
Sequential catalytic nanomedicine augments synergistic chemodrug and chemodynamic cancer therapy†
Ruijie
Liang
a,
Yu
Chen
 *b,
Minfeng
Huo
b,
Jun
Zhang
c and
Yongsheng
Li
*a
*b,
Minfeng
Huo
b,
Jun
Zhang
c and
Yongsheng
Li
*a
aLab of Low-Dimensional Materials Chemistry, Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: ysli@ecust.edu.cn
bState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: chenyu@mail.sic.ac.cn
cDepartment of Radiology, Huashan Hospital Affiliated to Fudan University, Shanghai 200040, China
First published on 20th February 2019
Abstract
The tumor microenvironment (TME) provides intriguing features/indications for rational design of diverse therapeutic protocols with high tumor specificity and therapeutic efficacy. In this work, we report the introduction of the sequential catalytic concept into theranostic nanomedicine for cancer-specific therapy, which has been achieved by the construction of glucose oxidase (GOD) and Mitomycin C (MMC) co-loaded superparamagnetic iron oxide nanoparticles (designated as SMG nanocatalysts). Based on the large amounts of glucose in tumors, the GOD component in SMG catalyzes glucose to convert into hydrogen peroxide (H2O2) and gluconic acid with the simultaneous consumption of oxygen. The post-produced H2O2 is further catalyzed by iron oxide in SMG to produce large amounts of highly toxic hydroxyl radicals for cancer therapy, and the generated gluconic acid enhances such a Fenton-based catalytic reaction. On the other hand, the loaded MMC drug is activated because of the consumption of oxygen and enhanced hypoxia in tumors, causing high chemotherapeutic efficacy. Based on the high synergistic chemodrug and chemodynamic therapeutic efficacy in combating cancer, cancer cells are efficiently killed and tumor growth is thus significantly suppressed. This work paves a new way for cancer therapy by taking into account the full features and advantages of the TME and the physiochemical properties of the chosen nanocatalysts, which also links nanocatalytic science and nanomedicine for proposing new efficient tumor-therapeutic modalities.
Conceptual insightsWith specific conditions including mild acidity, unique vascular distribution and hypoxia, the tumor microenvironment (TME) severely decreases the therapeutic efficacy of the major therapeutic modalities such as radiation therapy, reactive oxygen species-related therapy and the most conventional chemotherapy. Therefore, it is highly promising to take into account the full features of the TME for the design of therapeutic systems for tumor-specific therapy with concurrent high therapeutic efficacy and low side effects in combating cancer. Herein, a synergistic chemodrug and chemodynamic therapeutic strategy has been introduced for efficient cancer therapy by designing a simple biocompatible sequential nanocatalyst augmented with a hypoxia-activated chemodrug, which is able to in situ catalyze oxygen and nutrients (glucose) to toxic ROS responsive to the specific TME. The elaborately designed synergistic chemodrug and chemodynamic nanocatalysts with high biocompatibility (SMG nanocatalysts) exhibit a highly expected tumor-inhibition effect towards 4T1 breast tumor xenografts (77.96%), simultaneously with high therapeutic biosafety. |
As one of the most conventional antitumor strategies, chemotherapy suffers several critical demerits such as insufficient bioavailability and non-specificity to tumor regions, causing severe side impacts towards the healthy organs/tissues of patients.1–7 In contrast, physical therapies, including radiotherapy,8–11 ultrasound therapy,12,13 photothermal therapy14–16 and photodynamic therapy,17–19 exhibit satisfactory curative effects accompanied by the competencies of locating the therapeutic sites with the aid of diverse imaging modalities such as magnetic resonance imaging (MRI), ultrasound (US) imaging, photoacoustic (PA) imaging, computerized tomographic (CT) imaging, etc. Nevertheless, the above-mentioned therapies usually cause some serious damage to normal tissues, or suffer from limited tissue-penetrating depth or incur miserable tumor metastasis.20 Therefore, the development of tumor-specific therapies with concurrent high therapeutic efficacy and low side effects are highly urgent and necessary.
It has been well-demonstrated that tumors feature a unique microenvironment, known as the tumor microenvironment (TME), in which the pH value, vascular distribution, chemical composition and oxygen level differ from those in normal tissues, which has been extensively explored for designing diverse tumor-specific theranostic modalities.21–23 For instance, specific-responsive aggregation,24 drug release25,26 and imaging27 in the TME have been investigated broadly. In addition, its pharmacology,28 simulated models29 and specific tumor models30 have been studied for cancer therapy and drug development.31 Moreover, various cytokines, enzymes and responsive systems such as hypoxia factors,32 legumain33 and acidity34 have been applied for TME-responsive therapy. Therefore, taking the full features of the TME into account for the design of therapeutic systems for combating cancer is highly promising for tumor-specific therapy.
Triggering an in situ chemical reaction for tumor-specific therapy is now one of the research frontiers for theranostic nanomedicine, such as chemical reaction-induced chemodynamic therapy,35 gas therapy,36,37 diagnostic imaging,27 and chemotherapy,6 which is mainly based on some specific substances in the TME. For instance, the hydrogen peroxide (H2O2)-based Fenton reaction has been triggered for chemodynamic therapy, which kills the cancer cells by generating toxic hydroxyl radicals (˙OH).38 However, the H2O2 concentration in the TME is still rather low,39 which means that it would not produce sufficient reactive oxygen species (ROS) in vivo when the Fenton reaction was triggered. In addition, it has been considered that the acidity of solid tumors (extracellular pH 6.5–7.0)40 is still not sufficient to guarantee the effective Fenton reaction where the typical acidic condition is required. It should also be noted that the oxygen level in the intratumoral microenvironment (20–30 mmHg)41 is much lower as compared to normal tissues (well-known as hypoxia), which severely decreases the therapeutic efficacy of the major therapeutic modalities such as radiation therapy, ROS-related therapy and chemotherapy.42–48
To simultaneously address the series of aforementioned critical issues for tumor-specific and TME-responsive therapy, this work proposes the new concept of catalytic nanomedicine (CNM) by taking the full reactant and product features of a representative enzyme-catalytic reaction for subsequently enhanced chemodynamic therapy and hypoxia-enhanced chemotherapy. The fast growth of the tumor consumes a large amount of glucose; therefore, glucose is abundant and important in the TME.49 The introduction of glucose oxidase (GOD) can efficiently catalyze the reaction of glucose and oxygen (O2) to produce H2O2 and gluconic acid.50 The further introduced catalysts of Fenton reactions (e.g., Fe-based catalysts) could react with the produced H2O2 to generate highly toxic hydroxyl radicals (˙OH) for killing the cancer cells, and the produced gluconic acid could increase the TME acidity to accelerate the Fenton reaction and improve the production efficacy of hydroxyl radicals.51 In particular, such an enzyme-catalyzed reaction by GOD would decrease the oxygen level and accelerate the hypoxia in the TME.50 The further introduction of a hypoxia-activated chemotherapeutic drug could further induce cancer-cell death, therefore synergistically enhancing the Fenton reaction-based chemodynamic therapy.52 This work has successfully designed and constructed a simple biocompatible nanoplatform to fully achieve the above-mentioned goal, which is based on superparamagnetic biocompatible iron oxide nanoparticles (Fenton catalysts) co-loaded with GOD (enzyme) and Mitomycin C (MMC, a hypoxia-activated drug). Systematic evaluations of the in vitro intracellular level and an in vivo tumor xenograft in nude mice have been conducted to demonstrate the highly efficient and synergistic therapeutic outcome of this new nanocatalytic concept for combating cancer.
