
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergy and antagonism in natural product extracts: when 1 + 1 does not equal 2
Lindsay K.
Caesar
 and
Nadja B.
Cech
*
and
Nadja B.
Cech
*
Department of Chemistry & Biochemistry, University of North Carolina at Greensboro, Greensboro, North Carolina, USA. E-mail: nadja_cech@uncg.edu
First published on 12th June 2019
Abstract
Covering: 2000 to 2019
According to a 2012 survey from the Centers for Disease Control and Prevention, approximately 18% of the U.S. population uses natural products (including plant-based or botanical preparations) for treatment or prevention of disease. The use of plant-based medicines is even more prevalent in developing countries, where for many they constitute the primary health care modality. Proponents of the medicinal use of natural product mixtures often claim that they are more effective than purified compounds due to beneficial “synergistic” interactions. A less-discussed phenomenon, antagonism, in which effects of active constituents are masked by other compounds in a complex mixture, also occurs in natural product mixtures. Synergy and antagonism are notoriously difficult to study in a rigorous fashion, particularly given that natural products chemistry research methodology is typically devoted to reducing complexity and identifying single active constituents for drug development. This report represents a critical review with commentary about the current state of the scientific literature as it relates to studying combination effects (including both synergy and antagonism) in natural product extracts. We provide particular emphasis on analytical and Big Data approaches for identifying synergistic or antagonistic combinations and elucidating the mechanisms that underlie their interactions. Specific case studies of botanicals in which synergistic interactions have been documented are also discussed. The topic of synergy is important given that consumer use of botanical natural products and associated safety concerns continue to garner attention by the public and the media. Guidance by the natural products community is needed to provide strategies for effective evaluation of safety and toxicity of botanical mixtures and to drive discovery in botanical natural product research.
 Lindsay K. Caesar | Dr Lindsay Caesar completed her PhD in analytical chemistry and natural product discovery from the University of North Carolina at Greensboro. She is currently finishing her work in the laboratory of Professor Nadja Cech at UNCG where she is developing bioinformatic approaches to understand the complexities of botanical medicines. Dr Caesar is a National Institutes of Health T32 Fellow. Her research focuses on the development of mass spectrometric and multivariate statistical approaches to evaluate synergy in complex mixtures to facilitate the discovery of bioactive compounds. |
 Nadja B. Cech | Dr Nadja Cech is Patricia A. Sullivan Distinguished Professor of Chemistry at the University of North Carolina at Greensboro (UNCG). She has had a lifelong interest in the use of plants for medicine, stemming from her involvement as a child in establishing one of the world's largest medicinal plant and seed companies, a company still operated by her family today. Dr Cech leads a dynamic research group at UNCG, for which a major focus in the development of metabolomics as a tool to understand synergy and complexity in biologically active botanical natural products. This work has been continuously funded by the National Institutes of Health for more than 15 years, and was awarded the Jack L. Beal Award from the Journal of Natural Products in 2011. Dr Cech is a member of the Center of Excellence for Natural Product Drug Interaction Research, and Co-Director of the Medicinal Chemistry Collaborative (https://mcsquared.uncg.edu/). |
1. Introduction
Plants have been used as medicine since the beginning of human history.1 Texts from ancient Sumeria, India, Egypt, China, and others contain recipes for medicinal plant preparations for the treatment of disease.1,2 Today, medicinal plant use remains widespread, and a significant portion of the world's population utilizes herbal natural products and supplements as the primary mode of healthcare.3–5 In the United States, nearly 20% of adults and 5% of children utilize botanical supplements to treat disease.6Despite centuries of use, the activity of botanical medicines is only partially understood, and for most natural products on the market, there is a lack of knowledge as to which constituents are responsible for the purported biological activity. Scientific investigation of botanical natural products is challenging because of their immense complexity and variability.7–9 Natural products chemistry efforts are typically devoted to reducing complexity and identifying single “active” constituents for drug development. However, given that complex plant extracts, and not single molecules, are often administered for medicinal purposes, interactions between constituents could be of great importance.
Understanding how mixtures work in concert to achieve a given biological effect may address the ever-increasing threat of disease resistance. Indeed, many diseases are not regulated by a single molecular target, but often have a multi-factorial causality.7,9 It has been shown in numerous studies that disease resistance is less likely to occur against a combination of compounds than to single active constituents.8,10 Plants have evolved over millennia to address the multifactorial nature of disease pathogenesis by targeting pathogens through the combined action of structurally and functionally diverse constituents.7,11 As such, complex natural product mixtures offer an important resource for drug development, and to ensure future success in natural products research, understanding interactions within and between the constituents of natural product mixtures is paramount.
Pharmacological investigations into combination effects can be examined at the level of the molecular targets, disease pathways, cellular processes, and patient responses.12 As such, in vitro, in vivo, pre-clinical, and clinical research can all provide valuable insight into combination effects. Considerable progress has been made in the clinical arena in terms of investigating drug synergy, reviewed extensively in several publications.12–15 While much research is being conducted in this realm, this review will focus primarily on methodology to interpret combination effects using molecular and cellular methods.
Botanical extracts may contain hundreds or even thousands of individual constituents at varying abundance16 (Fig. 1) and identifying the compounds responsible for a given biological effect represents a significant challenge. Too often, it is assumed that the behaviour of a mixture can be described by the presence of just a few known constituents. However, a number of studies have shown that the overall activity of botanical extracts can result from mixtures of compounds with synergistic, additive, or antagonistic activity,9,17–20 and those who work in the field of botanical natural products research will be quick to admit that it is very often the case that isolation efforts on a botanical extract fail because activity is lost upon fractionation.9,17,20 While there are multiple possible explanations for this failure (including irreversible adsorption of compounds to the column packing),21 it is certainly true that in some cases loss of activity occurs because multiple constituents are required to observe the biological effect. Many investigators recognize the multi-factorial nature of botanical medicines. However, research methodology as applied to botanical mixtures still tends, in most cases, either to take a reductionist approach (focusing on just one or two “marker compounds”) or to ignore the question of chemical composition altogether, testing the biological effects of complex mixtures for which active constituents are unknown. The problem in the latter case is that results tend to be difficult both to interpret and to reproduce. Herein, we seek to provide an overview of the methodology that currently exists to understand combination effects within complex mixtures. We will highlight existing technologies for studying combination effects, placing particular emphasis on – Omics technologies and other Big Data approaches that have developed significantly in the last several years. We aim to provide practical advice to investigators seeking to comprehensively evaluate the constituents and mechanisms responsible for the biological activity of botanical mixtures.
