
MOF nanoleaves as new sacrificial templates for the fabrication of nanoporous Co–Nx/C electrocatalysts for oxygen reduction†
Jingjing
Li‡
a,
Wei
Xia‡
ab,
Jing
Tang
*bcd,
Haibo
Tan
 b,
Jiayu
Wang
b,
Yusuf Valentino
Kaneti
b,
Yoshio
Bando
be,
Tao
Wang
*a,
Jianping
He
*a and
Yusuke
Yamauchi
*df
b,
Jiayu
Wang
b,
Yusuf Valentino
Kaneti
b,
Yoshio
Bando
be,
Tao
Wang
*a,
Jianping
He
*a and
Yusuke
Yamauchi
*df
aCollege of Materials Science and Technology, Jiangsu Key Laboratory of Materials and Technology for Energy Conversion, Nanjing University of Aeronautics and Astronautics, 210016 Nanjing, China. E-mail: wangtao0729@nuaa.edu.cn; jianph@nuaa.edu.cn
bInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: TANG.Jing@nims.go.jp
cSchool of Chemistry and Molecular Engineering, Shanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai, 200062, China
dSchool of Chemical Engineering and Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: y.yamauchi@uq.edu.au
eInstitute for Superconducting and Electronic Materials, Australian Institute for Innovative Materials University of Wollongong, Squires Way, North Wollongong, NSW 2500, Australia
fDepartment of Plant & Environmental New Resources, Kyung Hee University, 1732 Deogyeong-daero, Giheunggu, Yongin-si, Gyeonggi-do 446-701, South Korea
First published on 1st March 2019
Abstract
Although traditional three-dimensional (3D) zeolitic-imidazolate framework (ZIF) particles have been explored as promising precursors for preparing carbon-based electrocatalyst for oxygen reduction reaction (ORR), their natural tendency to agglomerate impedes the exposure of the active sites and significantly reduces their ORR performance. Herein, novel Co/Zn-containing bimetallic ZIF nanoleaves are synthesized by the “shape-transforming method in an aqueous system”, in which the nitrogen atoms in amines form hydrogen bonds with hydrogen atoms from H2O molecules, which induce the formation of sodalite layers to generate the ZIF nanoleaves. By directly pyrolyzing the ZIF nanoleaves, the obtained cobalt-embedded nitrogen-doped mesoporous carbon (Co–N/C) product possesses refined leaf-like two-dimensional (2D) morphologies. Moreover, the obtained 2D catalyst with a high mass loading of cobalt nanoparticles (31.17 wt%) shows an excellent electrocatalytic performance for the ORR in an alkaline electrolyte. The half-wave potential of the Co–N/C catalyst is 0.825 V versus the reversible hydrogen electrode, which is 14 mV more positive than that of the commercial Pt/C (0.811 V). In addition, the resulting zinc–air battery assembled using the Co–N/C air cathode with a liquid electrolyte exhibits both high open-circuit potential (1.446 V) and high energy density (837.5 W h kgzn−1).
Conceptual insightsRecently, three-dimensional (3D) zeolitic-imidazolate framework (ZIF) derived electrocatalysts have shown great potential as electrocatalyst for oxygen reduction reaction (ORR); however, their natural tendency to agglomerate impedes the exposure of the active sites and significantly reduces their ORR performance. In this case, we report a “shape-transforming via aqueous solution-mediated method” to rationally synthesize novel leaf-like ZIFs which achieve similar morphological advantages as 2D MOFs. The developed strategy for synthesizing ZIF nanoleaves will have enormous potential for developing various novel transition metal-based electrocatalysts with highly mesoporous structure and accessible active sites to enhance the overall ORR performance and durability. |
Introduction
Electrochemical power generators, such as proton exchange membrane fuel cells and metal–air batteries, require highly efficient electrocatalysts for accelerating the oxygen reduction reaction (ORR) to achieve an efficient electrochemical performance.1–4 Intensive efforts have been carried out to develop high-performance ORR catalysts from earth-abundant elements to replace the rare and expensive Pt-based electrocatalysts.5–8 As a matter of fact, transition metal (TM) and nitrogen (N)-doped carbon (TM–N/C) nanostructures have attracted significant attention due to their high ORR activity, excellent stability and ease of large-scale preparation.9–13 Metal–organic frameworks (MOFs) have emerged as attractive precursors to prepare non-precious electrocatalysts due to their highly porous structure, controllable composition, and high carbon/nitrogen content. Recently, MOFs have been utilized as templates to prepare nanoporous carbons, which usually exhibit large specific surface area and high electrochemical performance.14 Zeolitic-imidazolate frameworks (ZIFs) are a sub-class of MOFs made up of imidazolate or its derivatives with tetrahedrally coordinated with metal cations (e.g., Zn2+, Co2+). They are commonly employed as precursors for the fabrication of nanoporous carbons towards energy storage applications. For instance, functionalized porous carbon with large surface area, high degree of graphitization, and uniform Co, N dopant was previously prepared via direct pyrolysis of ZIF-67, which exhibited high ORR activity.15 However, ZIFs typically exhibit bulk 3D polyhedral morphology. As a result, ZIF-derived carbon-based electrocatalysts typically suffer from major inner-pore mass-transport resistance and long diffusion length, leading to reduced active surface area and covered active sites.16–19Recently, 2D MOFs have attracted enormous research interest for the derivation of 2D porous carbon materials. Compared to bulk 3D MOF crystals, 2D MOF materials exhibit higher surface areas and richer active sites.20–23 Furthermore, 2D MOFs are less likely to aggregate during the pyrolysis process, which is favorable for the construction of multiple pores or channels.24 Zhang and co-workers25 fabricated ultrathin 2D MOF nanosheets via a surfactant-assisted method which showed a thickness of less than 10 nm. Bu and co-workers24 reported the synthesis of 2D Mn-based MOF nanostructures by utilizing monodentate ligands to enable the planar growth of MOF nanosheets, which were then thermally treated under inert atmosphere to generate nitrogen-doped porous carbon foils. Despite some success, the current synthetic strategies for achieving 2D MOFs are quite complex, time-consuming and energy-intensive. Therefore, it is highly desirable to develop a simple and cost-effective way to fabricate MOF precursors that have similar morphological advantages as 2D MOFs. The rational design of leaf-like MOFs and their derived carbon-based catalysts with high surface area and highly accessible active sites is fundamentally important for achieving new electrocatalysts with high ORR performance.
