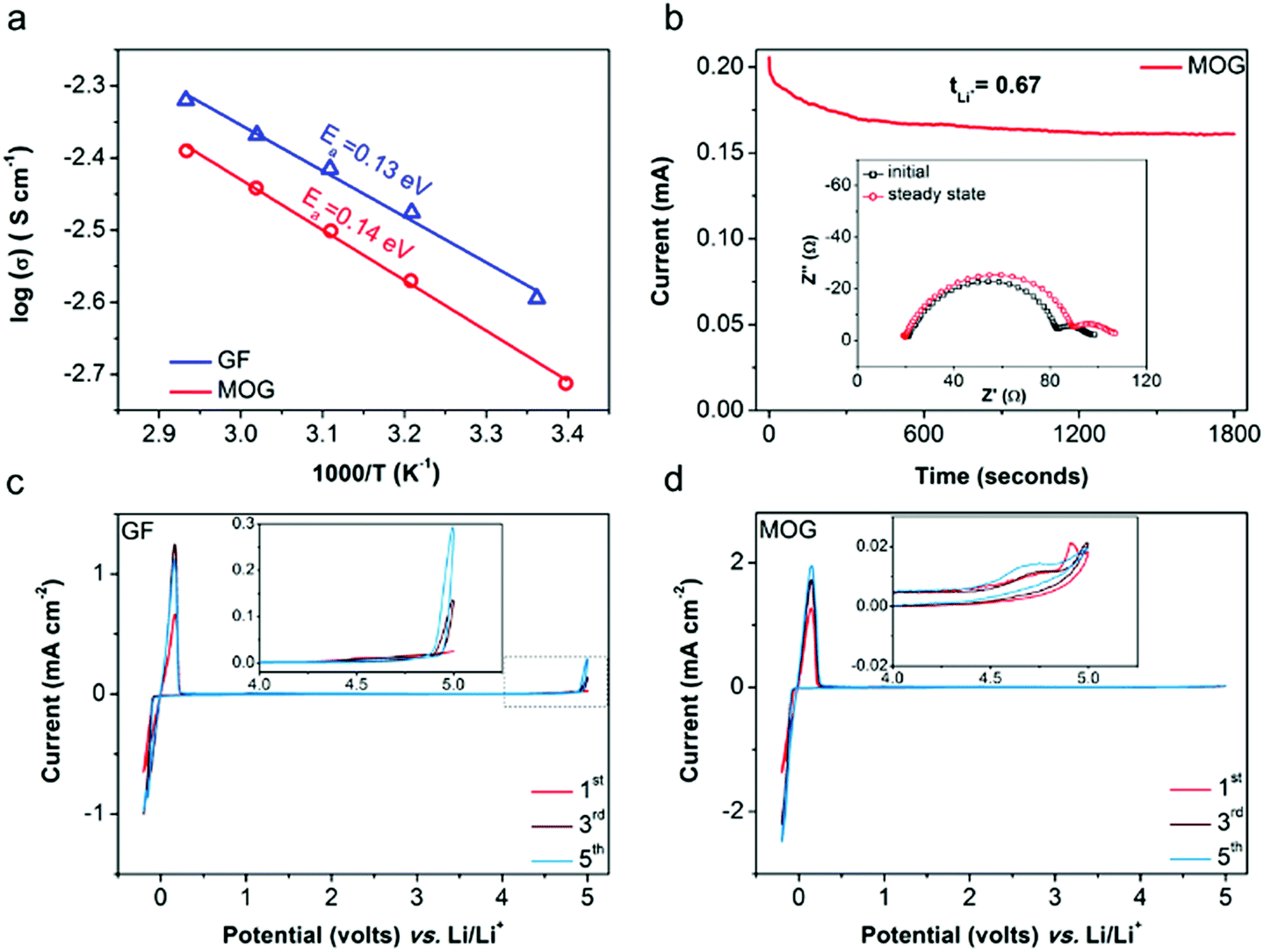 | ||
| Fig. 3 (a) Temperature dependent conductivities of LPC-saturated GF and MOG. The activation energies (Ea) are obtained from linear fittings of Arrhenius equation. (b) Measurement of Li+ transference number (tLi+) by potentiostatic polarization of Li|Li symmetric cell with MOG (inset shows the EIS plots before and after polarization). CV curves of SS|Li cells using (c) GF and (d) MOG between −0.2 to 5 V (vs. Li/Li+) at a sweep rate of 1 mV s−1 (insets show enlarged curves above 4 V). | ||
To evaluate the electrochemical stability window, cyclic voltammetry (CV) was performed in asymmetric cells with stainless-steel plates (SS) as the working electrode and Li as the counter/reference electrode (SS|Li). Fig. 3c and b show the representative CV curves of SS|Li cells using GF and MOG separators, respectively. Notable redox peaks near 0 V (vs. Li/Li+) are attributed to Li+ stripping and plating processes on the SS electrodes. Moreover, the reaction peak current from the cell with MOG (≈2 mA cm−2) is about twice that of the cell using GF (≈1 mA cm−2), which suggests faster reaction kinetics of Li+ stripping/plating in the presence of MOG, as indicated by the Randles–Servick equation.34 During the anodic sweep, the cell using GF exhibits increasing peak current (up to 0.3 mA cm−2) above 4.4 V (vs. Li/Li+), indicating the progressive oxidative decomposition of electrolyte.35 In contrast, the cell using MOG maintains a substantially smaller peak current (consistently below 0.02 mA cm−2), implying greatly suppressed electrolyte breakdown at the interphase between the electrode and LPC-saturated MOG. The advantage of MOG in mitigating electrolyte decomposition can be interpreted by the partial electrolyte confined within the MOF pore channels, leading to fewer side reactions between the electrolyte and the electrodes.