 | ||
| Fig. 1 Chromatograms (obtained with liquid-chromatography coupled to mass spectrometry) of a complex extract of the botanical Salvia miltiorrhiza (Chinese red sage or Danshen). The full chromatogram is shown in (A), while (B) shows a zoomed in version of the baseline that demonstrates the immense complexity of the mixture. Counts for numbers of ions detected are shown at the right, and it is observed that the number of ions detected increases by ∼10-fold with each 10-fold decrease in the cutoff for peak area. Notably, each mixture component may be represented by more than one ion, making it difficult to assign specifically the number of mixture components. Nonetheless, the data indicate the immense complexity of the botanical extract. | ||
2. Terminology and identification of combination effects
2.1 Definitions of synergy and antagonism
Several reviews have been written on the topic of combination effects in recent years that provide valuable commentary on defining combination effects in complex mixtures.7–9 Although the evaluation of interactions between multiple bioactive constituents has gained popularity in many scientific disciplines,17,22–25 it remains difficult to give a undisputable definition for the term synergy.9,12,26 It is generally agreed, however, that interactions between multiple agents can be classified as antagonistic, additive/non-interactive, or synergistic. Additive and non-interactive combinations indicate that the combined effect of two substances is a pure summation effect, while an antagonistic interaction results in a less than additive effect. Positive interactions, known as potentiation or synergy, occur when the combined effect of constituents is greater than the expected additive effect.7–9,12,27–292.2 Assays for gathering biological data
To successfully acquire useful data for understanding combination effects in complex mixtures, one must first choose an appropriate biological assay for combination testing. Because combination effects can present themselves through myriad mechanisms (including changes to absorption and metabolism, affecting multiple cell targets, etc.), in vivo model systems provide the most comprehensive assessment of the overall effects on a living organism.30 The development of high-throughput in vivo testing of mixture-based libraries shows promise for identifying multi-target constituents within mixtures.30 Despite this, it remains challenging to address the complexity of in vivo systems, which require the sacrifice of test animals and maintenance of animal facilities. Additionally, results may not successfully translate from one animal model to another. Even when evaluating drug effectiveness in human patients, cell-to-cell variability and patient-to-patient variability in drug responses are common.12 Because of this, it is possible that patients receiving combination treatments have improved treatment efficacy because their disease is sensitive to at least one of the drugs in the combination (i.e. independent drug action), rather than because of true combination effects.12,31To overcome some of these challenges, many researchers work with in vitro systems instead. However, many cell-free, high-throughput assays that search for molecular targets do not accurately model the biology of an intact cell, making the discovery of relevant combination effects unfeasible.32 As such, cell-based assays can be employed that strike a balance between efficiency and preservation of molecular pathway interactions.33 Many useful cellular systems for identifying combination effects in vitro have been discussed in a recent publication by Pemovska et al.12 In addition to choosing relevant cellular systems for conducting biological testing, it is important to mimic physiological conditions in the assay itself. Indeed, most media used to grow cells for biological testing do not mimic physiological conditions, influencing the metabolism and phenotypic response of the cells under study.34 Similarly, biological assay conditions can result in “dynamic residual complexity,” in which the sample is subjected to an environmentally-induced chemical change, making interpretation of results challenging.35 In their recent publication, Vande Voorde et al.34 illustrated that the utilization of a complex culture medium designed to mimic the physiological environment of cancer cells prevented the formation of unwanted phenotypic artifacts and improved the translatability between in vitro assay results and in vivo tumor models. The utilization of physiologically relevant media also improves the likelihood that components that elicit a biological response during biological testing will be soluble and stable in a biological system, facilitating identification of active constituents. Primary tissue assays comprised of multiple cell types, such as those used to screen drug combinations for anti-inflammatory activity in mixed cultures of lymphocytes, can also be used to reveal combination effects that work through multi-target mechanisms.32
When screening for biological activity in vitro, however, researchers should be aware of potential false-positive results originating from pan-assay interference compounds, commonly referred to as PAINS, which are often identified as hits in biological screens.35 These false positive results may occur through a variety of mechanisms, including fluorescence quenching, aggregation effects, chemical reactivity, oxidation/reduction, membrane disruption, and residual complexity.35 Synergy results are often identified in aqueous media due to aggregation effects, which can be minimized by the addition of detergent to the media.36 While the promiscuous nature of PAINS compounds may in some cases be cause for concern, numerous examples of clinical drugs contain substructures that fall into this category.37 For example, many quinone-based drugs have been approved by the FDA for their antineoplastic, immunosuppressant, and antiprotozoal activities.37 As such, biological assay results should be seen as hypothesis-generating tools, and further verification is required to identify true leads (or to eliminate “PAINS” compounds from consideration).
In addition to carefully choosing the biological system to study for combination effects, data enabling the efficient comparison of a drug combination to agents in isolation must be gathered.8,32 Combination effects including synergy and antagonism can occur over a broad range of concentrations, so various ratios of the samples under study must be tested.8,32,33,38–41 Numerous methodologies have been developed to acquire data to discover combination effects in vitro, including checkerboard assays and time-kill methods, many of which are quite labor- and material-intensive.8,32,42 One of the simplest methods for identifying potential combination effects is through testing samples alone and in combination, and determining if the combined effect of the samples is greater, equal, or less than the expected sum of the two samples in isolation. Although simple, assays employing this approach cannot claim synergy without further study because they lack the range of concentration combinations required to fully assess combination effects, and should be used only to prioritize samples for more in-depth studies.43 These in-depth studies can be achieved using a dose–response matrix design,33 also known as a checkerboard assay, in which a series of dose–response curves using different dose combinations of the agents under study are acquired and compared.8,32
In addition to concentration-based approaches to evaluate combination effects, time-based approaches have also been developed and applied to identify antimicrobial synergy and to describe the relationship between bactericidal activity and sample concentration.44 This method involves exposing a selected pathogen to an inhibitor (or combination of inhibitors), sampling cultures at regular time intervals, serially diluting and incubating aliquots, and comparing the colony forming units produced. The resulting dose–response curve can be used to define additive, synergistic, and antagonistic effects.44 Importantly, several of these methods have been compared using the same datasets,38–41 revealing a lack of consistency between conclusions met using these approaches.38–40,42,45 Not only do in vitro tests often result in conflicting results, but it is very often the case that reproducible hits in vitro lack efficacy in vivo.30 Because of this inconsistency, preliminary screening efforts should be used to prioritize candidates with potential synergy but should not be used to unequivocally define combination effects.