Typically, organic solvents (such as methanol and N,N-dimethylformamide (DMF)) are used in the preparation of ZIFs, which are environmentally hazardous, toxic and unsuitable for large-scale production. Some researchers have reported the synthesis of ZIFs in water. For instance, ZIF-90 nanoparticles with a small particle size of 270 nm can be easily synthesized in a water-based solution,26 and well-defined ZIF-8 can be also rapidly obtained in an aqueous solution.27 However, these previously reported ZIFs were limited to 3D particles. Herein, we report a “shape-transforming method in an aqueous solution” to rationally synthesize Co/Zn-containing bimetallic ZIF nanoleaves (named as CoxZn1−x-ZIF nanoleaves). With the assistance of H2O molecules, the nitrogen atoms in 2-methylimidazole (2-mlm) form hydrogen bonds with hydrogen atoms from H2O molecules instead of coordinating with the transition metals, leading to the generation of sodalite layers linked to the formation of the nanoleaves. Following the carbonization of the bimetallic ZIF nanoleaves at high temperatures under inert atmosphere, the obtained cobalt-embedded nitrogen-doped mesoporous carbon nanoleaves (Co–N/C) display 2D leaf-like shape and show high electrocatalytic activity for the ORR in an alkaline electrolyte. In addition, the Zn–Air batteries fabricated with the Co–N/C as the cathode exhibit high electrochemical performance and good long-term stability.
Results and discussion
A schematic illustration depicting the synthesis procedure of the ZIF nanoleaves and their subsequent conversion to Co–N/C nanoleaves is given in Scheme 1. The ZIF nanoleaves were synthesized in an aqueous solution instead of methanol. The connecting 2-mlm ligands between the sodalite layers are thought to be destroyed by the H2O molecules which leads to the creation of periodic layers. Such 2D networks are stabilized by interactions with the assistance of 2-mlm, in which nitrogen atoms in 2-mlm form hydrogen bonds with hydrogen atoms from H2O molecules instead of coordinating with transition metals. These bonds promote the formation of N–H⋯N hydrogen bonds with other 2-mlm, through which the sodalite layers are connected to form ZIF nanoleaves.28–31 Co–N/C catalysts were obtained by direct pyrolysis of the ZIF nanoleaves at 800 °C under N2 atmosphere followed by acid etching to remove the inactive cobalt particles, other cobalt species (e.g., cobaltous oxide) produced during pyrolysis and the remaining Zn species (e.g., zinc oxide). As a matter of fact, the Zn is atomically dispersed in the ZIF precursors. The physical properties of the atomically dispersed Zn are different from the bulk Zn (boiling point is 907 °C), causing the atomic-scale Zn to evaporate at a much lower pyrolysis temperature (e.g., 800 °C).32 Besides, the residual Zn could be further removed by acid etching at 80 °C for 8 hours. The resulting precursors and the corresponding derived catalysts are denoted as CoxZn1−x-ZIF nanoleaves and Cox–N/C-T, respectively, where x indicates the molar ratio of Co2+/(Co2+ + Zn2+) in the initial ZIF nanoleaves and T is the pyrolysis temperature.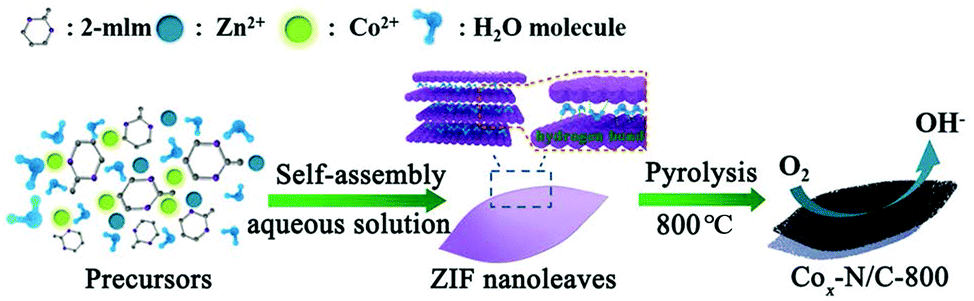 | ||
| Scheme 1 Schematic illustration showing the synthesis process of the ZIF nanoleaves in an aqueous solution and the subsequent conversion to cobalt-embedded N-doped mesoporous carbon (Co–N/C) nanoleaves. | ||
The morphology of the ZIF nanoleaves was characterized by scanning electron microscopy (SEM), as presented in Fig. 1a. It can be observed that the Co0.6Zn0.4-ZIF sample has a leaf-like morphology with a lateral dimension of approximately 5 μm. As expected, the resulting Cox–N/C-T retains the morphology of the ZIF nanoleaf precursor after the thermal treatment under nitrogen atmosphere at 800 °C (Fig. 1b), with an average lateral size of around 3 μm. When the aqueous solution is replaced by methanol as the solvent, Co0.6Zn0.4-ZIF dodecahedrons with an average size of around 500 nm were achieved (Fig. S1a, ESI†). The shrinkage of both Co0.6Zn0.4-ZIF nanoleaves and 3D Co0.6Zn0.4-ZIF dodecahedrons was unavoidable during pyrolysis (Fig. 1b and Fig. S1b, ESI†). Further morphological analysis of Co0.6–N/C-800 was performed using transmission electron microscopy (TEM). As shown in Fig. 1c, the Co0.6–N/C-800 sample shows a delicate leaf-like morphology. The porous structure can be observed from the circle in the inset of Fig. 1c. Both Co0.2–N/C-800 and Co–N/C-800 show leaf-like morphology in Fig. S2 (ESI†). As shown in Fig. 1d, abundant graphitic carbon structures with the interplanar spacing of the (002) crystal lattice (3.4 Å) are observed,33 with the Co nanoparticles (NPs) being wrapped tightly by these graphitic carbon layers. The TEM elemental mapping indicates that the Co0.6–N/C-800 sample is composed of C, Co and N, which are dispersed uniformly throughout the carbon matrix (Fig. 1e). The thickness of the nanoleaves was estimated to be 160 nm based on the AFM image (Fig. 1f). The precise mass loading of cobalt in Co0.6–N/C-800 was determined by ICP-OES to be 31.17 wt%.