In lithium batteries, the degradation of the lithium metal anode is the major factor limiting cycle life. Thus, Li symmetric cells (Li|Li) were tested with different LPC-saturated separators to evaluate the long-term cycling stability of the lithium metal anode. Galvanostatic tests were performed at 1.5 mA cm−2 with an areal capacity of 3 mA h cm−2 for each cycle to mimic the practical situation in lithium batteries. As shown in Fig. 4a, the cell using GF suffers from escalating polarization; the cell voltage drastically increases from 50 to 800 mV after operation for 200 h, which is a typical sign of the growth of Li dendrites and the increase in interfacial resistance.36 After 200 h, a sudden drop in overpotential suggests a short circuit in the cell due to lithium dendrite penetration. In comparison, the cell using MOG manifests a steady and smooth curve for up to 350 h with a small overpotential of ∼55 mV. This demonstrates the advantages of MOG in blocking Li dendrites and ameliorating interfacial resistance with Li. The Coulombic efficiency (CE) of Li plating and stripping on Cu electrode (Cu|Li cell) is shown in Fig. S7 (ESI†) with a fixed areal capacity of 0.5 mA h cm−2 at 0.5 mA cm−2. The cell with MOG retains a higher CE (93%) than the cell using GF (86%). At a higher areal capacity of 2 mA h cm−2 and current density of 1 mA cm−2 (Fig. 4b), the cell with bare GF shows a rapid decay in CE, and only ∼36% of the plated capacity is reversible at the 25th cycle, indicating the occurrence of severe parasitic reactions between LPC and Li metal. In contrast, the cell using MOG exhibits a much superior performance with an average CE of 95%. This CE is remarkable for a carbonate-based electrolyte, which is known to form an unstable solid–electrolyte interphase (SEI) on Li metal. Thus, the presence of MOF moieties at the interphase between Li metal and MOG is expected to assist in the formation of a more robust SEI that blocks the continuous parasitic reactions between LPC and Li metal.