2.3 Models for assessing combination effects
To identify if an interaction exists between individual compounds or complex samples, the observed combination response must be compared to the expected effect using a “null reference model.”12,46,47 Much of the confusion around categorizing interactions as antagonistic, additive, or synergistic results from the use of different reference models that are used to define the “expected” outcome of a given combination.27,48–51 Several reference models, as well as their biological assumptions and their limitations, are summarized in a recent paper by Pemovska et al.12 As described in a recent paper by Tang et al.,26 the two major reference model classes are the Bliss independence model52 and the Loewe additivity model,53 each of which relies on a different set of biological assumptions. The Bliss independence model, for example, assumes that each sample has independent, yet competing effects, while the Loewe additivity model defines the expected effect as a sample combined with itself.47 Recently, an additional reference model, the zero interaction potency (ZIP) model, was developed that takes advantage of both Loewe and Bliss models.47 The ZIP model is based on the assumption that two non-interacting samples will cause minimal changes to the dose–response curves, both in terms of the slope of the curve and in the half maximal effect.47 This model shows particular promise for high-throughput drug combination screenings and shows potential for identifying the variety of combination effects that can occur across different concentration ranges.47 These models, and other lesser utilized models, are discussed in depth in several publications.26,27,49,54Despite the existence of numerous reference models, the general isobole equation, based on the assumptions of the Loewe additivity principle, remains the most popular for studying combination effects.8,9,27–29,55 As described elsewhere, an isobole, or an “isobologram,” is a graphical representation of the combination effects between two samples.8,9,27–29,55 The axes of the plot represent the doses of individual agents, and the points plotted indicate the combination of concentrations of the two treatments required to reach a particular fixed effect (i.e. 50% inhibition of cell growth).8 If the two samples have no interaction, the line joining the axes will be a straight line. Synergy will result in a concave curve, and antagonism will result in a convex curve (Fig. 2).8,9,27–29 In a recent publication, Lederer et al.55 scrutinized the implicit assumptions of the Loewe additivity model (and with it the general isobole equation), and found that the consistency of the model only holds if the two samples under study do not differ in the slopes nor the maximal effects of their dose–response curves.55 In cases where one sample reaches an effect that cannot be reached by the other sample, the Loewe additivity consistency condition is violated.54,56 To overcome this limitation with the Loewe additivity consistency condition, Lederer et al.55 developed an adaptation of the general isobole equation termed the explicit mean equation. The explicit mean equation is equivalent to the isobole equation in cases where the two samples meet the Loewe additivity consistency condition and is capable of identifying combination effects even if this condition is violated. In a follow up study, Lederer et al.46 compared six models built on either Loewe additivity or Bliss independence principles using existing, high-throughput datasets57,58 and found that Loewe additivity models performed better than Bliss independence at separating synergy relationships from other combination effects and that the explicit mean equation was the overall best performing model.46
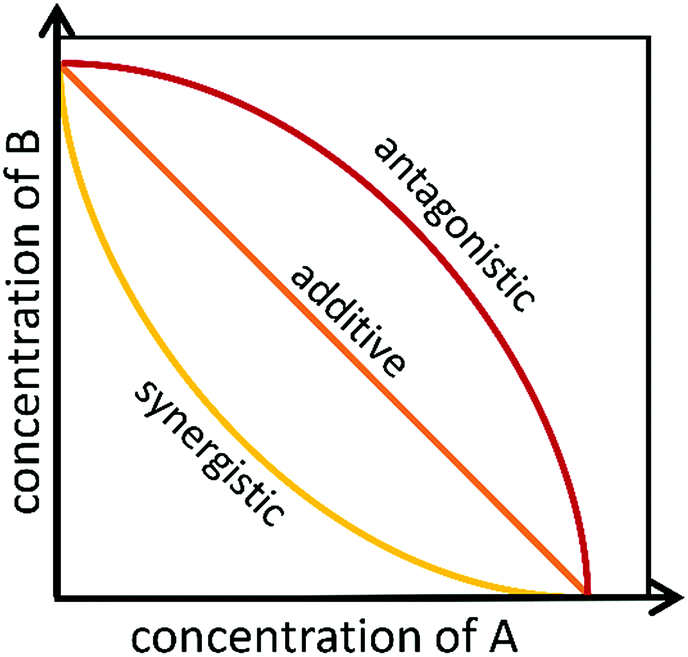 | ||
| Fig. 2 Example of isobolograms for antagonistic, additive, and synergistic components. Axes represent the doses of individual agents, and the points represent the combination of concentrations of the two agents required to reach a particular fixed effect. | ||
In recent years, variants of the Loewe additivity model and the Bliss independence model have been developed.56,59–63 However, because the expected responses from these different models are often disparate,27,46 it is challenging to draw biological conclusions from the resulting data. In some instances, combination effects have been identified as synergistic by one model but antagonistic by another.57 As such, researchers should be clear about which model they have chosen to adopt, as stated in the Saariselkä agreement.64 Tang et al.26 have expanded upon this suggestion and have proposed the use of terminology that incorporates results from both Bliss independence and Loewe additivity models. The authors argue that the level of consistency between models should be used to designate the degree of synergy or antagonism. For example, if both models identify a given interaction as synergistic, that interaction should be considered “strong synergy,” and if the combination is identified as synergistic by one model only, it should be considered “weak synergy”.26 By utilizing both models, this proposal minimizes the incorporation of bias into predictions and provides more informative definitions for the combination effects described. While in principle this proposal makes sense, it also relies on the assumption that the models are equally valid. While Loewe additivity models have been shown to perform better than Bliss independence models on numerous occasions,46 Russ and Kishony65 found that the Bliss independence models are more consistent when interactions between more than two samples are evaluated. As such, the use of any synergy model should be seen only as a hypothesis-generating tool to prioritize potential interaction effects for further study. Indisputable definitions of synergy and antagonism remain elusive, and a wider agreement on the terminology used for interaction assessment is still required to standardize future research initiatives.
2.4 Scoring and interpreting biological data
In addition to a lack of consensus among the theoretical models to utilize for defining combination effects, there are challenges on how to apply and interpret existing models to analyze drug combinations.47 As stated earlier, most synergy analyses focus on the differences in isobologram shapes at fixed effects, and summary interaction scores such as the fractional inhibitory concentration (∑FIC) index have found wide application.8,47,48,66,67 The ∑FIC index is calculated using eqn (1):8 | (1) |
In this equation, A and B represent the samples under study, IC50A and IC50B represent the concentrations of A or B in isolation to reach 50% inhibition, [A] is the IC50 of A in the presence of B, and [B] is the IC50 of B in the presence of A. Notably, any fixed effect can be used to calculate ∑FIC indices, but IC50 values are perhaps the most common metric.
Despite the popularity of this method, the interpretation of ∑FIC scores for defining combination effects varies considerably from author to author. In their recent publication, van Vuuren and Viljoen8 provide an excellent commentary on ∑FIC score interpretation, improving upon the earliest interpretations proposed by Berenbaum in which synergistic interactions were considered to be any value below one, additive/indifferent interactions focused on one, and antagonistic interactions above one.27 However, because of the inconsistency across null reference models, and because fixed effects can often be placed within a three-dilution range using in vitro assays,68 a more conservative approach is warranted. Taking this into consideration, van Vuuren and Viljoen8 and the authors of this review suggest that synergistic interactions be defined as interactions having ∑FIC ≤ 0.5, additive interactions range from 0.5 to 1.0, non-interactive effects range from 1.0 to 4.0, and antagonistic effects fall above 4.0 (Table 1).