 | ||
| Fig. 1 (a) Typical SEM image of the Co0.6Zn0.4-ZIF nanoleaves; (b) SEM image, (c) TEM image (inset: magnified image of the red marked area; typical pore structure is marked by the red circle), (d) HRTEM image, and (e) EDX-mapping of Co0.6–N/C-800; (f) AFM images of Co0.6Zn0.4-ZIF nanoleaves. | ||
To further explore the structural properties and chemical compositions of the electrocatalysts, XRD, XPS, and Raman spectroscopy were employed. The powder XRD patterns of Zn-ZIF and Co0.6Zn0.4-ZIF nanoleaves (Fig. 2a) show the same diffraction peaks as the calculated ZIF nanoleaves, indicating that the chemical addition of Co does not change the lattice structure of the ZIF nanoleaves, due to the close atomic sizes of Co and Zn. The XRD pattern of N/C-800 displays two peaks at 25° and 44° indexed to (002) and (101) diffractions of amorphous carbon (Fig. 2b). The Co0.6–N/C-800 sample shows a sharper (002) diffraction peak at 26°, compared with the broad peak at 25° of N/C, suggesting the formation of a graphitic carbon structure (Fig. 2b).34–36 The peaks located at around 44°, 51° and 75° are assignable to (111), (200) and (022) diffractions of fcc Co (PDF 15-9026) originated from the Co NPs dispersed throughout the carbon matrix.37–39
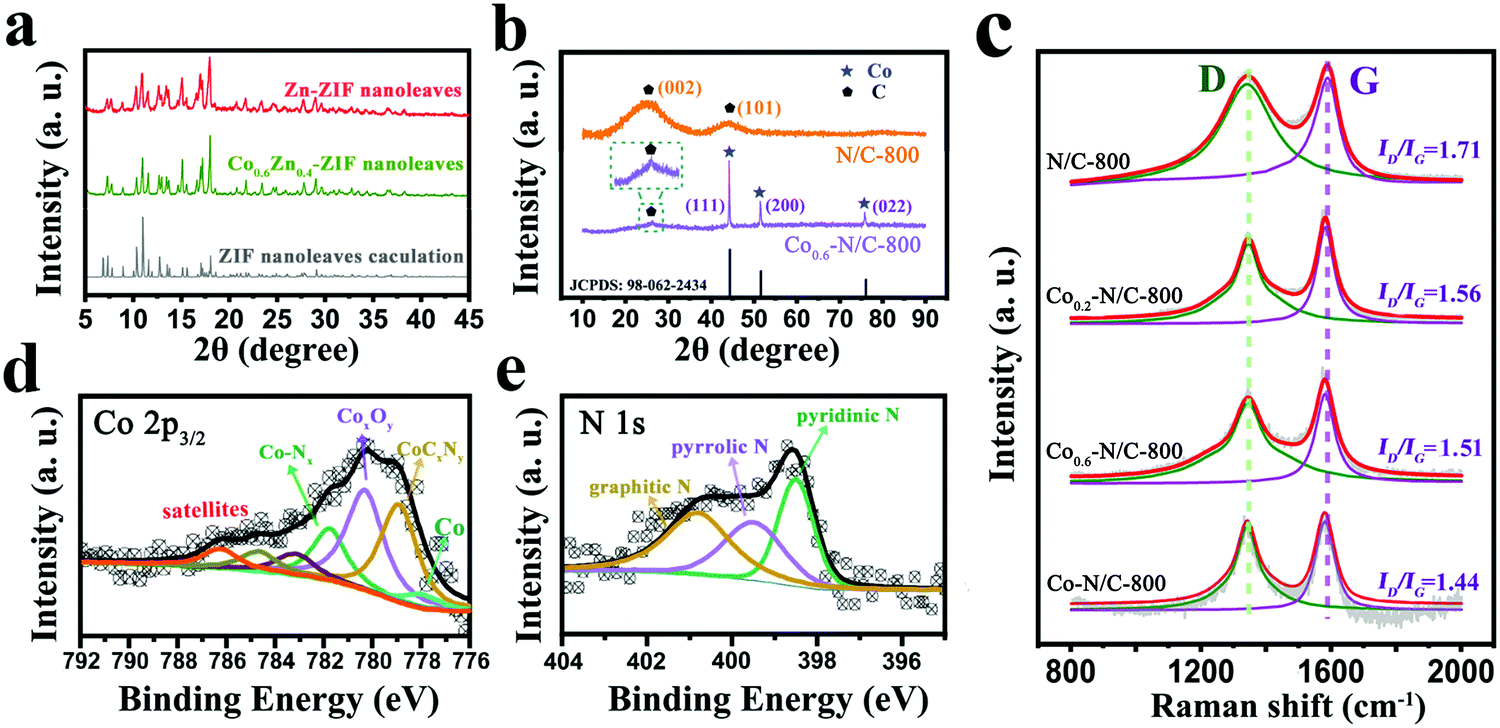 | ||
| Fig. 2 XRD patterns of (a) Zn-ZIF and Co0.6Zn0.4-ZIF nanoleaves and (b) N/C-800 and Co0.6–N/C-800 nanoleaves. (c) Raman spectra of N/C-800, Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800 (from top to bottom). (d) Co 2p3/2 and (e) N 1s high-resolution XPS spectra of Co0.6–N/C-800. | ||
The Raman spectra of Cox–N/C with different molar ratios of Co and Zn are shown in Fig. 2c. The four samples show two peaks, at 1345 and 1582 cm−1, which correspond to the first-order D band and G band, respectively.40 Here, the D band represents the defects at the edges of the carbon structure and the G band originates from the presence of carbon. The ratio of the calculated integrated areas for the D band to the G band is related to the density of defects in the lattice.10,41 The values of ID/IG gradually decrease from N/C-800 to Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800 (Fig. 2c). This trend indicates that the Co content in the ZIF precursors plays an important role in controlling the degree of graphitization due to the catalytic effect of Co. With the increased Co content in the ZIF precursors, the graphitization degree of the ZIF-derived carbons is increased.