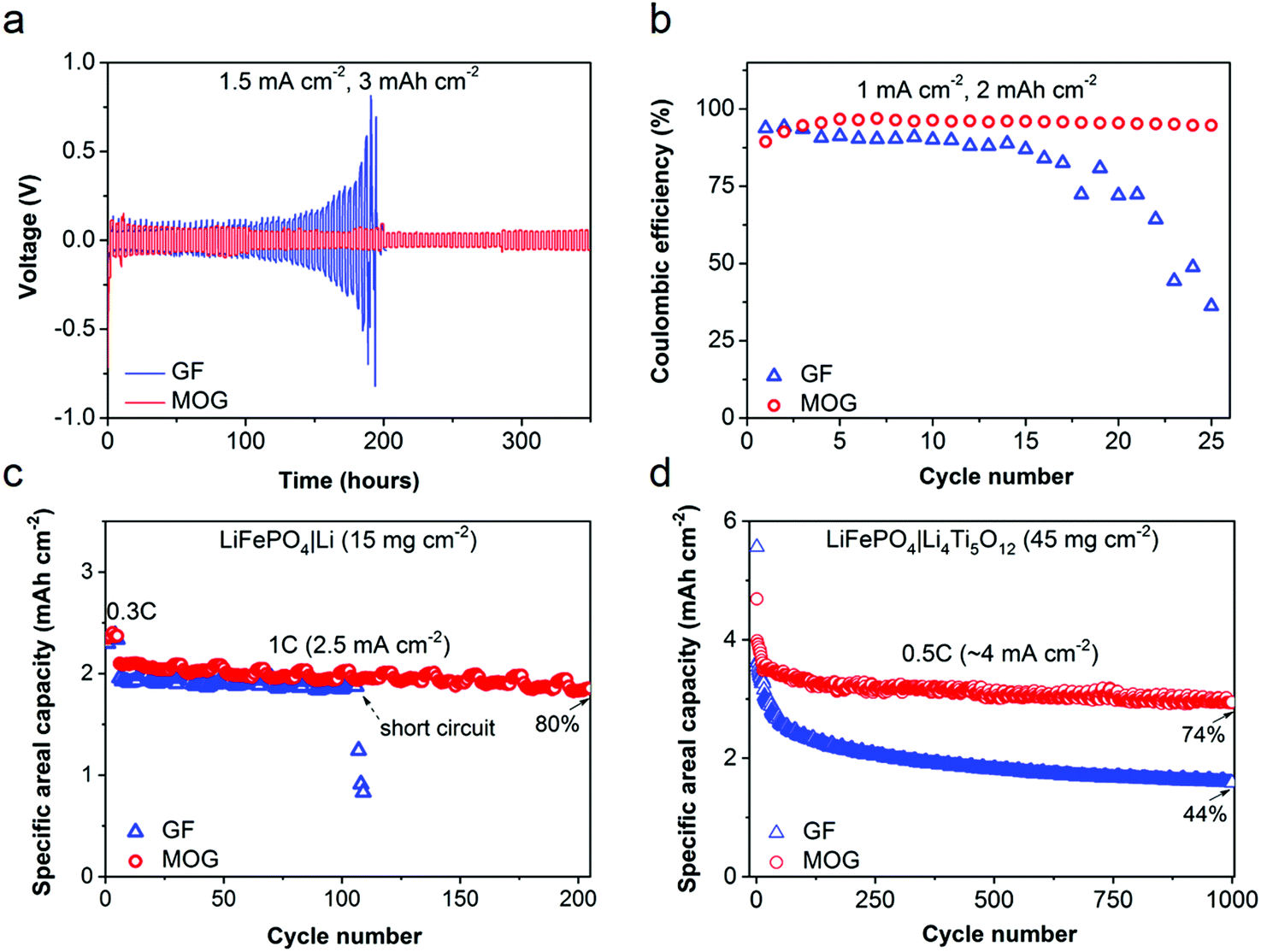 | ||
| Fig. 4 (a) Li+ stripping and plating tests in Li|Li cells using GF and MOG (3 mA h cm−2 for each cycle at 1.5 mA cm−2). (b) Coulombic efficiency of Li+ stripping and plating in Cu|Li cells (2 mA h cm−2 for each discharge process at 1 mA cm−2). (c) Galvanostatic cycling of LiFePO4|Li cells using GF and MOG at a current density of 1C (2.5 mA cm−2) between 2.4 and 4 V. (d) Long-term cycling of LiFePO4|Li4Ti5O12 full cells using GF and MOG at a current density of 0.5C (4 mA cm−2). | ||
To demonstrate the potential use of MOG separators in lithium batteries, prototype cells were assembled using LiFePO4 as a cathode with an active material loading of 15 mg cm−2 and Li metal as anode (LiFePO4|Li). The rate performance of the LiFePO4|Li cell was evaluated at various rates from 0.2 to 5C. As depicted in Fig. S8 (ESI†), the cell using MOG retains 31% of its original capacity (0.2C) at 5C, which is much higher than the cell using GF (4.6%) under the same conditions. The superior rate capability of the cell using MOG demonstrates the high Li+ transport efficiency and alleviated concentration polarization under high-rate operation. Fig. 4c compares the cycling stability of cells using MOG and GF separators. After first 5 cycles at 0.3C (0.75 mA cm−2), the cell with MOG displays an areal capacity of 2.1 mA h cm−2 at a current density of 1C (2.5 mA cm−2) and maintains ∼80% of the value after 200 cycles. The control cell with GF shows a slightly lower capacity of 2.0 mA h cm−2, and an abrupt capacity decline is observed at the 106th cycle, indicating the penetration of dendritic Li (Fig. S9, ESI†). This phenomenon can be interpreted by the ion concentration gradient across the cell upon polarization at high current density.37 Due to the lower mobility of Li+ compared to anions (low tLi+) in liquid electrolyte, the depletion of Li+ near the electrode surface leads to local space charge, decomposition of anions/solvent molecules, and the growth of dendritic Li. This is evidenced by the SEM images of post-cycled Li electrodes harvested from LiFePO4|Li cells at 1C (100 cycles). As shown in Fig. S10 (ESI†), the surface of Li using GF exhibits a porous and dendritic morphology. In contrast, the surface of Li using MOG is smooth and dense.
Accelerating effective Li+ transport with MOG would effectively stabilize the interface between liquid electrolyte and Li metal. In addition, the utilization of active material in conventional intercalation-type electrodes with large thickness would also be notably improved. This was exemplified by cycling full cells comprising high-loading LiFePO4 cathodes and Li4Ti5O12 anodes (45 mg cm−2), as shown in