| Combination effect | ∑FIC range |
|---|---|
| Synergy | ∑FIC ≤ 0.5 |
| Additivity | 0.5 < ∑FIC ≤ 1.0 |
| Indifference | 1.0 < ∑FIC ≤ 4.0 |
| Antagonism | 4.0 < ∑FIC |
Despite its popularity, the ∑FIC index, like the isobologram upon which it is based, is insufficient to effectively capture the combination effects that may occur across multiple dose regions.46,47 An inherent limitation of the ∑FIC index is the focus on a single interaction parameter. In a recent publication, Lederer et al.46 compared multiple synergy measurements and found that the “lack of fit” model,69 where synergy scores are defined by the volume spanned between the null reference model and the measured response, performed better than parametric models in its ability to identify synergistic effects.46
Similarly, Yadav et al. developed a score that enables the use of an interaction landscape over the full dose–response matrix to identify combination effects across multiple dosages and response levels.47 Rather than relying on a single parameter such as the IC50 measurement, the delta-score utilized in this study was calculated by assessing changes in both the shape parameter and the midpoint of each dose–response curve for individual samples and combinations thereof. The delta scores were visualized using a response surface plot to visualize the combination effect landscape over all tested dosage combinations, enabling identification of potency changes and differences in combination effects even within the same sample pair (Fig. 3). There appears to be value in using these different methods to explore synergy; however, these approaches have not yet been applied to understand synergy in complex natural products and discussion of their merit for this purpose remains hypothetical. Despite the aforementioned limitations, isobole analysis and the ∑FIC index have found the widest utility in natural products research.8
 | ||
| Fig. 3 Example of synergistic (top) and antagonistic (bottom) interaction landscapes using delta scores (δ) calculated with the zero interaction potency model of compounds in combination with ibrutinib, an approved anti-cancer drug targeting Bruton's tyrosine kinase. (A) Interaction map between anti-cancer activity of ispinesib (a selective kinesin spindle protein inhibitor) and ibrutinib. Average delta across the dose–response matrix (Δ) is 17.596, indicative of overall synergy. (B) Interaction map between canertinib (an epidermal growth factor receptor family inhibitor) and ibrutinib. The Δ value is −14.038, indicative of overall antagonism. Figure is reprinted with permission from Yadav et al. 2015.47 | ||
3. Documented examples of synergism and antagonism
Proponents of the health benefits of plant-based medicines often proclaim that whole plant preparations are more effective than isolated compounds due to the beneficial interactions between constituents within them.9,18,70,71 While this claim is sometimes disputed,72–75 considerable evidence exists that combination effects within complex extracts can alter the biological activity of a mixture.7–9,76 Here, we provide a few case studies in which synergy and/or antagonism within botanical preparations have been discussed. Additional examples of synergy within and between botanical extracts have been extensively reviewed in several publications,8,9,20,28,76 providing compelling evidence that at least in some cases, the combined effect of botanical mixtures is not simply a summation of their individual constituents. However, explorations into phytosynergy are only in their infancy. The vast majority of complex natural product mixtures still await chemical investigation, representing an untapped resource with considerable potential for future scientific exploration.3.1 Anti-plasmodium activity of Artemisia annua
Artemisia annua L. (Asteraceae) has gained considerable popularity over the last few years since the award of the 2015 Nobel Prize in Physiology or Medicine to Youyou Tu for her discovery of artemisinin, an antimalarial sesquiterpene lactone produced by this plant.77,78 Artemisinins have been established as potent and safe antimalarial agents,79 and artemisinin-based combination therapies are now the front-line treatment recommendation by the World Health Organization.80 The replacement of ineffective malaria treatments such as chloroquine with artemisinin-based combination therapies has decreased malaria-associated morbidity and mortality worldwide.81–83 Several researchers have suggested that artemisinin acts to destroy Plasmodium falciparum parasites through the activation of a trioxane bridge in the P. falciparum food vacuole in a heme-dependent manner.84,85 This disruption causes the production of free radicals that interrupt heme detoxification, ultimately generating more reactive oxygen species and killing the parasite.In addition to artemisinin, there are approximately 30 other flavonoids and sesquiterpenes within A. annua, some of which have minor anti-plasmodial activities.86 As might be expected, since botanical preparations are multi-factorial rather than monospecific in nature, both in vitro and in vivo studies evaluating the activity of A. annua extracts have found that the amount of artemisinin in the extracts does not fully explain the extract's efficacy against P. falciparum parasites.87,88 Indeed, various combination therapies including artemisinin and its derivatives are utilized as antimalarial treatments.89,90 In a recent study, Suberu et al.91 aimed to identify the compounds within A. annua tea extract contributing to its anti-plasmodial efficacy. Building upon the work of previous studies which found that several flavonoids potentiated the activity of artemisinin against P. falciparum,92,93 Suberu et al.91 tested the tea extract, purified standards from the extract, and various combinations of artemisinin with purified compounds against both chloroquine-sensitive and chloroquine-resistant strains of P. falciparum. Interestingly, the type of combination effect observed, whether it be synergistic, additive, or antagonistic, often differed depending on the dosage of the combined constituents and/or the resistance profile of the parasite under analysis.91
Using isobologram analysis and calculating ∑FIC indices, Suberu et al.91 found several compounds that enhanced or antagonized the activity of artemisinin against P. falciparum. Two compounds that contained anti-plasmodial activity, 9-epi-artemisinin and artemisitene, were found to antagonize the efficacy of artemisinin against both chloroquine-sensitive and chloroquine-resistant strains. Although the mechanism by which these compounds antagonize artemisinin's activity is unknown, it is reasonable to assume these compounds, which have only minor structural differences, compete for the same molecular target, reducing the overall efficacy of the compounds in combination.91 Several additional compounds contained within the extract, however, did not demonstrate the same combination effect at all concentrations tested. For example, 3-caffeoylquinic acid showed a summation effect in combination with artemisinin at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (artemisinin to 3-caffeoylquinic acid) when tested against the chloroquine-sensitive strain, but at higher combination ratios (1:10–100), synergistic interactions were observed. Similarly, casticin, which possessed antagonistic activity at the 1:3 ratio, has been reported to be synergistic in other studies using higher combination ratios (1:10–1000).92,93 The reason for this discrepancy is unknown, but it is possible that these compounds act as either anti-oxidant or pro-oxidant species depending on the dosage level.94,95 When combined at a low concentration with artemisinin, they may have counteracted artemisinin activity through anti-oxidative interaction, minimizing the oxidative stress resulting from the reactive oxygen species formed through artemisinin's activity, while at higher concentrations they were pro-oxidative, increasing the oxidative stress and leading to increased efficacy of artemisinin.91
:3 (artemisinin to 3-caffeoylquinic acid) when tested against the chloroquine-sensitive strain, but at higher combination ratios (1:10–100), synergistic interactions were observed. Similarly, casticin, which possessed antagonistic activity at the 1:3 ratio, has been reported to be synergistic in other studies using higher combination ratios (1:10–1000).92,93 The reason for this discrepancy is unknown, but it is possible that these compounds act as either anti-oxidant or pro-oxidant species depending on the dosage level.94,95 When combined at a low concentration with artemisinin, they may have counteracted artemisinin activity through anti-oxidative interaction, minimizing the oxidative stress resulting from the reactive oxygen species formed through artemisinin's activity, while at higher concentrations they were pro-oxidative, increasing the oxidative stress and leading to increased efficacy of artemisinin.91
Other compounds, including rosmarinic acid and arteannuin B, showed differential combination effects when tested against sensitive and resistant strains of P. falciparum. Rosmarinic acid was synergistic against the sensitive strain, but showed antagonistic activity in the resistant strain.91 Similarly, arteannuin B had an additive/indifferent interaction in the chloroquine sensitive strain, but a synergistic interaction with the resistant strain, leading to a three-fold improvement in artemisinin's activity.91 Because arteannuin B selectively potentiates the activity of artemisinin in the chloroquine-resistant strain, it likely targets the parasite's chloroquine resistance mechanism, illustrating the promise of combination treatments not only for developing therapeutics against drug-resistant pathogens, but also for providing insight into the mechanisms by which parasites gain resistance as a whole.