Superparamagnetic iron oxide nanoparticles (SPIONs) have been approved by the Food and Drug Administration (FDA) for clinical T2-weighted magnetic resonance imaging (MRI),53,54 indicating their high biocompatibility for biomedical use. In particular, research work has recently focused on the therapeutic application of SPIONs based on their specific catalytic capability for the Fenton reaction, which is easily triggered in the TME.55 As the classical Fenton nanoagents, SPIONs feature higher biocompatibility without the hazard of toxic heavy metal ions, which makes them more appropriate than other Cu-based nanoparticles serving as the triggering agents in this sequential catalytic system. This work engineered monodispersed SPIONs for tumor-specific and MRI-guided synergistic therapy based on triggering a unique sequential reaction just in the TME (Fig. 1). Considering the stability of the nanoagents and the difficulties in the synthesis and surface engineering, herein highly biocompatible iron oxide nanoparticles with appropriate size and adequate catalytic capability were chosen for the sequential nanocatalytic reaction. Initially, monodispersed 12 nm-sized oleic-capped hydrophobic SPIONs were synthesized using a typical high-temperature pyrolysis protocol.56 Then, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino-(polyethylene glycol)2000] (DSPE-PEG2000-NH2) was employed to transfer the hydrophobic SPIONs into aqueous solution (SPION@PEG). A hypoxia-activated chemotherapeutic drug, Mitomycin C (MMC), was encapsulated into the polymeric shell of SPION@PEG (SPION-MMC@PEG). Finally, GOD was anchored onto the surface of SPION-MMC@PEG via the typical amide reaction between the amino group of DSPE-PEG2000-NH2 and the carboxyl group of GOD (SPION-MMC@PEG-GOD, abbreviated as SMG, Fig. 1a). It is well known that the immobilization of natural enzymes is one of the factors hindering the large-scale industrial enzymatic catalytic applications. The immobilization of GOD would slightly depress its catalytic performance; meanwhile, its stability would be strengthened. To keep the balance between activity and stability, we introduced GOD with high catalytic capability, in order to guarantee that the catalytic efficiency of slightly reduced GOD could still meet the need of the sequential nanocatalysts. In addition, there are many alternatives which could replace GOD for triggering this sequential reaction, such as calcium peroxide,57 the acetylcholine-acetylcholinesterase combination,58 and titania-coated gold nanorods.59 This design is a general approach where the critical components of the composite nanocatalysts could be substituted by their analogues for synergistic chemodynamic therapy and hypoxia-activated chemotherapy.
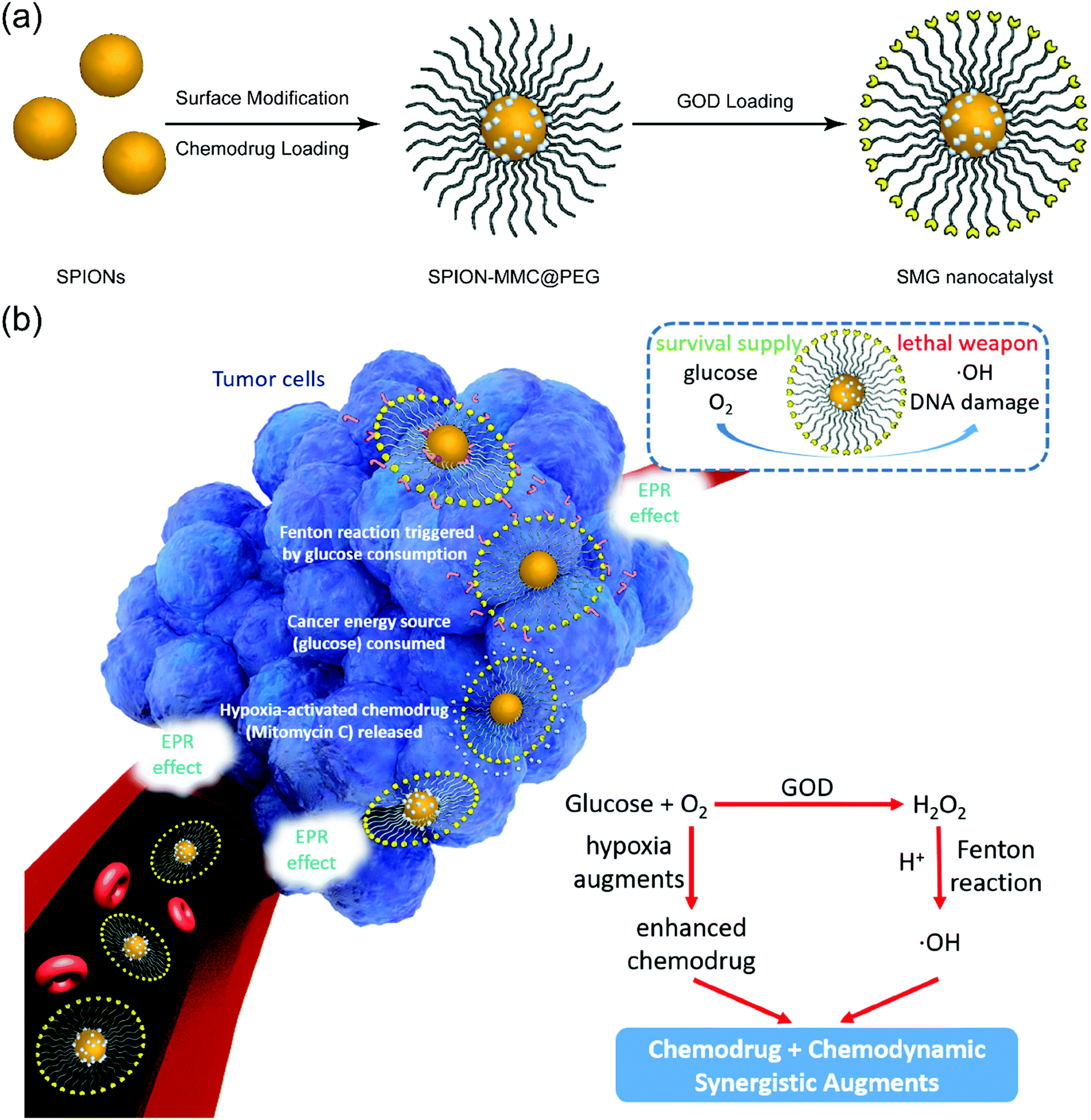 | ||
| Fig. 1 Schematic illustration of the synthetic procedure for SMG nanocatalysts and corresponding synergistic chemodrugs and chemodynamic therapy of cancer as augmented by sequential catalytic nanomedicine. (a) Stepwise drug-loading and enzyme conjugation on SPIONs. (b) In vivo accumulation of SMG nanocatalysts into the tumor via the typical EPR effect and TME-responsive sequential catalytic reactions for killing cancer cells, and further hypoxia-activated chemotherapy for synergistic treatment efficacy. | ||
The as-designed SMG nanoparticles (NPs) with small sizes could efficiently accumulate in tumors via the typical enhanced permeability and retention (EPR) effect (Fig. 1b).60 Upon arriving at the tumor, the GOD component in SMG initially catalyzes the transformation of glucose into H2O2 and gluconic acid along with the simultaneous consumption of oxygen. The Fe3O4 component in SMG then catalyzes the Fenton reaction by transforming the as-produced H2O2 into toxic hydroxyl radicals (˙OH), which is also enhanced by the increased acidity because of the contribution from post-produced gluconic acid. In particular, the loaded MMC drug is activated for chemotherapy by the accelerated hypoxia because of the oxygen consumption during the GOD catalysis process. Therefore, the SMG nanoplatforms for cancer-specific and synergistic therapy are mainly based on the unique TME and the full use of the GOD-catalyzed chemical reactions.
Uniform particle-size distribution and high dispersity of the original oleic-capped hydrophobic SPIONs were characterized by transmission electron microscopy (TEM, Fig. 2a and Fig. S1a, Fig. S2, ESI†). The high crystallinity of the as-synthesized SPIONs was demonstrated via high-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) (Fig. 2b, inset of Fig. 2b). In particular, the morphology and dispersity of the NPs were not changed after multi-step modification including PEGylation (Fig. 2c), MMC loading and further GOD conjugation (Fig. 2d and Fig. S1b–e, ESI†), and no obvious discrepancy could be observed against the high crystallinity nature of the nanocatalyst matrix. In addition, the particle-size distribution shows that the GOD conjugation slightly enlarges the particle size (from 34 nm to 90 nm) of the initial SPION@PEG as determined by dynamic light scattering (DLS, Fig. 2e). The magnetization curve of the SPIONs shows negligible coercivity and remanence, suggesting the superparamagnetic nature of the as-prepared NPs, and in addition, consistent with their diameter, the specific saturation magnetization was up to 42 emu g−1 (Fig. 2f). X-ray diffraction (XRD) patterns of SPIONs show the characteristic peaks at 30.1°, 35.4°, 43.1°, 56.9°, and 62.5°, which are indexed to the (220), (311), (400), (511), and (440) lattice planes of the magnetite phase of the SPIONs (Fig. 2g).