The X-ray photoelectron spectroscopy (XPS) of Co0.6–N/C-800 shows four peaks of C 1s, N 1s, O 1s and Co 2p (Fig. S3, ESI†), with the corresponding atomic contents of 82.7%, 4.1%, 5.0% and 5.4%, respectively. As presented in Fig. 2d, the high-resolution Co 2p3/2 spectrum is deconvoluted into satellite components (similar satellite components have been reported in the previous literature42) and the four distinct peaks can be assigned to Co–Nx (781.8 eV), CoxOy (780.2 eV), CoCxNy (778.8 eV) and cobalt metal (777.9 eV). This result suggests the presence of Co–Nx, which has been identified as active sites for ORR.18 Moreover, the high-resolution N 1s spectrum (Fig. 2e) can be deconvoluted into three types, pyridinic N (398.5 eV), pyrrolic N (399.3 eV) and graphitic N (400.8 eV).43 Obviously, pyridinic N and graphitic N are the dominant peaks in the N 1s spectrum. Previously, it was proposed that pyridinic N could promote the ORR on nitrogen-rich carbon surface along the efficient four-electron reaction path and graphitic N can greatly enhance the catalytic current for ORR.44 Therefore, optimizing the amounts of pyridinic N to graphitic N is beneficial for maximizing the catalytic performance of nitrogen-enriched carbon materials for ORR. To further understand the effect of carbonization temperature, XPS analysis of Cox–N/C-T catalysts are carried out (Fig. S4b, ESI†). As for Co0.6–N/C-T (T = 700, 800, 900 °C), the optimum contents of pyridinic N and graphitic N were achieved at 800 °C.
The N2 adsorption-desorption isotherms for N/C-800, Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800 display similar type-IV hysteresis loop, characteristic of micro/mesoporous materials (Fig. 3a and b). The specific surface areas of N/C-800, Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800 were calculated to be 924.7, 311.8, 283.2, and 179.1 m2 g−1, respectively. As shown in Fig. 3c, the pore size distribution curves show that all the samples exhibit mesopores centered at around 3.8 nm. The pore volume of Co0.6–N/C-800 is 0.421 cm3 g−1, which is higher than those of Co0.2–N/C (0.244 cm3 g−1) and Co–N/C (0.377 cm3 g−1), but lower than that of N/C-800 (0.553 cm3 g−1). These results imply that the sublimation of Zn during carbonization prevents the destruction of micropores in the Zn-ZIF derived carbons. However, the Co metal in bimetallic CoxZn1−x-ZIF and Co-ZIF displays a totally different role during the carbonization process. During carbonization, Co migrates and aggregates into Co nanoparticles, and the micropores were destroyed to form some mesopores. After the removal of the Co nanoparticles by acid treatment, mesopores were created. Therefore, the CoxZn1−x-ZIF and Co-ZIF derived carbons would possess both micropores and mesopores. Further, the surrounding carbons were graphitized by the Co nanoparticles during pyrolysis and the micropores and mesopores were both sacrificed.45–48 Therefore, the specific surface area of N/C-800 is higher than those of Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800.
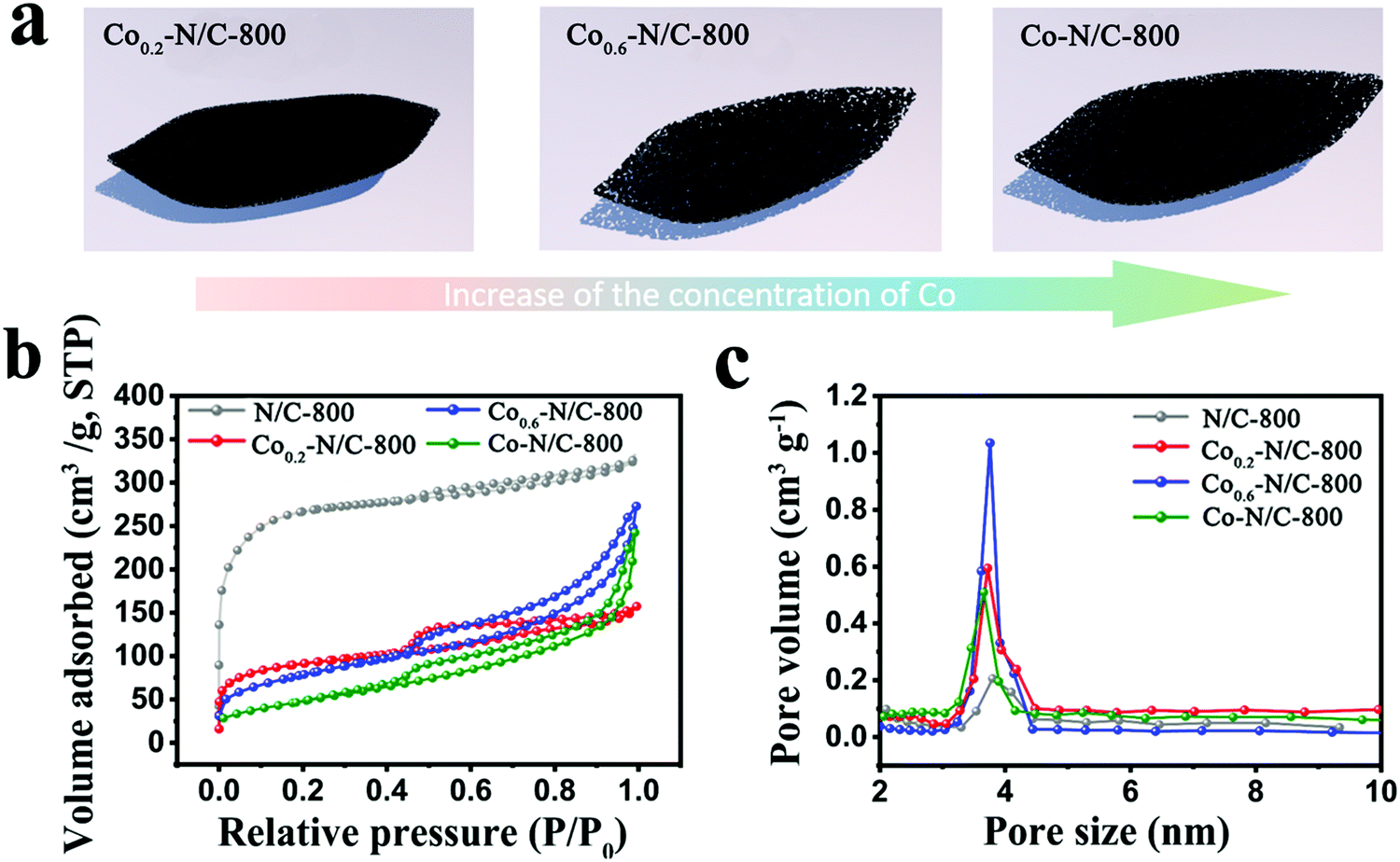 | ||
| Fig. 3 (a) Schematic of the surface pore structures of Co0.2–N/C-800, Co0.6–N/C-800, and Co–N/C-800. (b) Nitrogen adsorption–desorption isotherms of N/C-800, Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800. (c) The pore size distribution plots of N/C-800, Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800. | ||