Fig. 4d. Under a constant current density of 4 mA cm−2 (corresponding to 0.5C), the cell using MOG maintains an areal capacity of ∼3 mA h cm−2 at the 1000th cycle, which is 74% of the value at second cycle. However, aggressive capacity fading is observed for the cell with bare GF, with only 44% capacity retained at the 1000th cycle. This notable difference is related to the polarization at high areal current density, as verified by the severe voltage hysteresis in the cell with GF compared to the cell with MOG (Fig. S11, ESI†). Thus, the facile Li+ transport assisted by the MOG separators would enable the use of thick intercalation-type electrodes, which are critical to the development of advanced lithium batteries with high energy/power density.
Conclusions
In summary, we developed novel functional separators based on nanocomposite membranes of MOF-functionalized GF to tackle the intrinsic limitations of liquid electrolytes. By taking advantage of the unsaturated OMSs in MOFs, the anions in the liquid electrolyte are immobilized by the MOG separator, leading to enhanced Li+ transport, as verified by the doubled tLi+ value compared to the bare GF separator. With the MOG separator, the issues related to Li metal anodes, including large overpotential, low Coulombic efficiency and dendrite penetration, were alleviated, even under high current density. Moreover, thick intercalation-type electrodes (45 mg cm−2) have been successfully demonstrated with a lifespan of over 1000 cycles at 4 mA cm−2. The MOG separator functionalized with MOFs enables the stable operation of Li metal anodes and thick electrodes at high current density, which is critical to the development of advanced lithium batteries with high energy and power density. This work also provides an alternative strategy to improve electrochemical devices via the proper functionalization of ancillary components.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work financially supported by the HK-UCLA technical center for graphene technology and energy storage. H. B. W. acknowledges funding from the “Thousand Young Talents Program” of China, “Hundred Talents Program” of Zhejiang University, and the International Joint Laboratory of the Chinese Education Ministry on Resource Chemistry.Notes and references
- M. A. Hannan, M. S. H. Lipu, A. Hussain and A. Mohamed, Renewable Sustainable Energy Rev., 2017, 78, 834–854 CrossRef.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194 CrossRef CAS PubMed.
- K. G. Gallagher, S. E. Trask, C. Bauer, T. Woehrle, S. F. Lux, M. Tschech, P. Lamp, B. J. Polzin, S. Ha, B. Long, Q. Wu, W. Lu, D. W. Dees and A. N. Jansen, J. Electrochem. Soc., 2016, 163, A138–A149 CrossRef CAS.