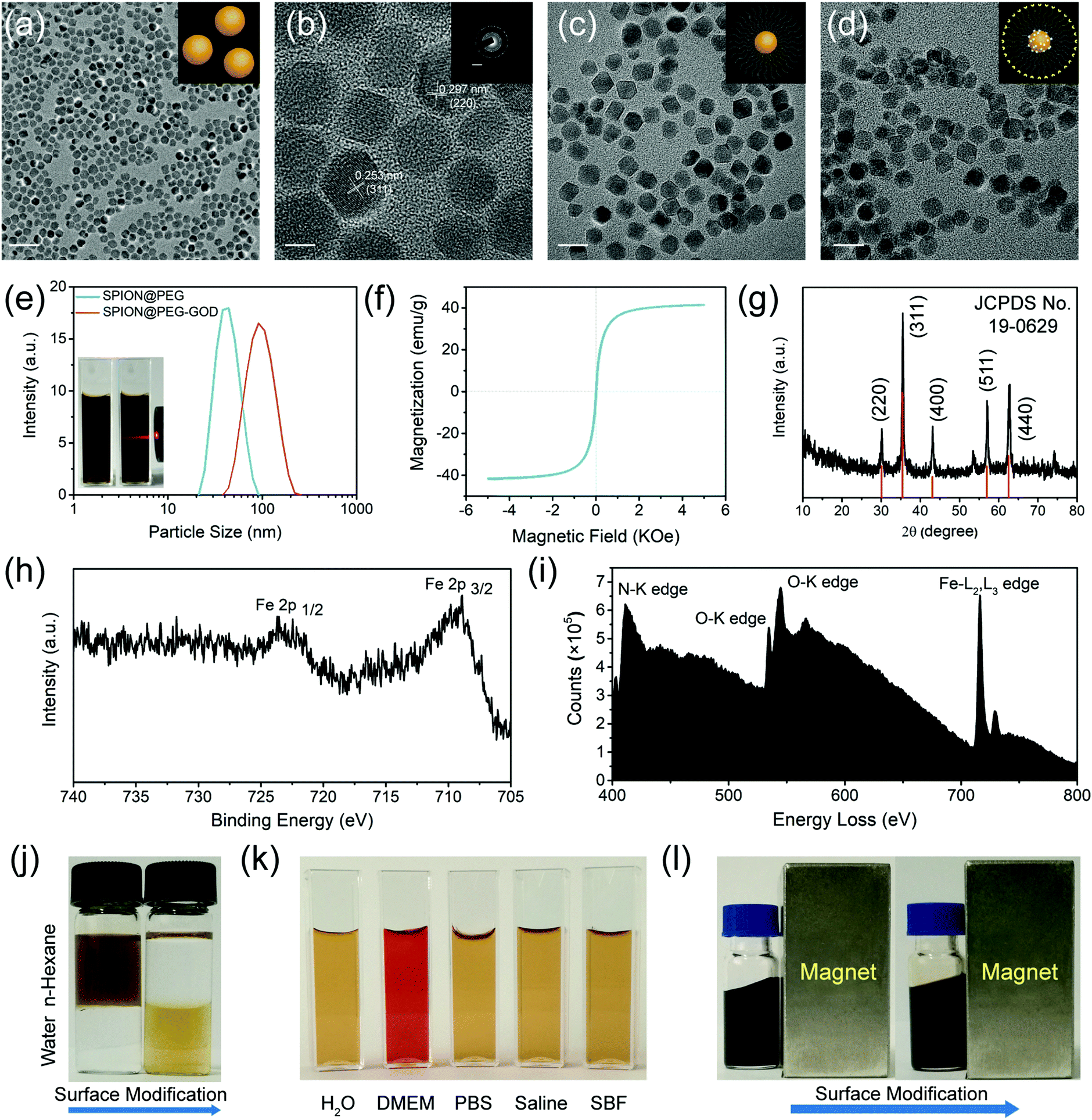 | ||
| Fig. 2 Characterizations of SPION, SPION@PEG, SPION@PEG-GOD and SMG nanocatalysts. (a) TEM image and (b) HRTEM image of SPIONs. Scale bar, (a) 100 nm; (b) 5 nm; inset: 5 nm−1. Inset: The SAED pattern of SPIONs, indicating the high crystallinity feature of the NPs. TEM images of (c) SPION@PEG NPs and (d) SMG nanocatalysts. Scale bar, (c) and (d) 20 nm. (e) Hydrodynamic diameter distribution of SPION@PEG NPs and SPION@PEG-GOD NPs. The inset shows digital photographs of SPION@PEG NPs dispersed in deionized water, exhibiting the typical Tyndall effect. (f) Field-related magnetization curve (M–H) of SPIONs measured at 300 K showing the superparamagnetic nature. (g) XRD pattern of SPIONs corresponding to the typical magnetite structure. (h) Local XPS spectrum of the Fe 2p region from the whole spectrum of SMG nanocatalysts. (i) EELS of SMG nanocatalysts. (j) A digital photograph of SPIONs dispersed in water–hexane mixed solution before (left images) and after (right images) surface PEGylation. (k) A digital photograph of SPION@PEG NPs dispersed in several representative solvents including deionized water, DMEM, PBS, saline and SBF. (l) Digital images of SPIONs with a macroscopic magnetic response feature before (left images) and after (right images) surface PEGylation. | ||
Fig. S3 (ESI†) and Fig. 2h show the X-ray photoelectron spectroscopy (XPS) and the corresponding Fe 2p region of the SMG nanocatalysts. The core level binding energies at 708.7 eV and 723.9 eV are assigned to the typical doublets of Fe 2p1/2 and Fe 2p3/2 of Fe3+, respectively. Furthermore, the loading of GOD was confirmed by the typical Fourier Translation Infrared spectroscopy (FT-IR). The obvious characteristic peak attributed to the carbonyl stretching vibration for the GOD conjugation appears at 1680 cm−1, indicating the formation of an amide bond between GOD and DSPE-PEG2000-NH2 (Fig. S4a, ESI†). Although the characteristic peak was red-shifted to 1650 cm−1 due to the loading of hypoxia-sensitive chemodrug MMC, the successful conjugation could be verified as well (Fig. S4b, ESI†). The electron energy loss spectrometer (EELS) analysis of SMG nanocatalysts exhibits the presence of obvious N, O, and Fe signals, demonstrating the desirable SMG composition (Fig. 2i). The conjugation of GOD occupied the amino groups on the surface of the SPION@PEG NPs, reducing the zeta potential from −25.9 mV to −45.6 mV (Fig. S5, ESI†).
The thermogravimetric analysis (TGA) was carried out to quantify the loading amounts of GOD and MMC. According to the TGA curves and corresponding normalized mass loss distribution plots of the SPION@PEG, SPION-MMC@PEG and SMG nanocatalysts, the loading amounts of GOD and MMC are calculated to be 4.73% and 4.99%, respectively (Fig. S6, ESI†). As shown in Fig. 2j, oleic-capped hydrophobic SPIONs were transformed into water-dispersive NPs. In particular, the surface PEGylation of SPIONs guarantees their high dispersity in physiological solutions, which has been demonstrated by their high dispersity in different solvents including Dulbecco's Modified Eagle's Medium (DMEM), phosphate buffered solution (PBS), saline and simulated body fluid (SBF) (Fig. 2k). The desirable superparamagnetic property of SPIONs endows them with magnetic modulation behavior by responding to externally introduced magnetic fields (Fig. 2l).
It has been revealed that the appropriate pH range of GOD is 4–7, while the optimal pH value is 5.5,61 and this natural enzyme takes effect at 20–50 °C. Therefore, the mild acidic condition (pH 6.5–7.0) in the tumor microenvironment is in favor of the sequential catalytic system.62 Furthermore, the generated gluconic acid (pKa = 3.60) enhances such a Fenton-based catalytic reaction under the catalysis of GOD. Hence, the in vivo tumor microenvironment would strengthen its catalytic performance. Due to the catalytic nature of GOD as an organic enzyme and SPIONs as the nanocatalysts, such a sequential catalytic procedure of the SPION@PEG-GOD nanocatalysts exhibits glucose and pH dependence, which could decrease the dissolved oxygen level and the pH value, and enhance the production efficacy of hydroxyl radicals (˙OH) (Fig. 3a).50,51 Hydroxyl radicals with strongly oxidizing nature, the ultimate product of the sequential catalytic procedure, could easily be characterized by a typical dye depigmentation protocol. To validate the generation of ˙OH during the sequential catalytic procedures, methylene blue (MB) was applied to detect ˙OH signals. The depigmentation of MB was attributed to a series of oxidation processes, in which the carbon–nitrogen bonds were disrupted and N-dimethylamino groups were oxidized, leading to a decline in the absorption peak at 664 nm.63 Following a series of distinct procedures, the products of MB oxidative degradation turned to benzoic acid, acetic acid, oxalic acid formic acid and achromatous organic acids, resulting in the fading of MB. As a strong oxidant, ˙OH could oxidatively decompose MB to achromatous acids as well.