The ORR activities of Co0.6–N/C-T (T = 700, 800, and 900) in alkaline conditions were investigated by rotating disk electrode (RDE) experiments (Fig. S4a, ESI†). The results showed that 800 °C is the optimal pyrolysis temperature, which is in good agreement with the XPS results (Fig. S4b, ESI†). Thus, the electrocatalytic activities of Cox–N/C-800 samples with different Co content were evaluated by cyclic voltammetry (CV) in a 0.1 M KOH solution saturated with N2 and O2, respectively. As shown in Fig. 4a, no redox peak is observed for N/C-800, Co0.2–N/C-800, Co0.6–N/C-800, Co–N/C-800 and 20 wt% Pt/C in the potential range of 0 to 1.2 V in N2 saturated electrolyte. In O2 saturated electrolyte, these catalysts show an obvious oxygen reduction peak, except for N/C-800. Among the Cox–N/C samples, Co0.6–N/C-800 presents a distinct oxygen reduction peak at 0.796 V, superior to N/C-800 and Co–N/C-800 and is much closer to that of 20 wt% Pt/C, indicating its superior ORR activity. These results were further confirmed by steady-state linear sweep voltammetry (LSV) on an RDE in a 0.1 M KOH solution saturated with O2. As shown in Fig. 4b, Co0.6–N/C-800 possesses the best ORR activity with an onset potential (Eonset, calculated when the current density reaches 5% of the limiting current density) of 0.916 V vs. RHE and a half-wave potential (E1/2) of 0.825 V vs. RHE, which are quite similar to those of 20 wt% Pt/C (Eonset = 0.926 V, E1/2 = 0.811 V, respectively). As shown in Table S1 (ESI†), the electrocatalytic performance of the Co0.6–N/C-800 catalyst is superior to those of previously reported Co-containing and MOF-derived catalysts. The ORR performance of the bimetallic ZIF nanoleaf-derived carbon materials, especially for Co0.6–N/C-800, is much better than that of traditionally synthesized N/C-800 derived from ZIF nanoleaves. These findings clearly reveal that the doping of Co into the N-enriched carbon matrix can greatly improve its ORR activity, which might be attributed to the combined effects of abundant active sites including the pyridinic-N, graphitic-N, and Co–Nx coordination based active sites, as well as the 2D leaf-like morphology.49 The Koutecky–Levich (K–L) plots obtained from the RDE polarization curves of Co0.6–N/C-800 at various rotation speeds (Fig. S5, ESI†) display an almost constant slope, indicating first-order kinetics of the ORR on the Co0.6–N/C-800 catalyst. In addition, based on eqn (3) and (4) (ESI†), the electron transfer number (n) of the Co0.6–N/C-800 catalyst is calculated to be 3.90, revealing that the Co0.6–N/C-800 catalyst follows a dominant four-electron pathway. In addition, the Co0.6–N/C-800 electrocatalyst exhibits a much higher E1/2 as well as onset potential than the 3D Co0.6–N/C-800 electrocatalyst (Fig. S1b, ESI†), thus confirming the advantages of the 2D structure for enhancing the ORR activity (Fig. S6, ESI†). The detailed ORR performance of the as-synthesized 3D Co0.6–N/C-800 electrocatalysts is shown in Fig. S7a (ESI†). The electron transfer number (n) of the 3D Co0.6–N/C-800 catalyst is calculated to be 3.70 according to the K–L equation, which is lower than that of the 2D Co0.6–N/C-800 (3.90) catalyst. This indicates that the 2D morphology favors the full exposure of the active sites to the reactants and enables the rapid transfer of mass/electrons during the ORR process. As shown in inset of Fig. 4c, the Tafel slope of Co0.6–N/C-800 at low overpotentials is determined to be 77 mV per decade, which is lower than that of commercial Pt/C (96 mV per decade), further revealing the excellent electrocatalytic activity of Co0.6–N/C-800 for ORR, indicating that the catalyst promotes electron transfer during the catalytic oxygen reduction process, and effectively improves the slow kinetic process of oxygen reduction.