It is important to note that Suberu et al. chose somewhat liberal ranges for the ∑FIC indices used to define their combination effects,91 and other researchers, depending on the models chosen, may have categorized some of the synergistic and antagonistic interactions as “additive” or “indifferent”.8 Even if one were to re-categorize interactions based on conservative estimates, however, all three types of combination effects (synergy, additivity, and antagonism) were witnessed during the course of this study. While the specific categorizations of synergy, additivity, and antagonism chosen by Suberu et al. may be disputed, it is clear that the nature of combination effects did often change depending on both the dosage and the parasite strain under study.91
3.2 Endotoxin from bacterial endophytes in Echinacea species
Few botanicals have been the subject of as much research or as much controversy as plants from the genus Echinacea. This botanical, which is widely used for the treatment of upper respiratory infections, has been the subject of several clinical trials. Although these trials had conflicting results,96,97Echinacea species remain one of the most popular and best-selling botanical medicines in the United States,98 and preparations from this plant are popular in Europe as well.99The constituents responsible for the activity of Echinacea purpurea (L.) Moench (Asteraceae) extracts and the mechanisms by which these constituents exert their purported beneficial effects have been studied extensively. Early research on Echinacea attributed its purported health benefits to its ability to “activate” or “stimulate” immune cells. These findings were based upon early work by Wagner and co-workers, in which isolated Echinacea polysaccharides were observed to stimulate phagocytosis and induce TNF-alpha secretion by macrophages.100 Later research demonstrated that much of the immunostimulatory activity originally attributed to Echinacea polysaccharides could instead be linked to the lipopolysaccharides and lipoproteins. These lipoproteins and lipopolysaccharides are components of bacterial cell walls, and can be attributed to the presence of bacterial endophytes, bacteria living asymptomatically within the Echinacea plant tissues.101–104 Even very minute quantities of certain lipoproteins and lipopolysaccharides induce pronounced immunostimulatory effects in macrophages, so the presence of these compounds as contaminants can confound in vitro assay data.
An alternative narrative about the immunomodulatory activity of Echinacea preparations focused on alkylamide constituents. Contrary to the research on polysaccharides, lipoproteins, and lipopolysaccharides, these alkylamides were observed to suppress the production of pro-inflammatory cytokines by macrophages.105–108 Such activity could translate to a beneficial anti-inflammatory effect in vivo. The apparently contradictory activity of various classes of compounds, both isolated from Echinacea, suggested the possibility that the activity of some constituents might be masked by others in the context of complex Echinacea extracts. This was shown in a study by Todd et al.,104 in which complex E. purpurea extracts possessed little to no cytokine-suppressive activity, but could be separated to produce sub-fractions with opposing effects. Some fractions, those containing alkylamides, suppressed cytokine and chemokine production by macrophages, while others, those containing lipopolysaccharides, induced cytokine production. Thus, it was demonstrated that lipopolysaccharides (and likely other compounds of bacterial origin) masked the anti-inflammatory effect of complex Echinacea preparations, effectively acting as antagonists. It was not until these fractions were separated that the individual activities of the various constituents could be observed.
3.3 Combination effects in Chinese Herbal Medicines
Chinese Herbal Medicine, a branch of Traditional Chinese Medicine, has been used for over 1000 years to promote health and to treat various illnesses in China and other Asian countries.109 In this field of medicine, multiple herbs are combined in order to take advantage of combination effects that improve the efficacy of active constituents and/or minimize side effects associated with treatment. The complexity of such formulations, however, poses a great challenge to researchers attempting to validate the effectiveness of herbal preparations. Zhou et al. have recently written an excellent review on the state of synergy research as it relates to Chinese Herbal Medicine, outlining methods to understand combination effects in multi-herb preparations as well as providing examples of specific herbal formulations.109Salvia miltiorrhiza Bunge (Lamiaceae) and Pueraria lobata (Willd.) Ohwi (Fabaceae), known as Danshen and Gegen, respectively, are often combined to treat coronary heart disease.110 In a recent study, Danshen and Gegen were tested both alone and in combination in order to confirm the presence of anti-atherogenic effects and to identify potential combination effects between herbal constituents. Biological effects of the three extracts (Danshen and Gegen alone and in combination) were tested for their anti-inflammatory, anti-foam cell formation, and anti-vascular smooth muscle cell (vSMC) proliferation effects.110 The biological assay results were evaluated using both fixed-ratio experimental design and fractional inhibitory concentration indices (alternatively named “combination indices” in this publication). The Danshen–Gegen combination was characterized as synergistic in the anti-inflammation assay, additive in the foam cell formation assay, and antagonistic in the vSMC proliferation assay, with ∑FIC indices of 0.75, 1.03, and 2.02, respectively.110 When categorizing these combination effects, however, the authors of this study chose quite lenient values. Using the recommended values in this review and others,8 the combination effects witnessed in these three assays would be re-categorized as additive (for the anti-inflammation assay) and indifferent (for the foam-cell formation and vSMC proliferation assays). Nonetheless, this study highlights the applicability of multi-herb formulas to treat disease, and provides rationale for the combination of Danshen and Gegen for the treatment of atherogenesis.110
4. Underlying mechanisms of synergy
Synergy can occur through a variety of mechanisms, including (i) pharmacodynamic synergism through multi-target effects, (ii) pharmacokinetic synergism through modulation of drug transport, permeation, and bioavailability, (iii) elimination of adverse effects, and (iv) targeting disease resistance mechanisms.8,9,76,77,111,112 While the general mechanisms by which synergy can occur are relatively well studied, the mechanisms by which specific botanical preparations exert synergistic effects remain largely unknown,76,113 stymying efforts to standardize and optimize them for therapeutic purposes. Only through understanding the nature of synergistic activity within botanical extracts will we be able to optimize safe and efficacious preparations for the treatment of disease.4.1 Pharmacodynamic synergism