 | ||
| Fig. 3 In vitro catalytic performances of SPION@PEG-GOD NPs for Fenton-like reactions. (a) Schematic illustration of the catalytic procedure of SPION@-PEG-GOD NPs used in Fenton-like reactions. (b and c) Methylene blue depigmentation curves of the experimental group (SPION@PEG-GOD and glucose), GOD + glucose groups and glucose only groups within (b) 1 h and (c) 3 d. (d) Time-dependent absorbance of TMB under the catalysis of SPION@PEG-GOD NPs with the addition of glucose at elevated concentrations (0.375 mM, 0.75 mM, 1.5 mM, and 3 mM). (e) Linear sections of absorption curves in the period of time from 90 s to 150 s. (f) Michaelis–Menten kinetic fitting curves of TMB under the catalysis of SPION@PEG-GOD NPs. Inset: Digital photographs of SPION@PEG-GOD before (left) and after (right) the chromogenic reaction of TMB. (g) ESR spectra of SPION@PEG-GOD NPs in mildly acidic solution (pH = 6.5) with or without the addition of glucose. Dissolved oxygen analysis of SPION@PEG-GOD NPs under (h) mildly acidic conditions (pH = 6.5) with the addition of glucose at elevated concentrations (10 mM, 20 mM, 40 mM, and 80 mM) or (i) in various buffers (pH = 4.5, 5.5, and 6.5) at the fixed glucose concentration of 40 mM. | ||
According to the short-term degradation curves of MB (Fig. 3b), the fading rate of the control group (GOD + glucose groups) was enormously higher (6 folds) than that of the experimental group in a short period (10 min), which indicates that the production of hydrogen peroxide (H2O2) could result in the depigmentation of MB as well. However, the coloration of the GOD + glucose group was restored over time. The negative impact of hydrogen peroxide on MB was eliminated within 6 h (Fig. 3c). In contrast, the SPION@PEG-GOD and glucose groups generated hydroxyl radicals (˙OH) persistently, leading to the depigmentation of MB with non-restoring nature.
The GOD component in SPION@PEG-GOD could catalyze β-D-glucose to produce hydrogen peroxide (H2O2), which was further catalyzed by the SPION component to generate highly toxic hydroxyl radicals under the mildly acidic conditions of the TME. To characterize such a sequential catalytic reaction for producing hydroxyl radicals, the typical chromogenic agent 3,3′,5,5′-tetramethyl-benzidine (TMB) was used for in vitro kinetic characterization in this cascade reaction. Because of the ˙OH production by the decomposition of H2O2 as catalyzed by SPIONs, the colorless TMB could be oxidized by ˙OH to produce TMB oxide, a chromogenic reagent which can be used to determine the relative concentration of ˙OH in solution at the wavelength of 650 nm (E = 3.9 × 104 M−1 cm−1) via spectrophotometry.
Glucose at elevated concentrations (0.375 mM, 0.75 mM, 1.5 mM and 3 mM) was applied as the reactant under the catalysis of SPION@PEG-GOD. In this assay, GOD acted as the natural organic enzyme and SPION functioned as the peroxidase-like nanoenzyme.50 Therefore, the representative Michaelis–Menten steady-state kinetics were chosen to estimate the catalytic activity of SPION@PEG-GOD in this cascade reaction (Fig. 3d–f).64 The time-dependent absorbance is displayed in Fig. 3d and e and the initial velocities corresponding to glucose concentrations were acquired. In accordance with Beer–Lamberts Law (eqn (1)), average initial velocities (v0) originating from absorbance varieties could be converted to initial velocities (v0) of hydroxyl radical or cation free radical formation, which were protracted to corresponding glucose concentrations and fitted with Michaelis–Menten curves (eqn (2) and Fig. 3f). Photographs (inserted in Fig. 3f) exhibit the chromatic changes before and after glucose addition. The values of the Michaelis–Menten constant (KM) and the maximum velocities (Vmax) of SPION@PEG-GOD were calculated to be 1.56 mM and 1.81 × 10−7 M s−1, respectively.
The KM value of SPION@PEG-GOD NPs reveals that the sequential nanocatalysts would exhibit half of the maximum catalytic performance with 1.56 mM β-D-glucose. Hence, sufficient therapeutic efforts could be achieved with SMG nanocatalysts since the endogenous concentrations of glucose in the cytoplasm of cancer cells are ordinarily below 3 mM.65 Notably, provided all the active sites of the sequential nanocatalysts were occupied, SMG nanocatalysts could catalyze β-D-glucose under the maximum velocity of 1.81 × 10−7 M s−1, exhibiting satisfying sustained therapeutic effects against cancer.
To qualitatively detect and analyze the formation of short-lived hydroxyl radicals (less than 10−8 s), radical scavenger, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was used to trap radicals to produce radical-DMPO adducts with relatively long life-time, which could be identified by electron spin resonance (ESR) spectroscopy.66 It has been found that the appearance of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 hydroxyl radical signals in the ESR spectrum of SPION@PEG-GOD evidences the characteristics of the DMPO-OH adduct under weak acid conditions (pH = 6.5) (Fig. 3g), indicating that glucose-triggered sequential catalytic reactions as catalyzed by SPION@PEG-GOD nanocatalysts could be triggered in the mildly acidic surrounding microenvironment. No radical signals were detected in the ESR spectrum under similar conditions in the absence of glucose.
:2:2:1 hydroxyl radical signals in the ESR spectrum of SPION@PEG-GOD evidences the characteristics of the DMPO-OH adduct under weak acid conditions (pH = 6.5) (Fig. 3g), indicating that glucose-triggered sequential catalytic reactions as catalyzed by SPION@PEG-GOD nanocatalysts could be triggered in the mildly acidic surrounding microenvironment. No radical signals were detected in the ESR spectrum under similar conditions in the absence of glucose.
The pH-dependent dissolved oxygen scavenging was further monitored by in vitro testing the dissolved oxygen in aqueous solution via an oxygen electrode (Fig. 3h). After the co-incubation of glucose at elevated concentrations (80 mM, 40 mM, 20 mM and 10 mM) with the SPION@PEG-GOD nanocatalysts, it has been found that the dissolved oxygen level decreased significantly at the glucose concentration of 20 mM where the dissolved oxygen level substantially declined to approximately 50% in 180 s, indicating the efficient catalytic performance of the SPION@PEG-GOD nanocatalysts. To further assess the pH influence on the catalytic performance, the dissolved oxygen level was evaluated in a series of buffer solutions with different pH values (Fig. 3i). The SPION@PEG-GOD nanocatalysts exhibited optimal catalytic performance in a mildly acidic environment (pH = 6.5), which means that mild acidity could improve the catalytic efficiency of GOD. As the pH of the environment transformed to a further more acidic degree (pH = 5.5), the enzymatic activity of the SPION@PEG-GOD nanocatalysts showed a slight decrease. As the pH value further decreased to 4.5, departing from the optimum pH scope of GOD, the enzymatic activity of the SPION@PEG-GOD nanocatalysts was overwhelmingly restrained, leading to extremely low dissolved oxygen scavenging performance. Furthermore, mildly acidic conditions are beneficial for the Fenton reaction.38 Hence, the mildly acidic TME in solid tumors is a favorable condition for the sequential catalytic therapeutic performance of the SMG nanocatalysts.
| Beer–Lamberts law: A = kbc | (1) |
 | (2) |
Confocal laser scanning microscopy (CLSM) images distinctly show the efficient cellular uptake of fluorescein isothiocyanate (FITC)-labeled SPION@PEG NPs within 2 h of co-incubation (Fig. S7, ESI†). Then, the cytotoxicities of carriers with or without the loading of the hypoxia-activated chemodrug and the TME-specific enzyme were further evaluated. The potential in vitro cytotoxicity was evaluated against 4T1 mammary tumor cells using the cell-counting kit-8 (CCK-8) assay. In the absence of the substrate (H2O2), the SPION@PEG NPs exhibit catalytic inactivity and have negligible cytotoxicity against the proliferation of 4T1 cells at concentrations as high as 800 μg mL−1 after 24 h of co-incubation (Fig. 4a), indicating the high biocompatibility of the nanocatalysts.