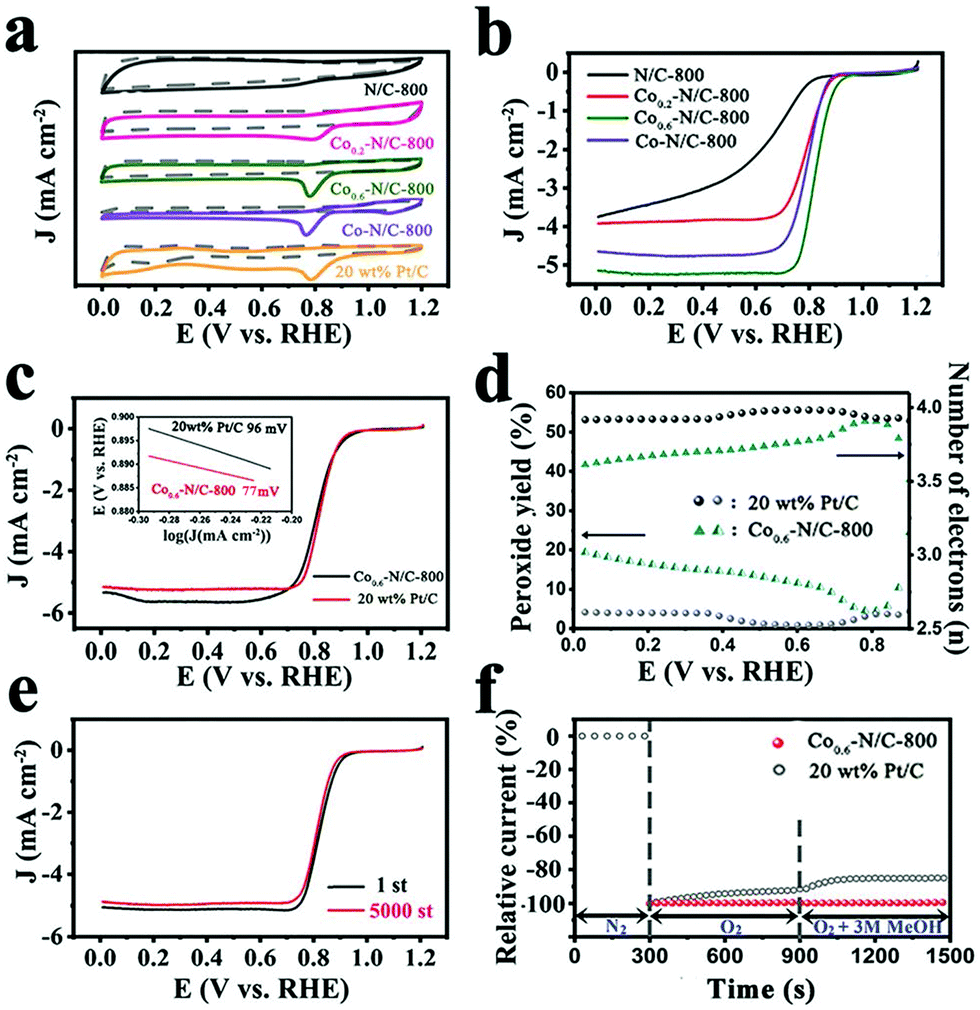 | ||
| Fig. 4 (a) Cyclic voltammograms (CV) of N/C-800, Co0.2–N/C-800, Co0.6–N/C-800, Co–N/C-800 and 20 wt% Pt/C in O2-saturated and N2-saturated 0.1 M KOH with a scan rate of 5 mV s−1 at 1600 rpm. (b) LSV curves of N/C-800, Co0.2–N/C-800, Co0.6–N/C-800 and Co–N/C-800. (c) RRDE polarization curves of Co0.6–N/C-800 and 20 wt% Pt/C. The inset shows the corresponding Tafel plots (1600 rpm, 5 mV s−1). (d) Peroxide yield (bottom) and electron transfer number (n: top). (e) RDE polarization curves of Co0.6–N/C-800 before and after 5000 cycles in O2-saturated 0.1 M KOH. (f) Chronoamperometric response with 3 M methanol of Co0.6–N/C-800 and 20 wt% Pt/C at 0.4 V vs. RHE and rotation speed of 1600 rpm. | ||
The rotating ring-disk electrode (RRDE) measurements were used to study the reaction pathways of the as-prepared catalysts. As shown in Fig. 4d, based on eqn (1) and (2) (ESI†), the Co0.6–N/C-800 catalyst has a peroxide yield below 20% over the potential range of 0–0.85 V and a high electron transfer number of around 3.85, which is close to that of 20 wt% Pt/C (n ≈ 3.99), indicating the high selectivity of Co0.6–N/C-800 towards the four-electron reduction of O2. In contrast, the 3D Co0.6–N/C-800 electrocatalyst exhibits a low electron transfer number of around 3.45 and a high peroxide yield above 30% (Fig. S7b, ESI†). The results suggest that the hierarchical porous 2D structure of Co0.6–N/C-800 favors quick mass transfer and full exposure of the active sites, thereby avoiding the temporary or permanent deactivation of these sites due to the blocking or collapse of the pores. Additionally, Co0.6–N/C-800 exhibits good stability and better methanol tolerance compared to 20 wt% Pt/C. The half-wave potential of Co0.6–N/C-800 is almost maintained with only a negative shift of 8 mV after 5000 continuous cycles (Fig. 4e). After injection of 3 M methanol into the electrolyte, the current of Co0.6–N/C-800 shows a negligible change, indicating a good tolerance to methanol oxidation compared with the commercial Pt/C catalyst (Fig. 4f).
To evaluate the practical applicability of the as-prepared catalysts in Zn–air batteries, a homemade primary Zn–air battery with a two-electrode system was assembled (Fig. 5a). A zinc plate was used as an anode, the ink of Co0.6–N/C-800 was deposited on carbon paper and used as an air-cathode with a catalyst loading of about 0.5 mg cm−2 and a 6 M KOH solution was employed as an electrolyte. The discharge polarization and power density curves for the liquid Zn–air battery are shown in Fig. 5b. The Co0.6–N/C-800 catalyst exhibits an open-circuit potential of 1.446 V, a little higher than that of 20 wt% Pt/C (1.4 V). Furthermore, this potential is also lower than the equilibrium potential of 1.65 V as it is difficult for the oxygen electrode reaction to reach thermodynamic equilibrium in the standard state, and its stable potential value is more negative than the equilibrium potential.50 Furthermore, the Co0.6–N/C-800 catalyst displays a slightly slower decrease in potential with increasing current densities than the 20 wt% Pt/C catalyst, suggesting its good discharge performance. The power density of Co0.6–N/C-800 is 176 mW cm−2 at 250 mA cm−2, which is higher than that of 20 wt% Pt/C (133 mW cm−2, the highest power density of Pt/C), meaning that the Zn–air battery assembled by Co0.6–N/C-800 can be discharged at high current densities or high rates. The galvanostatic discharge curves in Fig. 5c show the excellent durability of the Co0.6–N/C-800 catalyst with an almost smooth discharge curve in output voltage at 5 mA cm−2 for 31.5 h and a slight loss of output voltage at 20 mA cm−2. The working voltage of the battery gradually decreases with the extension of the discharge time mainly due to the polarization of the two electrodes. The gradual increase in electrochemical polarization is due to poor mass transfer during discharge with the conversion of active substances and the gradual decrease in the active surface area of the electrode available for reactions. At a current density of 5 mA cm−2, the specific capacity of the Co0.6–N/C-800-based battery is over 670 mA h g−1 (corresponding to an energy density of ∼837.5 W h kgzn−1 after normalization with the mass of consumed Zn). The discharge curves of Co0.6–N/C-800-based and 20 wt% Pt/C-based batteries are similar, and the energy density of both the batteries can reach as high as 760 W h kgzn−1. To verify the stability of the Co0.6–N/C-800 catalyst, the Zn–air battery was mechanically charged at a current density of 5 mA cm−2. As shown in Fig. 5d, the output voltage of the battery remains relatively stable after 3 cycles of mechanical charge and no significant voltage drop is observed, demonstrating the excellent discharge stability of the Co0.6–N/C-800 based Zn–air battery.