Cancerous cells and pathogenic organisms can quickly gain resistance to drugs containing a single compound, and many cancers and resistant bacterial infections are now treated with complex drug combinations affecting multiple targets to overcome the development of resistance.114,115 Plants have long had to defend themselves against multi-factorial diseases, and have evolved to produce multiple active constituents that can adhere to cell membranes, intercalate into RNA or DNA, and bind to numerous proteins.7,116–118 Pharmacodynamic synergism results from the targeting of multiple pathways, which can include enzymes, substrates, metabolites, ion channels, ribosomes, and signal cascades.9,119Oftentimes, disease targets are able to counteract the therapeutic effect of an active metabolite, resulting in its reduced efficacy.76 One type of pharmacodynamic synergism involves “anti-counteractive action” in which a synergistic compound binds to an anti-target, effectively inhibiting the disease target from counteracting the therapeutic effect of the active constituent.76 Pharmacodynamic synergy may also occur through complementary actions, in which synergists in a mixture interact with multiple points of a given pathway, resulting in positive regulation of a process affecting the drug target or in the negative regulation of competing mechanisms. Through the selective variation of target activity and expression through complementary actions, pharmacodynamic synergists can both augment beneficial effects of treatments and reduce adverse effects of the disease.76 For example, Ginkgo biloba L. (Ginkgoaceae) has been shown in numerous studies to have synergistic neuroprotective effects both in vivo and in vitro by inhibiting the formation of free radicals, scavenging reactive oxygen species, regulating gene expression of mitochondrial targets, and reducing excessive stimulation of nerve cells by neurotransmitters.66,120
4.2 Pharmacokinetic synergism
In addition to pharmacodynamic synergy, plants often contain compounds that do not possess specific pharmacological effects themselves, but increase the solubility, absorption, distribution, or metabolism of active constituents.7,9,76,121 These pharmacokinetic effects result in enhanced bioavailability of active constituents, enabling increased efficacy of the extract as compared to individual constituents in isolation.9 Several examples exist in which mixture constituents improve the solubility of active constituents. For example, hypericin from Saint John's Wort (Hypericum perforatum L. (Hypericaceae)), is poorly soluble in water. However, when hypericin is combined with H. perforatum mixture constituents including procyanidin B2 and hyperocide, solubility and oral bioavailability of hypericin are significantly improved.122 In a recent study, researchers interrogated the function of highly abundant sugars, amino acids, choline, and organic acids that are found commonly in microbial, mammalian, and plant cells.123 Through these studies, it was found that these abundant molecules likely play a role in the production of “natural deep eutectic solvents,” which may serve as a third liquid phase, intermediate in polarity between lipid and water phases, where biosynthetic products of intermediate polarity are produced and stored.123 The presence of compounds that improve the solubility of bioactive constituents, both within and between organisms, is a particularly important type of synergism that is often underappreciated.Absorption of active constituents can be improved through a variety of mechanisms, including the inhibition of drug exporters such as P-glycoproteins.111,124,125 Additionally, transport barriers may be disrupted or their recovery delayed, improving permeability of active constituents into target cells.76 For example, the absorption of baicalin, a constituent of the plant Scutellaria baicalensis Georgi (Lamiaceae), is synergistically enhanced by the addition of both coumarins and volatile oils from the botanical Angelica dahurica Bentham et Hooker (Apiaceae), likely by affecting transport systems independent of P-glycoproteins.126 Pharmacokinetic synergy also results from constituents that inhibit enzymes that convert drugs into excretable or inactive forms, or that activate enzymes that convert pro-drugs into active forms.7,76
4.3 Elimination of adverse effects
An additional type of synergy occurs when inactive mixture constituents serve to neutralize the unwanted side effects of a toxic, yet bioactive constituent. This type of synergy, if it can truly be called that, does not function to improve the efficacy of the active compound(s) per se, but rather functions to minimize the negative effects that the active agent may cause.9 Many potent chemotherapeutic agents, for example, while successful in targeting tumor cells, are often limited by severe side effects caused by action of active agents against healthy cells. In a recent study, an extract of staghorn sumac (Rhus hirta (L.) Sudw. (Anacardiaceae)) was combined with the chemotherapeutic drug 5-fluorouracil (5-FU) commonly used to treat breast and colon cancer.127 In combination with 5-FU, the R. hirta extract was found to protect normal cells from 5-FU toxicity in vitro. This chemoprotective effect may have be attributed in part to the presence of antioxidants in the R. hirta extract,128 which minimized oxidative stress and cell damage initiated by 5-FU treatment.1274.4 Targeting disease resistance mechanisms
Many diseases, such as cancers and infectious diseases, have evolved resistance to single-target drugs. In cancer, drug resistance to single chemotherapeutic agents has increased largely due enzymatic cross-talk129 and counteractive pathways.130,131 Combination chemotherapy is growing in popularity, in part due to the ability for multi-constituent mixtures to modulate different pathways and overcome drug resistance.132 Infectious diseases, including those caused by fungi,133 viruses,134 and bacteria,135 are also becoming more challenging to treat due to the development of drug resistance.136 Bacterial pathogens gain resistance to antibiotics due to three major reasons: (i) active site modification resulting in inefficient drug binding, (ii) metabolism of antibiotics into inactive forms, or (iii) efflux of antibiotics out of bacterial cells (Fig. 4).20,111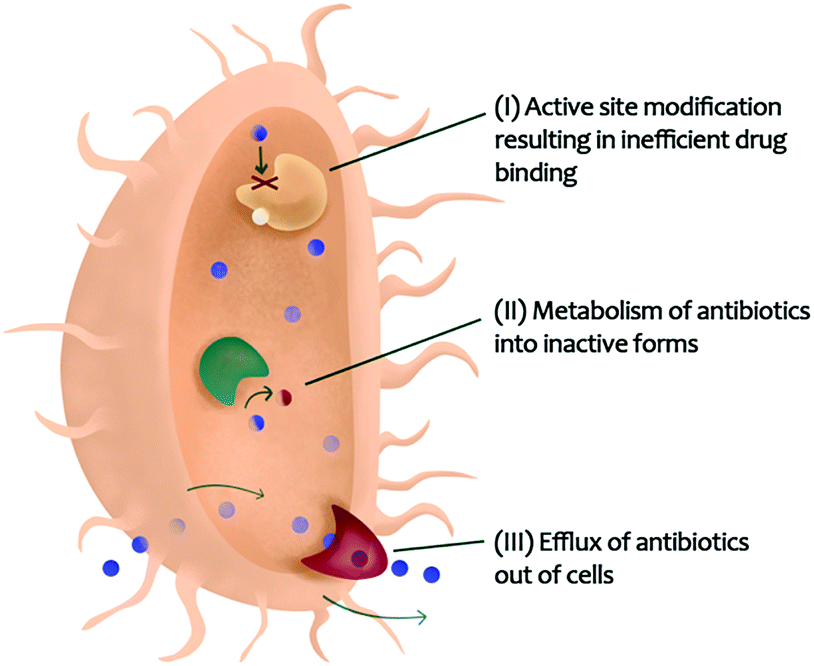 | ||
| Fig. 4 Bacterial resistance mechanisms that could be targeted with combination therapy enabling re-sensitization of resistant organisms to existing antibiotics. | ||