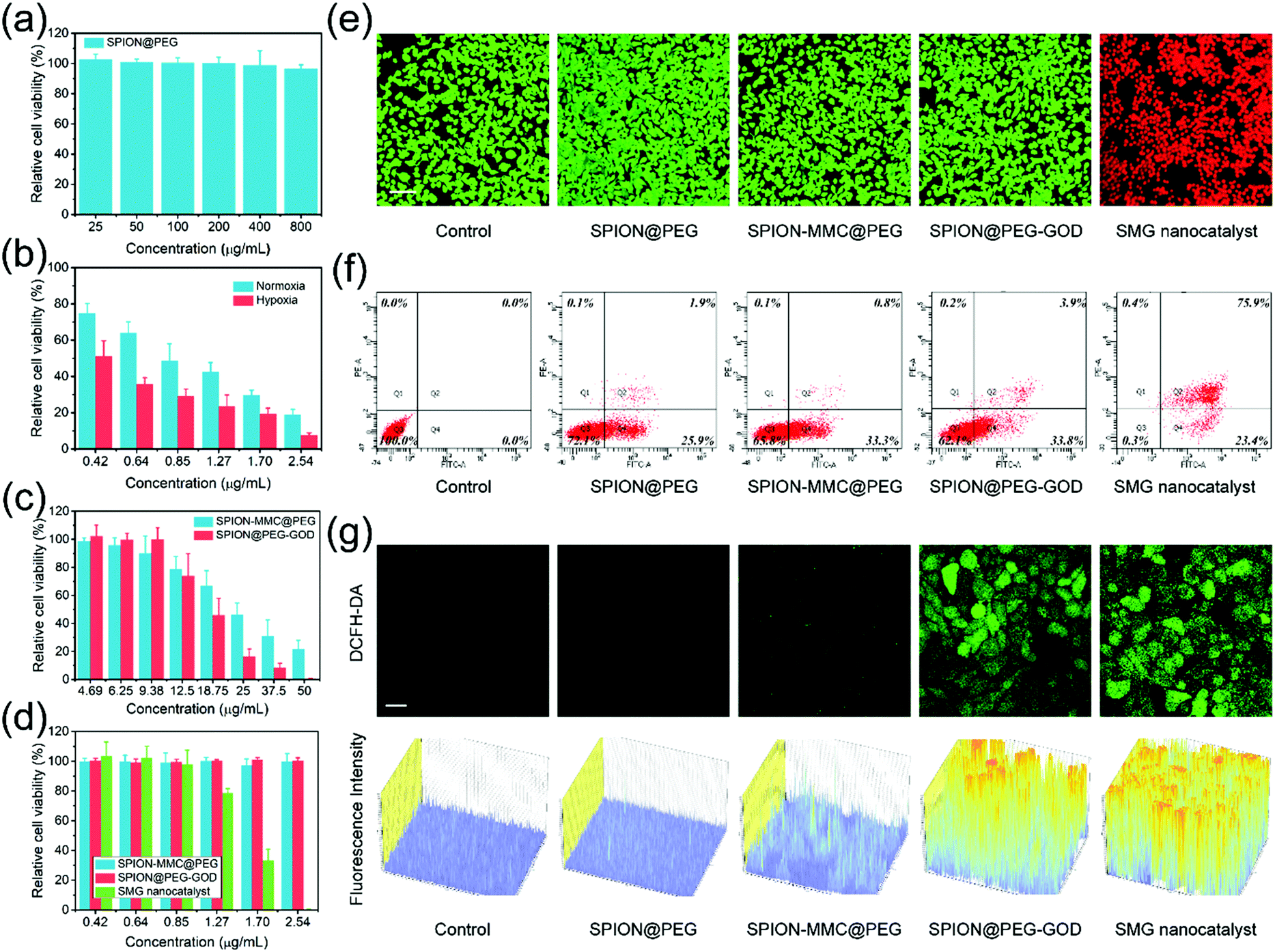 | ||
| Fig. 4 In vitro synergistic chemodrug and chemodynamic therapy as augmented by sequential catalytic reactions. In vitro cytotoxicity histograms of (a) SPION@PEG, (b) free MMC under normoxic and hypoxic conditions and (c and d) SPION-MMC@PEG, SPION@PEG-GOD and SMG catalysts. (e) CLSM images of live or dead cell distributions after co-incubation for 24 h with the corresponding nanoagents at the concentration of 2.54 μg mL−1, followed by staining with calcein-AM/PI reagents. Scale bar: 100 μm. (f) Flow cytometry analysis of 4T1 cells after co-incubated for 24 h with the corresponding agents and staining by Annexin-V/PI reagents. (g) CLSM images of 4T1 tumor cells after co-incubation with or without varied nanoagents for 4 h, followed by staining with fluorescent probe DCFH-DA for intracellular ROS detection. Scale bar: 30 μm. | ||
As a typical hypoxia-activated chemodrug with an alkylating function, MMC exhibits enhanced cytotoxicity in the hypoxic microenvironment of tumors.67 This chemodrug goes through one-electron reduction to semiquinone radical anions under the catalysis of NAD(P)H:cytochrome C (P450) reductase.68 By cross-linking with the double helix of DNA molecules, MMC may destroy the structure and function of tumor cells, inhibiting DNA replication in proliferating cells. Unfortunately, this process could be back-oxidized in the presence of molecular oxygen. Hence, decreasing the oxygen level in the TME would inevitably strengthen the intratumoral cytotoxicity of MMC. This specific property endows MMC, as well as SMG nanocatalysts with hypoxia-enhanced toxicity. The in vitro cytotoxicity profiles of MMC were obtained under both normoxic (21% O2) and hypoxic (1% O2) conditions. After 24 h of co-incubation with 4T1 mammary tumor cells, free MMC at various concentrations (0.42, 0.64, 0.85, 1.27, 1.70 and 2.54 μg mL−1) shows concentration-dependent hypoxia-activated cytotoxicity (Fig. 4b). In the CCK-8 assay, free MMC at low concentration down to 2.54 μg mL−1 presents a relative cell viability of 18.96% under normoxic conditions while much lower cell viability (7.57%) could be observed at identical concentration under hypoxic conditions. Furthermore, free MMC under hypoxic conditions shows a relative cell viability of approximately 60% that under normal oxygen content at all concentrations, indicating the hypoxia-activated feature of MMC.
Due to the loading of chemodrug MMC in the SPION-MMC@PEG NPs, they presented desirable in vitro concentration-dependent cytotoxicity. The obvious cytotoxicity was also observed in the CCK-8 assay of SPION@PEG-GOD because of the production of strongly oxidative hydroxyl radicals and the exhaustion of nutrients (Fig. 4c). Importantly, the co-loading of MMC and GOD combining sequential catalysis with hypoxia-activated chemotherapy enormously enhanced the in vitro cytotoxicity of the SMG nanocatalysts. The SMG nanocatalysts killed almost all 4T1 tumor cells even at extremely low concentration (2.54 μg mL−1), showing great potential in synergistic catalytic nanomedicine for cancer therapy (Fig. 4d).
The visualization of the aforementioned cytotoxicity was achieved with CLSM images of live or dead cell distributions. Stained with calcein-AM/PI reagents, 4T1 tumor cells co-incubated with the SMG nanocatalysts for 24 h exhibited obvious red fluorescence while the cells in other groups emitted green fluorescence, confirming the high therapeutic efficacy of the SMG nanocatalysts (Fig. 4e). Based on flow cytometry, the qualitative and quantitative assessments of the in vitro cell apoptosis as induced by the SMG nanocatalysts were assessed (Fig. 4f). After being co-incubated with the SMG nanocatalysts for 24 h and then stained with Annexin-V/PI reagents, the percentages of early apoptosis, late apoptosis and necrosis of 4T1 mammary cancer cells were measured to be 0.3%, 23.4%, and 75.9%, respectively, indicating the high therapeutic efficacy of the SMG nanocatalysts.
The whole cytotoxicity of the SMG nanocatalysts includes hypoxia-activated chemotherapy and TME-specific chemodynamic therapy. To visualize and quantify the ROS production in TME-specific chemodynamic therapy, intracellular ROS detection was conducted (Fig. 4g). 4T1 cancer cells were stained by a ROS fluorescence probe, 2′,7′-dichlorofluorescin diacetate (DCFH-DA), after co-incubation with SPION@PEG-GOD for 4 h. Barely no fluorescence was observed in the CLSM images of the control and SPION@PEG groups, while faint green fluorescence was distinguished in cells co-incubated with SPION-MMC@PEG, which could be attributed to the unoxidized semiquinone radical anions induced by MMC. In contrast to the control group, the strong green fluorescence was obviously observed in the CLSM images of the tumor cells co-incubated with the SPION@PEG-GOD NPs or SMG nanocatalysts, indicating substantial production of ROS intracellularly. Furthermore, the corresponding fluorescence intensity provides a quantitative assessment of the ROS-generating capability of the SPION@PEG-GOD NPs and SMG nanocatalysts.
Immunoblot (IB) analysis of hypoxia inducible factor-1α (HIF-1α) expression was performed for probing the intracellular hypoxia status while glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was chosen as an internal reference (Fig. S8, ESI†). 4T1 mammary cancer cells were co-incubated with SPION@PEG NPs, SPION-MMC@PEG NPs, SPION@PEG-GOD NPs and SMG nanocatalysts for 4 h before analysis. Based on the immunoblot assay, the relative contents of HIF-1α of SPION@PEG and SPION-MMC@PEG were found to be approximately 1, while those of the SPION@PEG-GOD and SMG nanocatalysts show an approximately 60% augment, indicating significant enhancement induced by intracellular HIF-1α in the presence of SPION@PEG-GOD NPs and SMG nanocatalysts.