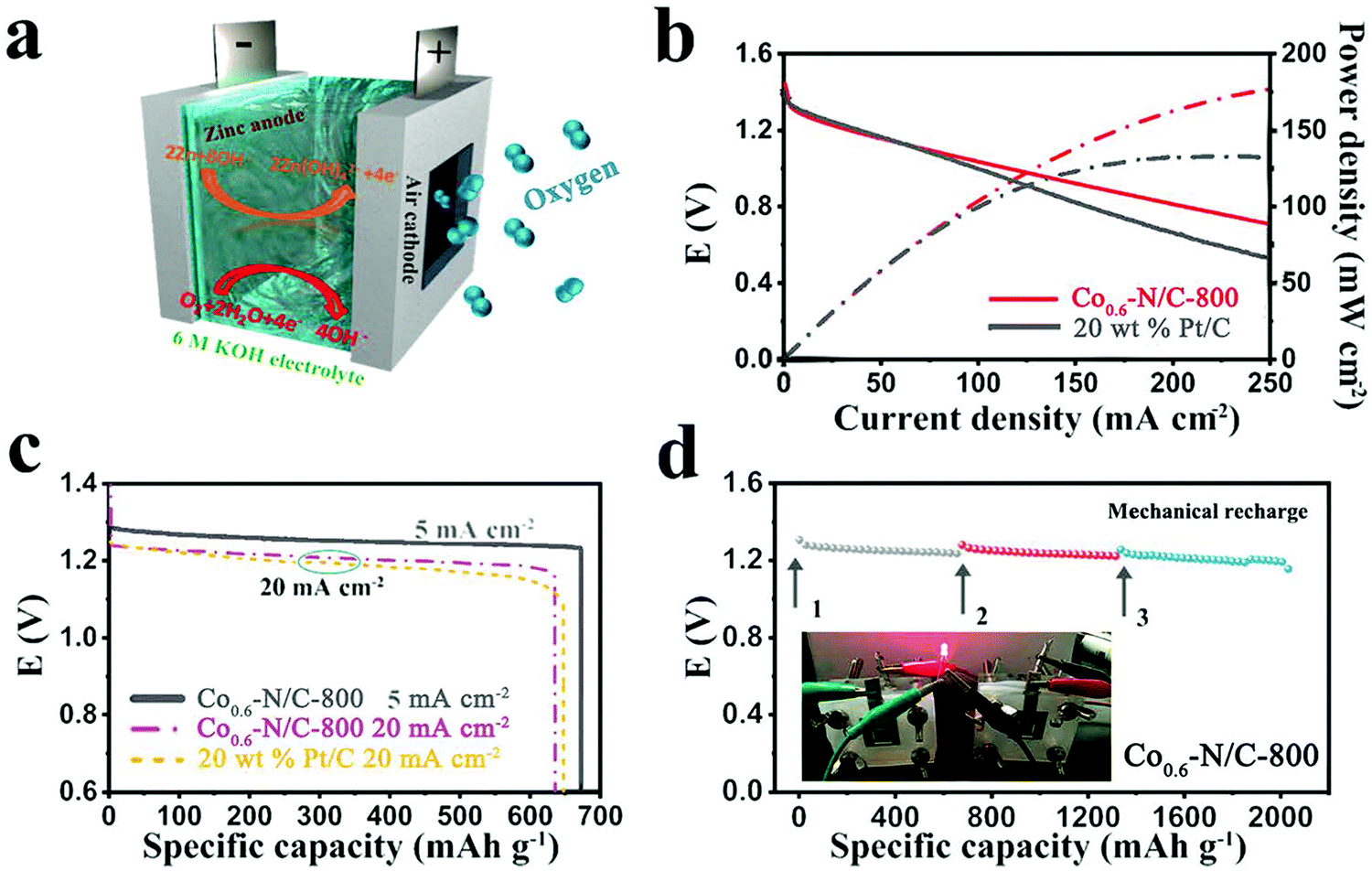 | ||
| Fig. 5 (a) Schematic of the primary liquid Zn–air battery. (b) The polarization and power density curves of Co0.6–N/C-800 or 20 wt% Pt/C as an ORR catalyst (mass loading of 0.5 mg cm−2). (c) The discharge curves of the liquid Zn–air battery using Co0.6–N/C-800 or 20 wt% Pt/C as an ORR catalyst at a current density of 5 and 20 mA cm−2. (d) Mechanical recharge of the primary Zn–air battery assembled from Co0.6–N/C-800; inset is a photograph of a red light-emitting diode (LED) powered by two Co0.6–N/C-800 based Zn–air batteries in series. | ||
Conclusions
Co/Zn-containing bimetallic ZIF nanoleaves were successfully synthesized via a simple but efficient “shape-transforming via aqueous solution-mediated method”. By pyrolyzing ZIF nanoleaves at high temperatures under nitrogen atmosphere, 2D Co, N embedded carbon (Co–N/C) electrocatalysts with open mesopores were obtained for Zn–air battery. Interestingly, controllable doping of more Co into the nitrogen-doped carbon nanoleaves facilitates the generation of mesoporous structure during the pyrolysis process, allowing for full exposure of the active sites and fast transfer of the mass/electrons during the ORR process. As a result, the optimized Co0.6–N/C-800 electrocatalyst shows good ORR performance in alkaline electrolyte and in the Zn–air battery. These findings show that highly mesoporous structure and accessible active sites are fundamentally important for promoting high ORR activity. The proposed “shape-transformation via aqueous solution-mediated method” will provide useful guidance for developing novel electrocatalysts with enhanced performance for energy storage and conversion applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (11575084 and 51602153), the Natural Science Foundation of Jiangsu Province (BK20160795), the Fundamental Research Funds for the Central Universities (No. NE2018104), the Foundation of Graduate Innovation Center in NUAA (No. kfjj20180615) and a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Jing Tang would like to thank the JSPS International Research Fellowship (No. 17F17080) for their support. This work was performed in part at the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities for the researchers of Australia.References
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- M. Breitwieser, M. Klingele, S. Vierrath, R. Zengerle and S. Thiele, Adv. Energy Mater., 2018, 8, 1701257 CrossRef.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 127, 598–603 CrossRef.