Bacterial resistance to beta-lactam antibiotics is achieved by the development of beta-lactamase enzymes that cleave the antibiotics into inactive forms.137 This resistance mechanism can be overcome by combining beta-lactam antibiotics with beta-lactamase inhibitors. In a recent study, Catteau et al.138 found that a dichloromethane extract of shea butter tree leaves (Vitellaria paradoxa C.F. Gaertn. (Saptoaceae)) synergized the activity of ampicillin, oxacillin, and nafcillin against methicillin-resistant Staphylococcus aureus by targeting PBP2a +/− beta-lactamase enzymes. V. paradoxa constituents ursolic acid and oleanolic acid were found identified as the constituents responsible for this synergistic activity.138
Similarly, promiscuous efflux pumps promote resistance by extruding a wide array of compounds from bacterial cells.139,140 In their hallmark paper, Stermitz et al.18 described the presence of an inhibitor of the norA efflux pump, 5′-methoxyhydnocarpin, in Berberis species that potentiated the activity of the efflux pump substrate berberine. More recently, the berberine-containing plant Hydrastis canadensis L. (Ranunculaceae) was found to possess synergistic norA efflux pump inhibitory activity.141 Many of these synergists have been characterized in subsequent publications.17,22,142
5. Identifying constituents responsible for combination effects
When working with complex natural product mixtures, constituents responsible for activity are often not known. Additionally, the composition of natural product extracts varies depending on how and where the source material is grown, prepared, processed, and stored,143 and as such, there is a lack of knowledge for many natural products about the composition and identity of what is being consumed. To address this safety risk, and to improve efficacy of natural product mixtures, bioactive mixtures should be comprehensively characterized and the concentrations and identities of constituents contributing to the biological activity (whether it be through additive, synergistic, or antagonistic means) should be determined. This task, while straightforward in theory, is quite challenging in practice since the biologically important constituents are often not known and are part of a complex matrix containing hundreds or thousands of unique constituents.165.1 Targeted approaches
Despite the historical effectiveness of bioassay-guided fractionation,150 loss of activity during fractionation is very common.145,148 Additionally, because structural information is not used to guide separations, this approach may result in the repeated isolation of previously described molecules.148 To avoid re-isolation of known molecules, preliminary structural assessment steps to identify and discard samples containing known active constituents can be taken.148,151–153 This process, termed “dereplication,” enables prioritization of samples likely to contain new biologically important entities, facilitating efficient use of resources for compound discovery.148,151–153 Dereplication is often achieved by comparing the spectral patterns of mixture constituents through mass spectrometry,151,154–156 NMR,157 or UV spectroscopy,151 and searching for known compounds with matching spectral fingerprints in a dereplication database. Recently, the Global Natural Product Social Molecular Networking (GNPS) platform has been developed that enables spectral annotation and identification of related compounds using MS/MS molecular networking.148,153,158 In addition, GNPS provides researchers the ability to share raw MS/MS spectra online, enabling crowdsourced spectra annotation and knowledge sharing between laboratories around the world.153
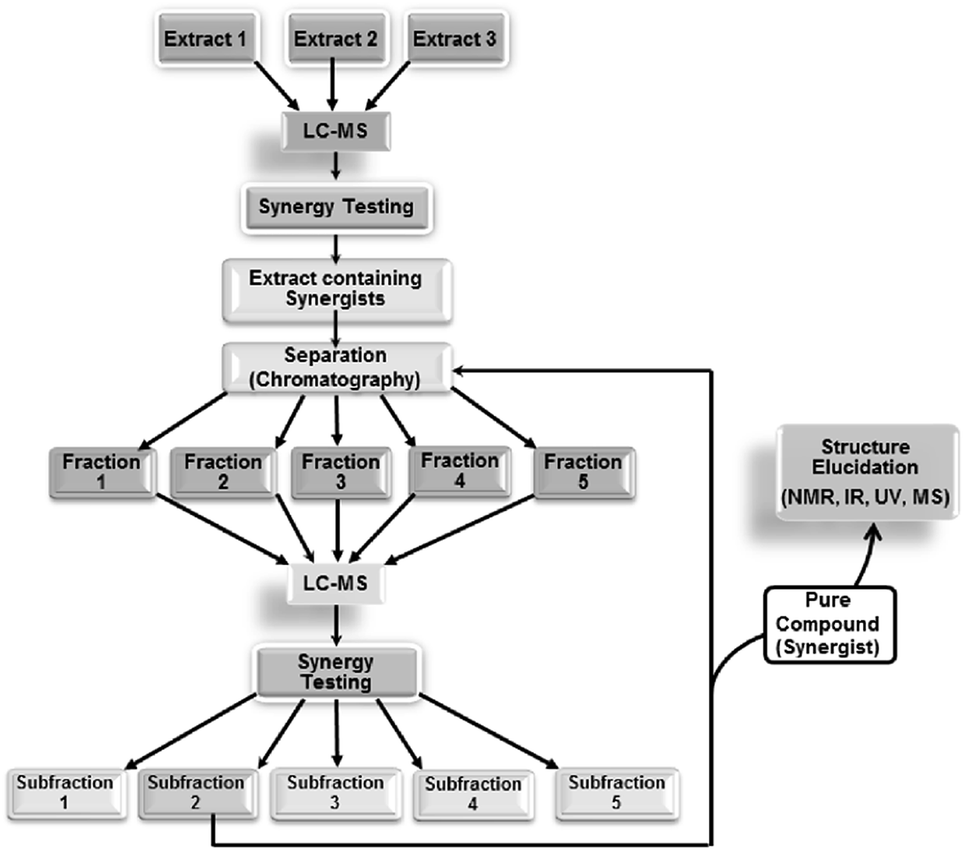 | ||
| Fig. 5 Synergy-directed fractionation workflow. Reproduced with permission from Junio et al. 2011.17 | ||
5.2 Metabolomics and biochemometrics
In mass spectrometry-based biochemometrics studies, the number of variables (ions detected) tends to greatly outnumber the number of samples analyzed (i.e. extracts or simplified fractions), posing a problem for many multiple regression models.163 Partial least-squares (PLS) analysis, however, due to its combination of principal component analysis (PCA) and multiple regression analysis, is less affected by this mismatch and is the most popular tool for modelling biochemometric data.163 The resulting PLS models, however, are often incredibly complex and difficult to decipher. Numerous data visualization tools have been developed to extract meaningful information from PLS datasets.146,163–165
One commonly used tool is the S-plot, in which correlation and covariance of variables with a given biological activity are plotted. In a recent study, S-plots were utilized to identify differences in metabolite profiles (detected using UPLC-QTOF-MS) of Garcinia oblongifolia Champ. Ex Benth. (Clusiaceae) leaves, branches, and fruits and to correlate those differences to differences in biological activity.162 Using this approach, 12 marker compounds were identified, primarily xanthones, that were likely responsible for the enhanced antioxidant and cytotoxic properties of the branch extract over other plant parts.162 In another study, S-plots were generated from bioactivity and chemical profiles of the fungus Ganoderma sinense to identify potential anti-tumor agents. This approach successfully identified five known cytotoxic compounds with significant antitumor potential.161 A recent study compared the use of S-plot analysis with an additional data visualization tool, the selectivity ratio, to identify antimicrobial constituents from the fungal organisms Alternaria and Pyrenochaeta sp.146 In this study, both S-plot and selectivity ratio analyses identified macrosphelide A as the dominant bioactive constituent from Pyrenochaeta sp. However, when attempting to identify bioactive compounds from Alternaria sp., the selectivity ratio outperformed the S-plot in its ability to identify altersetin, a low abundance antimicrobial constituent, without being confounded by highly abundant (and only weakly active) constituents in the mixture.146 In a follow up study, an inactive mixture was spiked with known antimicrobial compounds to identify the impact of data acquisition and data processing parameters on biochemometric analysis using the selectivity ratio plot.159 This study found that data transformation, contaminant filtering, and model simplification tools had major impacts on the selectivity ratio models, emphasizing the importance of proper data processing approaches for extracting reliable information from biochemometric datasets.159 In all selectivity ratio studies applied to identify bioactive natural products,146,159,160 bioactive mixture constituents were identified early in the fractionation process, enabling chromatographic isolation efforts to be tailored to mixture constituents that were most likely to possess bioactivity.