In particular, the presence of SPION in SMG is highly desirable for contrast-enhanced T2-weighted MRI for potentially monitoring the therapeutic process of SMG in combating cancer, which was further systematically evaluated both in vitro and in vivo on a 3.0 T clinical MR scanner. Initially, the in vitro MRI assay was conducted by measuring the relaxation time of the SMG nanocatalysts in aqueous solution. It has been found that the SMG nanocatalysts show darker contrast in the T2-weighted MR images with elevated Fe concentrations from 0 to 0.50 mM as compared to pure water (Fig. 5a). To analyze quantitatively, the values of 1/T2 were linear with increased Fe concentrations, based on which the r2 value of the SMG nanocatalysts was calculated to be 60.14 mM−1 s−1 (Fig. 5b), indicating the desirable T2-MRI performance of SMG.
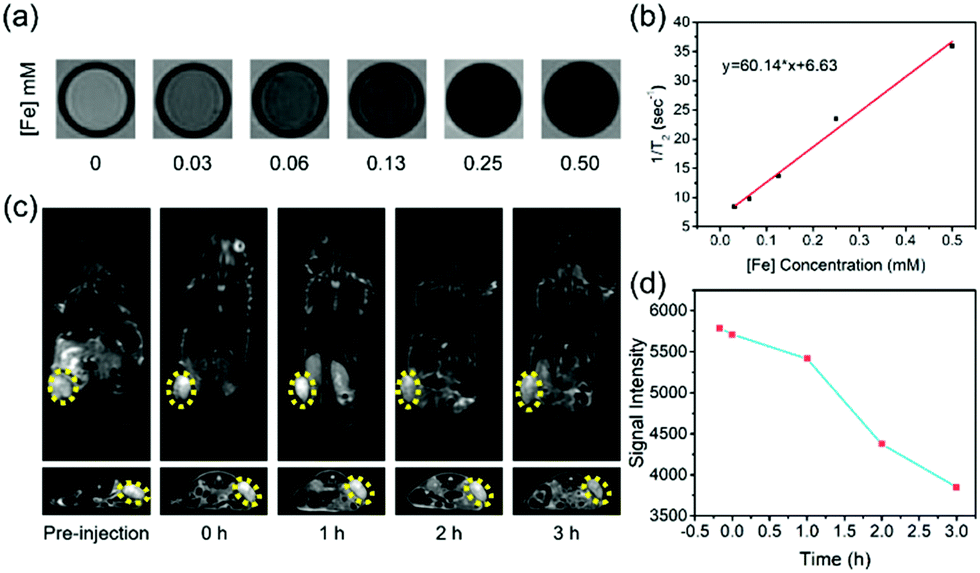 | ||
| Fig. 5 In vitro and in vivo T2-weighted MR imaging performance of the SMG nanocatalysts. (a) In vitro MRI images of SMG NPs in aqueous solution and (b) the corresponding linear fitting of 1/T2 to Fe concentration for determining the r2 value of the SMG NPs. (c) In vivo contrast-enhanced T2-weighted MR imaging of tumor-bearing mice after intravenous administration of SMG NPs, and (d) corresponding signal-intensity variation in the tumor. | ||
The in vivo contrast-enhanced MR imaging capability of SMG was further evaluated on 4T1 tumor-bearing nude mice. SMG was injected intravenously into the tumor-bearing mice, and MR images of coronal and transverse sections were acquired at various time points. An obvious enhancement in the gray scale in the tumor region was observed within 2 h after the intravenous injection of SMG nanocatalysts, indicating the desirable performance of the SMG nanocatalysts in MR imaging (Fig. 5c). The contrast-enhanced MRI signals were attributed to the superparamagnetic property of the Fe3O4 component and efficient accumulation of the SMG nanocatalysts via the EPR effect. Furthermore, it has been found that the tumor shows a negative enhancement with a signal intensity reduction by approximately 34% as shown in the T2-weighted MR images (Fig. 5d). The favorable T2-MRI performance ensures that the SMG nanocatalysts can be used for efficient in vivo cancer theranostics.
The pharmacokinetics of the intravenously injected SMG nanocatalysts were assessed to reveal their in vivo behaviors. Once injected intravenously into the mice, SMG would circulate within plasma and gradually undergo phagocytosis into tissues and tumors. Due to the substantial PEG component in the SMG nanocatalysts, their in vivo terminal half-life (τ1/2) was calculated to be as high as 7.69 h, a quite persistent retention period (Fig. 6a). The long hemodynamic half-life is herein ascribed to the high stability achieved via the surface PEGylation modification and the ultrasmall nanoparticle size of these nanocatalysts. The SMG nanocatalysts could maintain their structural integrity and catalytic capacity within a long period, which could be confirmed by the supplemented DLS, zeta potential and catalytic analyses (Fig. S9, ESI†) of the SMG nanocatalysts stored for different periods. After the storage period, the hydrodynamic diameter of the SPION@PEG-GOD NPs was slightly enhanced to 103 nm, while the zeta potential was varied to −42.2 mV. Furthermore, the catalytic parameters KM and Vmax of the SPION@PEG-GOD NPs were fitted to be 1.13 mM and 1.796 × 10−7 M s−1, quite close to the original values of 1.56 and 1.81 × 10−7. Therefore, the SPION@PEG-GOD NPs with high stability could retain their structural integrity and catalytic properties for a long period. In the following bio-distribution assay, the SMG nanocatalysts (in Fe %ID) in major organs were found to distribute majorly in the liver and spleen, which was due to capture by the reticuloendothelial system. The high enrichment in the tumor (4.02% at 48 h) was attributed to the EPR effect in the solid tumor (Fig. 6b). The GOD component in SMG is biodegradable and the Fe3O4 component is difficult to biodegrade within a short time. However, they could easily be excreted out of the body via urine and feces, which has been demonstrated in the metabolic assay (Fig. 6c). Quite a number of SMG nanocatalysts (34.57%) were excreted in feces and merely 3.04% of them were excreted in urine within 24 h. The rapid metabolism feature of SMG guarantees its favourable in vivo biosafety.
 | ||
| Fig. 6 In vivo pharmacokinetics and synergistic therapeutic performance of SMG nanocatalysts. (a) Blood circulation curves of SMG nanocatalysts injected intravenously (n = 4). The half-life period (τ1/2) was calculated to be approximately 7.69 h. (b) Biodistribution of SMG nanocatalysts (%ID of Fe) in main tissues and tumors after being intravenously injected into tumor-bearing mice for 2 h, 24 h and 48 h (n = 4). (c) Excretion amount of Fe in urine and feces at 2 h, 6 h, 12 h and 24 h post the intravenous injection of SMG nanocatalysts. (d) Schematic illustration of the mechanism of efficient in vivo therapy using SMG nanocatalysts. (e) Time-dependent body-weight curves of 4T1 tumor-bearing BALB/c nude mice after the intravenous injection of the corresponding therapeutic agents (saline for the control group) including free MMC, SPION-MMC@PEG NPs, SPION@PEG-GOD NPs, and SMG nanocatalysts. (f) Time-dependent relative tumor volumes of 4T1 tumor-bearing BALB/c nude mice of five groups (the control group, the free MMC group, the SPION-MMC@PEG group, the SPION@PEG-GOD group, and the SMG nanocatalyst group) after intravenous injection with the corresponding therapeutic agents (n = 4). Statistical significance is assessed by the Student's two-sided t-test compared to the SMG nanocatalyst group. *p < 0.05, **p < 0.01, ***p < 0.001. (g) Homologous pathological sections (left) stained with H&E and Antigen Ki-67 immunofluorescence. Scale bar: 100 μm. (1) control, (2) free MMC, (3) SPION-MMC@PEG, (4) SPION@PEG-GOD and (5) SMG nanocatalysts. Digital photographs (right) of tumors from each group at the end of therapy. | ||
Based on the aforementioned in vitro evaluation and discussion, the potential in vivo therapeutic efficacy of the SMG nanocatalysts consists of two major contributions (Fig. 6d). Under the catalysis of GOD, β-D-glucose and oxygen in the solid tumor are transformed into gluconic acid and hydrogen peroxide (H2O2). This reaction not only decreases the tumor pH value and the oxygen level of the TME, but also provides a crucial reagent for the following acid-enhanced sequential chemodynamic therapy, causing cell apoptosis by the post-produced toxic hydroxyl radicals. Furthermore, thanks to the artificially enhanced hypoxic TME, hypoxia-activated chemotherapy could be implemented, resulting in the disaggregation of deoxyribonucleic acid (DNA). The augmented combination of chemodynamic therapy and chemotherapy of the SMG nanocatalysts would significantly suppress the tumor growth.