- J. Tang, J. Liu, N. L. Torad, T. Kimura and Y. Yamauchi, Nano Today, 2014, 9, 305–323 CrossRef CAS.
- G. Wu, K. L. More, P. Xu, H. L. Wang, M. Ferrandon, A. J. Kropf, D. J. Myers, S. Ma, C. M. Johnston and P. Zelenay, Chem. Commun., 2013, 49, 3291–3293 RSC.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- H. Tan, Y. Li, X. Jiang, J. Tang, Z. Wang, H. Qian, P. Mei, V. Malgras, Y. Bando and Y. Yamauchi, Nano Energy, 2017, 36, 286–294 CrossRef CAS.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- H. Wu, H. Li, X. Zhao, Q. Liu, J. Wang, J. Xiao, S. Xie, R. Si, F. Yang, S. Miao, X. Guo, G. Wang and X. Bao, Energy Environ. Sci., 2016, 9, 3736–3745 RSC.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W. C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 129, 16302–16306 CrossRef.
- J. Tang and Y. Yamauchi, Nat. Chem., 2016, 8, 638–639 CrossRef CAS PubMed.
- Y. Z. Chen, C. Wang, Z. Y. Wu, Y. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- H. Xia, J. Zhang, Z. Yang, S. Guo, S. Guo and Q. Xu, Nano-Micro Lett., 2017, 9, 43 CrossRef PubMed.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- G. Fang, J. Zhou, C. Liang, A. Pan, C. Zhang, Y. Tang, X. Tan, J. Liu and S. Liang, Nano Energy, 2016, 26, 57–65 CrossRef CAS.
- Y. V. Kaneti, S. Dutta, M. S. Hossain, M. J. A. Shiddiky, K.-L. Tung, F.-K. Shieh, C.-K. Tsung, K. C.-W. Wu and Y. Yamauchi, Adv. Mater., 2017, 29, 1700213 CrossRef PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed.
- Y. Wang, M. Zhao, J. Ping, B. Chen, X. Cao, Y. Huang, C. Tan, Q. Ma, S. Wu, Y. Yu, Q. Lu, J. Chen, W. Zhao, Y. Ying and H. Zhang, Adv. Mater., 2016, 28, 4149–4155 CrossRef CAS PubMed.
- L. Kong, J. Zhu, W. Shuang and X. H. Bu, Adv. Energy Mater., 2018, 8, 1801515 CrossRef.
- M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu and H. Xu, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed.
- F.-K. Shieh, S.-C. Wang, S.-Y. Leo and K. C.-W. Wu, Chem. – Eur. J., 2013, 19, 11139–11142 CrossRef CAS PubMed.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC.
- B. Motevalli, N. Taherifar, H. Wang and J. Z. Liu, J. Phys. Chem. C, 2017, 121, 2221–2227 CrossRef CAS.
- R. Chen, J. Yao, Q. Gu, S. Smeets, C. Baerlocher, H. Gu, D. Zhu, W. Morris, O. M. Yaghi and H. Wang, Chem. Commun., 2013, 49, 9500–9502 RSC.
- C. Guan, X. Liu, W. Ren, X. Li, C. Cheng and J. Wang, Adv. Energy Mater., 2017, 7, 1602391 CrossRef.
- H. Liang, X. Jiao, C. Li and D. Chen, J. Mater. Chem. A, 2018, 6, 334–341 RSC.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed.
- G. Gupta, D. A. Slanac, P. Kumar, J. D. Wigginscamacho, X. Wang, S. Swinnea, K. L. More, S. Dai, K. J. Stevenson and K. P. Johnston, Chem. Mater., 2009, 21, 4515–4526 CrossRef CAS.
- Z. Yan, M. Cai and P. K. Shen, J. Mater. Chem., 2012, 22, 2133–2139 RSC.
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed.
- X. Li, P. Cui, W. Zhong, J. Li, X. Wang, Z. Wang and J. Jiang, Chem. Commun., 2016, 52, 13233–13236 RSC.
- Q. Chen, Z. Cao, G. Du, Q. Kuang, J. Huang, Z. Xie and L. Zheng, Nano Energy, 2017, 39, 582–589 CrossRef CAS.
- G. Bepete, E. Anglaret, L. Ortolani, V. Morandi, K. Huang, A. Pénicaud and C. Drummond, Nat. Chem., 2017, 9, 347–352 CrossRef CAS PubMed.
- G. Nagaraju, S. C. Sekhar, G. S. R. Raju, L. K. Bharat and J. S. Yu, J. Mater. Chem. A, 2017, 5, 15808–15821 RSC.
- X. X. Wang, D. A. Cullen, Y. T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- S. Guo, P. Yuan, J. Zhang, P. Jin, H. Sun, K. Lei, X. Pang, Q. Xu and F. Cheng, Chem. Commun., 2017, 53, 9862–9865 RSC.
- Q. Lai, Y. Zhao, Y. Liang, J. He and J. Chen, Adv. Funct. Mater., 2016, 26, 8334–8344 CrossRef CAS.
- Q. Lai, J. Zhu, Y. Zhao, Y. Liang, J. He and J. Chen, Small, 2017, 13, 1700740 CrossRef PubMed.
- J. Xiang, J. Li, X. Zhang, Q. Ye, J. Xu and X. Shen, J. Mater. Chem. A, 2014, 2, 16905–16914 RSC.
- N. L. Torad, R. R. Salunkhe, Y. Li, H. Hamoudi, M. Imura, Y. Sakka, C. C. Hu and Y. Yamauchi, Chem. – Eur. J., 2014, 20, 7895–7900 CrossRef CAS PubMed.
- S. S. A. Shah, L. Peng, T. Najam, C. Cheng, G. Wu, Y. Nie, W. Ding, X. Qi, S. Chen and Z. Wei, Electrochim. Acta, 2017, 251, 498–504 CrossRef CAS.
- L. Huo, B. Liu, G. Zhang, R. Si, J. Liu and J. Zhang, J. Mater. Chem. A, 2017, 5, 4868–4878 RSC.
- J. Park, M. Risch, G. Nam, M. Park, T. J. Shin, S. Park, M. G. Kim, Y. S. Horn and J. Cho, Energy Environ. Sci., 2017, 10, 129–136 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00095j |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2019 |