- Z. Du, D. L. Wood, C. Daniel, S. Kalnaus and J. Li, J. Appl. Electrochem., 2017, 47, 405–415 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CrossRef CAS.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- F. Jiang and P. Peng, Sci. Rep., 2016, 6, 32639 CrossRef CAS PubMed.
- H. Lee, M. Yanilmaz, O. Toprakci, K. Fu and X. Zhang, Energy Environ. Sci., 2014, 7, 3857–3886 RSC.
- S. S. Zhang, J. Power Sources, 2007, 164, 351–364 CrossRef CAS.
- Z. Tu, M. J. Zachman, S. Choudhury, S. Wei, L. Ma, Y. Yang, L. F. Kourkoutis and L. A. Archer, Adv. Energy Mater., 2017, 7, 1602367 CrossRef.
- Z. Tu, S. Choudhury, M. J. Zachman, S. Wei, K. Zhang, L. F. Kourkoutis and L. A. Archer, Joule, 2017, 1, 394–406 CrossRef CAS.
- M. Chi, L. Shi, Z. Wang, J. Zhu, X. Mao, Y. Zhao, M. Zhang, L. Sun and S. Yuan, Nano Energy, 2016, 28, 1–11 CrossRef CAS.
- Z. Wang, F. Guo, C. Chen, L. Shi, S. Yuan, L. Sun and J. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 3314–3322 CrossRef CAS PubMed.
- X. Mao, L. Shi, H. Zhang, Z. Wang, J. Zhu, Z. Qiu, Y. Zhao, M. Zhang and S. Yuan, J. Power Sources, 2017, 342, 816–824 CrossRef CAS.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- H. B. Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- R. Ameloot, M. Aubrey, B. M. Wiers, A. P. Gómora-Figueroa, S. N. Patel, N. P. Balsara and J. R. Long, Chem. – Eur. J., 2013, 19, 5533–5536 CrossRef CAS PubMed.
- L. Shen, H. B. Wu, F. Liu, J. L. Brosmer, G. Shen, X. Wang, J. I. Zink, Q. Xiao, M. Cai, G. Wang, Y. Lu and B. Dunn, Adv. Mater., 2018, 30, 1707476 CrossRef PubMed.
- W. Liu, Y. Mi, Z. Weng, Y. Zhong, Z. Wu and H. Wang, Chem. Sci., 2017, 8, 4285–4291 RSC.
- J. Zhu, M. Yanilmaz, K. Fu, C. Chen, Y. Lu, Y. Ge, D. Kim and X. Zhang, J. Membr. Sci., 2016, 504, 89–96 CrossRef CAS.
- X.-B. Cheng, T.-Z. Hou, R. Zhang, H.-J. Peng, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Adv. Mater., 2016, 28, 2888–2895 CrossRef CAS PubMed.
- B. Zhang, Q. Wang, J. Zhang, G. Ding, G. Xu, Z. Liu and G. Cui, Nano Energy, 2014, 10, 277–287 CrossRef CAS.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- B. J. C. Thomas, M. S. P. Shaffer and A. R. Boccaccini, Composites, Part A, 2009, 40, 837–845 CrossRef.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. Doyle, T. F. Fuller and J. Newman, Electrochim. Acta, 1994, 39, 2073–2081 CrossRef CAS.
- R. Mukherjee, R. Krishnan, T.-M. Lu and N. Koratkar, Nano Energy, 2012, 1, 518–533 CrossRef CAS.
- D. Aurbach, Y. Talyosef, B. Markovsky, E. Markevich, E. Zinigrad, L. Asraf, J. S. Gnanaraj and H.-J. Kim, Electrochim. Acta, 2004, 50, 247–254 CrossRef CAS.
- Y. Lu, M. Tikekar, R. Mohanty, K. Hendrickson, L. Ma and L. A. Archer, Adv. Energy Mater., 2015, 5, 1402073 CrossRef.
- J. N. Chazalviel, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 7355–7367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00342d |
| This journal is © The Royal Society of Chemistry 2019 |