The efficient in vitro catalytic performance and high biocompatibility of the SMG nanocatalysts would guarantee their probably high in vivo therapeutic efficacy. During the therapeutic period within 18 days, no significant discrepancy was observed among the body weights of mice in the control group and various treatment groups, indicating that no obvious toxicity was induced by the injections of various therapeutic agents during the observation period (Fig. 6e). In the tumor-suppression evaluation, the tumor growth in the SMG nanocatalyst group was substantially suppressed, and the corresponding suppression rate was calculated to be 77.96%, indicating the satisfactory tumor suppression efficacy of the SMG nanocatalysts (Fig. 6f and Fig. S10, ESI†). This high therapeutic efficacy was attributed to the persistent half-time period, and definite enrichment efficiency in the solid tumor, along with the formidable in vitro cytotoxicity induced by SMG nanocatalysts in the specific TME. In the absence of MMC, the group of SPION@PEG-GOD NPs exhibits a relatively low suppression rate (57.15%). As for the free MMC group, due to its rapid metabolism, the suppression rate was quite low (16.50%), substantially lower as compared to the SPION-MMC@PEG group (41.26%). After the therapeutic processes, all the solid tumors of the mice were dissected, and stained by hematoxylin and eosin (H&E), and antigen Ki-67 for pathological analysis (Fig. 6g). In contrast to the control group, remarkable damage of tumor cells was observed in H&E-stained sections of tumors in the SMG nanocatalyst group. For the proliferation-related antigen Ki-67 staining, striking decreased Ki-67 expression was noticed in the SMG nanocatalyst group, indicating that SMG could substantially depress the tumor proliferation.
The in vivo biosafety assay was conducted on healthy six week-old female Kunming mice, which were intravenously injected with saline (control) and SPION@PEG-GOD NPs at the doses of 5 mg kg−1, 10 mg kg−1, 15 mg kg−1, 20 mg kg−1 and 30 mg kg−1, respectively. The recorded body weights of the mice within the observation period showed that the administration of SPION@PEG-GOD NPs has no obvious impact on mouse growth (Fig. S11a, ESI†). The blood indexes including hemoglobin, mean corpuscular hemoglobin (MCH), erythrocyte mean corpuscular volume (MCV), red blood cells, red blood cell specific volume (HCT), and kidney and liver functional indexes were further measured. The main indexes among the control group and SPION@PEG-GOD treated groups showed no obvious difference, indicating no critical organ or hematic toxicology profiles within the feeding period of one month (Fig. S11b–i, ESI†). The histopathologic analysis on major organs was conducted to verify the histocompatibility of the SPION@PEG-GOD NPs. In agreement with the previous results, no distinct tissue damage was observed among the control group and the groups injected with various doses of SPION@PEG-GOD NPs (Fig. S12, ESI†). The SMG nanocatalysts consist of sections with high biocompatibility and responsive-enhanced toxicity, which guarantees their high biosafety. By binding the catalysts with adequate targeting molecules, the biosafety of SMG nanocatalysts could be further improved with enhanced tumor accumulation and decreased effects on normal cells/tissues. These preliminary in vivo biosafety assessments demonstrate a relatively high biocompatibility of the SPION@PEG-GOD nanocatalysts, guaranteeing their potential clinical translations.
Conclusions
In summary, this work introduces the new concept of sequential catalytic nanomedicine for highly-efficient and cancer-specific therapy by taking the full reactant and product features of a representative enzyme-catalytic reaction, which has been further developed for chemodynamic therapy and improved chemotherapy. A multifunctional SMG nanocatalyst has been successfully constructed for the initial GOD-catalyzed glucose reaction to produce H2O2 and gluconic acid. The produced H2O2 was further catalyzed by the SPION component by the typical catalytic Fenton reaction to generate toxic hydroxyl radicals (˙OH) for killing cancer cells, which could be further enhanced by the produced gluconic acid. Because of the decreased oxygen level due to the GOD-catalyzed reaction, the loaded chemotherapeutic drug MMC was activated for hypoxia-enhanced chemotherapy. Both the systematic in vitro cellular level and in vivo tumor xenograft evaluations on nude mice have demonstrated the high synergistic chemodrug and chemodynamic therapeutic efficacy in combating cancer. This work provides an alternative but highly efficient strategy for cancer therapy by rationally building the linkage between nanocatalytic science and nanomedicine, which is also highly dependent on the unique features of the tumor microenvironment and the physiochemical property of various nanocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly acknowledge the financial support from the National Key Research and Development Program of China (Grant No. 2016YFA0203700), the National Nature Science Foundation of China (Grant No. 51672303, 51722211, 51461165202 and 51472085), the Young Elite Scientist Sponsorship Program by CAST (Grant No. 2015QNRC001), the National Natural Science Foundation of China for Innovative Research Groups (Grant No. 51621002) and the Fundamental Research Funds for Central Universities (Grant No. 222201718002).References
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021 RSC.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488 RSC.
- C. Liang, L. Xu, G. Song and Z. Liu, Chem. Soc. Rev., 2016, 45, 6250 RSC.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597 RSC.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457 RSC.
- A. Bergamo and G. Sava, Chem. Soc. Rev., 2015, 44, 8818 RSC.
- X. Li, F. Zhang and D. Zhao, Chem. Soc. Rev., 2015, 44, 1346 RSC.
- S. Siva, G. Kothari, A. Muacevic, A. V. Louie, B. J. Slotman, B. S. Teh and S. S. Lo, Nat. Rev. Urol., 2017, 14, 549 CrossRef PubMed.
- R. A. Sharma, R. Plummer, J. K. Stock, T. A. Greenhalgh, O. Ataman, S. Kelly, R. Clay, R. A. Adams, R. D. Baird, L. Billingham, S. R. Brown, S. Buckland, H. Bulbeck, A. J. Chalmers, G. Clack, A. N. Cranston, L. Damstrup, R. Ferraldeschi, M. D. Förster, J. Golec, R. M. Hagan, E. Hall, A. R. Hanauske, K. J. Harrington, T. Haswell, M. A. Hawkins, T. Illidge, H. Jones, A. S. Kennedy, F. McDonald, T. Melcher, J. P. O'Connor, J. R. Pollard, M. P. Saunders, D. Sebag-Montefiore, M. Smitt, J. Staffurth, I. J. Stratford and S. R. Wedge, Nat. Rev. Clin. Oncol., 2016, 13, 627 CrossRef CAS PubMed.
- A. R. Patel and A. J. Stephenson, Nat. Rev. Urol., 2011, 8, 385 CrossRef PubMed.
- H. E. Barker, J. T. Paget, A. A. Khan and K. J. Harrington, Nat. Rev. Cancer, 2015, 15, 409 CrossRef CAS PubMed.
- S. Mitragotri, Nat. Rev. Drug Discovery, 2005, 4, 255 CrossRef CAS PubMed.
- G. Leinenga, C. Langton, R. Nisbet and J. Gotz, Nat. Rev. Neurol., 2016, 12, 161 CrossRef PubMed.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436 CrossRef CAS PubMed.
- W. Yang, W. Guo, W. Le, G. Lv, F. Zhang, L. Shi, X. Wang, J. Wang, S. Wang, J. Chang and B. Zhang, ACS Nano, 2016, 10, 10245 CrossRef CAS PubMed.
- X. Cheng, R. Sun, L. Yin, Z. Chai, H. Shi and M. Gao, Adv. Mater., 2017, 29, 1604894 CrossRef PubMed.
- C. Zhang, K. Zhao, W. Bu, D. Ni, Y. Liu, J. Feng and J. Shi, Angew. Chem., Int. Ed., 2014, 53, 1408472 Search PubMed.
- Y. Cheng, H. Cheng, C. Jiang, X. Qiu, K. Wang, W. Huan, A. Yuan, J. Wu and Y. Hu, Nat. Commun., 2015, 6, 8785 CrossRef CAS PubMed.
- B. Q. Spring, R. Bryan Sears, L. Z. Zheng, Z. Mai, R. Watanabe, M. E. Sherwood, D. A. Schoenfeld, B. W. Pogue, S. P. Pereira, E. Villa and T. Hasan, Nat. Nanotechnol., 2016, 11, 378 CrossRef CAS PubMed.
- S. F. Bakhoum, B. Ngo, A. M. Laughney, J. A. Cavallo, C. J. Murphy, P. Ly, P. Shah, R. K. Sriram, T. B. K. Watkins, N. K. Taunk, M. Duran, C. Pauli, C. Shaw, K. Chadalavada, V. K. Rajasekhar, G. Genovese, S. Venkatesan, N. J. Birkbak, N. McGranahan, M. Lundquist, Q. LaPlant, J. H. Healey, O. Elemento, C. H. Chung, N. Y. Lee, M. Imielenski, G. Nanjangud, D. Pe'er, D. W. Cleveland, S. N. Powell, J. Lammerding, C. Swanton and L. C. Cantley, Nature, 2018, 553, 467 CrossRef CAS PubMed.
- N. Nagarsheth, M. S. Wicha and W. Zou, Nat. Rev. Immunol., 2017, 17, 559 CrossRef CAS PubMed.
- J. M. Taube, J. Galon, L. M. Sholl, S. J. Rodig, T. R. Cottrell, N. A. Giraldo, A. S. Baras, S. S. Patel, R. A. Anders, D. L. Rimm and A. Cimino-Mathews, Mod. Pathol., 2018, 31, 214 CrossRef CAS PubMed.
- N. Obermajer, J. Urban, E. Wieckowski, R. Muthuswamy, R. Ravindranathan, D. L. Bartlett and P. Kalinski, Nat. Protoc., 2018, 13, 335 CrossRef CAS PubMed.
- Z. Gao, Y. Hou, J. Zeng, L. Chen, C. Liu, W. Yang and M. Gao, Adv. Mater., 2017, 29, 1701095 CrossRef PubMed.
- J. Liu, Q. Chen, W. Zhu, X. Yi, Y. Yang, Z. Dong and Z. Liu, Adv. Funct. Mater., 2017, 27, 1605926 CrossRef.
- J. Fu and Y. Zhu, J. Mater. Chem. B, 2017, 5, 996 RSC.
- X. Liu, M. Wu, Q. Hu, H. Bai, S. Zhang, Y. Shen, G. Tang and Y. Ping, ACS Nano, 2016, 10, 11385 CrossRef CAS PubMed.
- M. A. Miller and R. Weissleder, Nat. Rev. Cancer, 2017, 17, 399 CrossRef CAS PubMed.
- D. M. Lewis, M. R. Blatchley, K. M. Park and S. Gerecht, Nat. Protoc., 2017, 12, 1620 CrossRef CAS PubMed.
- D. W. Scott and R. D. Gascoyne, Nat. Rev. Cancer, 2014, 14, 517 CrossRef CAS PubMed.
- J. L. Adams, J. Smothers, R. Srinivasan and A. Hoos, Nat. Rev. Drug Discovery, 2015, 14, 603 CrossRef CAS PubMed.
- G. Fluegen, A. Avivar-Valderas, Y. Wang, M. R. Padgen, J. K. Williams, A. R. Nobre, V. Calvo, J. F. Cheung, J. J. Bravo-Cordero, D. Entenberg, J. Castracane, V. Verkhusha, P. J. Keely, J. Condeelis and J. A. Aguirre-Ghiso, Nat. Cell Biol., 2017, 19, 120 CrossRef CAS PubMed.
- S. Ruan, C. Hu, X. Tang, X. Cun, W. Xiao, K. Shi, Q. He and H. Gao, ACS Nano, 2016, 10, 10086 CrossRef CAS PubMed.
- H. Zhou, J. Tang, J. Li, W. Li, Y. Liu and C. Chen, Nanoscale, 2017, 9, 3040 RSC.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928 CrossRef CAS PubMed.
- F. Yang, P. Chen, W. He, N. Gu, X. Zhang, K. Fang, Y. Zhang, J. Sun and J. Tong, Small, 2010, 6, 1300 CrossRef CAS PubMed.
- J. Gehring, B. Trepka, N. Klinkenberg, H. Bronner, D. Schleheck and S. Polarz, J. Am. Chem. Soc., 2016, 138, 3076 CrossRef CAS PubMed.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577 CrossRef CAS PubMed.
- C. Lennicke, J. Rahn, R. Lichtenfels, L. A. Wessjohann and B. Seliger, Cell Commun. Signaling, 2015, 13, 39 CrossRef PubMed.
- C. Corbet and O. Feron, Nat. Rev. Cancer, 2017, 17, 577 CrossRef CAS PubMed.
- J. Liu, W. Bu and J. Shi, Chem. Rev., 2017, 117, 6160 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539 CrossRef CAS PubMed.
- C. C. Huang, W. T. Chia, M. F. Chung, K. J. Lin, C. W. Hsiao, C. Jin, W. H. Lim, C. C. Chen and H. W. Sung, J. Am. Chem. Soc., 2016, 138, 5222 CrossRef CAS PubMed.
- H. S. Jung, J. Han, H. Shi, S. Koo, H. Singh, H. J. Kim, J. L. Sessler, J. Y. Lee, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 7595 CrossRef CAS PubMed.
- J. Kim, H. R. Cho, H. Jeon, D. Kim, C. Song, N. Lee, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2017, 139, 10992 CrossRef CAS PubMed.
- J. Bai, X. Jia, W. Zhen, W. Cheng and X. Jiang, J. Am. Chem. Soc., 2018, 140, 106 CrossRef CAS PubMed.
- C. Qian, J. Yu, Y. Chen, Q. Hu, X. Xiao, W. Sun, C. Wang, P. Feng, Q. D. Shen and Z. Gu, Adv. Mater., 2016, 28, 3313 CrossRef CAS PubMed.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29, 1700996 CrossRef PubMed.
- T. B. Rodrigues, E. M. Serrao, B. W. Kennedy, D. E. Hu, M. I. Kettunen and K. M. Brindle, Nat. Med., 2014, 20, 93 CrossRef CAS PubMed.
- D. Zhai, B. Liu, Y. Shi, L. Pan, Y. Wang, W. Li, R. Zhang and G. Yu, ACS Nano, 2013, 7, 3540 CrossRef CAS PubMed.
- C.-C. Huang, Z.-X. Liao, H.-M. Lu, W.-Y. Pan, W.-L. Wan, C.-C. Chen and H.-W. Sung, Chem. Mater., 2016, 28, 9017 CrossRef CAS.
- C. Vilcheze, T. Hartman, B. Weinrick and W. R. Jacobs, Jr., Nat. Commun., 2013, 4, 1881 CrossRef PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986 CrossRef CAS PubMed.
- R. Hachani, M. Lowdell, M. Birchall and N. T. Thanh, Nanoscale, 2013, 5, 11362 RSC.
- W. Fan, N. Lu, P. Huang, Y. Liu, Z. Yang, S. Wang, G. Yu, Y. Liu, J. Hu, Q. He, J. Qu, T. Wang and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 1229 CrossRef CAS PubMed.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273 CrossRef CAS PubMed.
- N. P. Noyma, L. Magalhaes, L. L. Furtado, M. Mucci, F. Oosterhout, V. L. M. Huszar, M. M. Marinho and M. Lurling, Water Res., 2016, 97, 26 CrossRef CAS PubMed.
- M. M. Liang, K. L. Fan, Y. Pan, H. Jiang, F. Wang, D. L. Yang, D. Lu, J. Feng, J. J. Zhao, L. Yang and X. Y. Yan, Anal. Chem., 2013, 85, 308 CrossRef CAS PubMed.
- M. C. Ortega-Liebana, J. L. Hueso, R. Arenal and J. Santamaria, Nanoscale, 2017, 9, 1787 RSC.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136 CrossRef CAS PubMed.
- C. F. Lourenco, A. Ledo, J. Laranjinha, G. A. Gerhardt and R. M. Barbosa, Sens. Actuators, B, 2016, 237, 298 CrossRef CAS.
- C. M. Wong, K. H. Wong and X. D. Chen, Appl. Microbiol. Biotechnol., 2008, 78, 927 CrossRef CAS PubMed.
- B. Yang, J. Zuo, X. Tang, F. Liu, X. Yu, X. Tang, H. Jiang and L. Gan, Ultrason. Sonochem., 2014, 21, 1310 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- R. A. Nascimento, R. E. Ozel, W. H. Mak, M. Mulato, B. Singaram and N. Pourmand, Nano Lett., 2016, 16, 1194 CrossRef CAS PubMed.
- W. Luo, C. Zhu, S. Su, D. Li, Y. He, Q. Huang and C. Fan, ACS Nano, 2010, 4, 7451 CrossRef CAS PubMed.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188 CrossRef CAS PubMed.
- W. A. Denny, Lancet Oncol., 2000, 1, 25 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c9nh00008a |
| This journal is © The Royal Society of Chemistry 2019 |