These numerous examples illustrate the efficacy of biochemometrics for distinguishing between active and inactive chemical entities in complex mixtures. However, these approaches do not provide structural information about putative unknown active constituents, hindering the ability to truly optimize isolation efforts. To address this gap, a recent study utilized a combination of selectivity ratio analysis and GNPS molecular networking to identify putative active constituents from the botanical medicine Angelica keiskei (Miq.) Koidz. (Apiaceae) and the molecular families to which they belonged.160 Using this approach, a subset of chalcone analogs were targeted for isolation, yielding two known antimicrobial constituents and an additional, low-abundance compound not previously known to possess antimicrobial activity.160 This concept was streamlined into a process called “bioactive molecular networking,” in which bioactivity predictions are directly visualized in molecular networks themselves, where the size of individual nodes correspond to the predicted bioactivity score for each ion (Fig. 6).148 By including both MS/MS fragmentation data and peak area data in the production of molecular networks, bioactive molecular networking enables dereplication, compound annotation, and identification of putative active compounds in one step.148
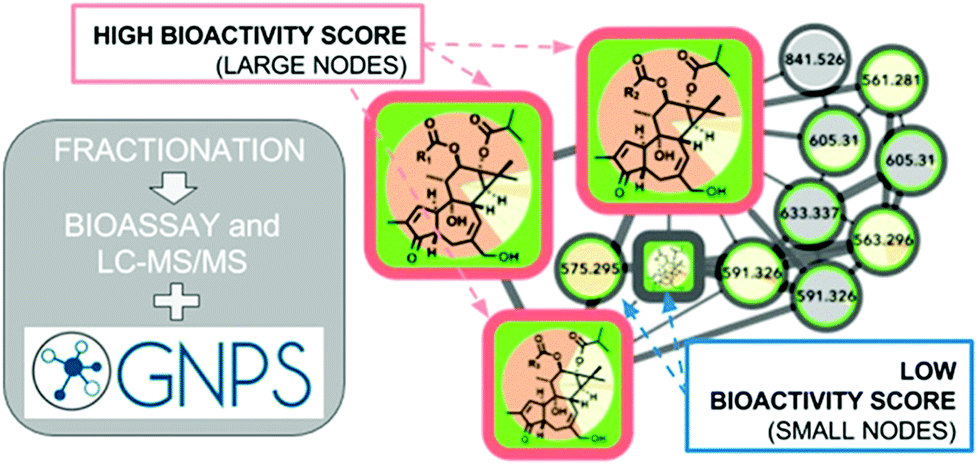 | ||
| Fig. 6 Bioactive molecular networking in which nodes connected in a network represent structurally related compounds based on MS/MS fragmentation patterns, and the size of nodes represents the correlation of compound peak areas with biological activity of interest. Figure is reprinted with permission from Nothias et al. 2018.148 | ||
An additional approach, Compound Activity Mapping, was developed by the Linington laboratory that utilizes image-based cytological screening data and high-resolution mass spectrometry-based metabolomics data to predict both the identities and biological functions of putative bioactive constituents early in the fractionation workflow.147 Using Compound Activity Mapping, biological and chemical datasets are integrated to identify putative bioactive constituents that show consistent positive correlation with phenotypes of interest (Fig. 7).147 The data are presented as a network display, enabling identification and prioritization of lead compounds, even those of low abundance, that likely contribute to a specific biological activity. The utility of this approach was demonstrated through the investigation of 234 extracts of actinobacterial origin.147 Using Compound Activity Mapping, biological and chemical datasets from these samples were combined to identify 13 clusters of bioactive fractions containing 11 known molecular families and four new compounds. Subsequent isolation efforts targeted towards these new compounds revealed the presence of a new natural product family, the quinocinnolinomycins, which were predicted to elicit a cytotoxic response through the induction of endoplasmic reticulum stress.147
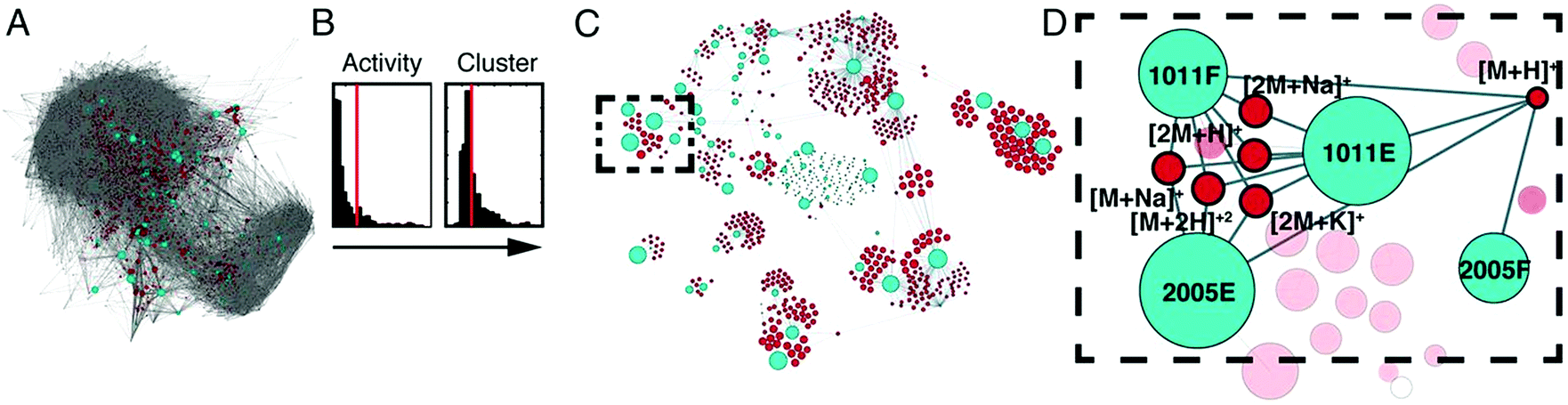 | ||
| Fig. 7 Compound activity mapping workflow. (A) Network analysis of the full chemical space of the tested actinobacterial extracts. Light blue nodes represent extracts connected to all m/z features (red), illustrating the immense chemical complexity of the extract library. (B) Activity histograms and cluster scores for all m/z features. (C) Compound activity map, displaying only extracts and m/z features predicted to be responsible for consistent phenotypes of interest. (D) Close up of a specific bioactive cluster, belonging to the staurosporine natural product family. This figure is reprinted with permission from Kurita et al. 2015.147 | ||
To identify synergists and additives in complex botanical mixtures, Britton et al. recently combined biochemometric analysis with synergy-directed fractionation to identify mixture components from Hydrastis canadensis that enhanced the antimicrobial efficacy of berberine through additive or synergistic mechanisms.22 In this study, mass-spectrometry datasets were combined with biological assay data to produce selectivity ratio plots predicting putative additives and synergists (Fig. 8). In these plots, negative selectivity ratios are indicative of biological activity, because growth inhibition data (where smaller values indicate biological activity) were used to guide the models. Unlike other biochemometric studies of its kind,146,160 the biological activity data used in this study did not measure of antimicrobial activity, per se, but was rather a measure of each sample's ability to improve the antimicrobial efficacy of berberine. Using this approach, six flavonoids not previously identified using synergy-directed fractionation approaches alone17 were identified as putative additives or synergists. Of these, one compound, predicted by selectivity ratio models to be the top contributor to activity, was isolated and characterized for the first time and its activity as a synergist confirmed. Notably, this compound possessed no antimicrobial activity on its own and may have been missed using biochemometric analyses guided by antimicrobial data alone.22
 | ||
| Fig. 8 Selectivity ratio plots for first, second, and third stages of fractionation [(A–C), respectively] of the botanical Hydrastis canadensis. Growth inhibition data were used to guide selectivity ratio analysis, so variables with negative selectivity ratio are most likely to possess additive or synergistic activity. Known flavonoids (likely to be synergists) are marked in green, while known alkaloids (likely to be additives) are marked in red. First-stage (A) and second-stage (B) models were not able to identify known compounds as contributing to activity. However, the third-stage model (C) predicted seven flavonoids (1, 2, 3, 5, 6, 8, 29) and three alkaloids (10, 22, 23) to possess additive or synergistic activity. With this approach, a new synergistic flavonoid (29) was identified in H. canadensis, and known flavonoids and alkaloids not previously known to possess additive or synergistic activity were prioritized for future studies. This figure is reprinted with permission from Britton et al. 2017.22 | ||
6. Elucidating mechanisms that underlie synergy and antagonism
In addition to identifying putative active constituents contributing to biological effects of complex mixtures and recognizing the type of interactions in which they are involved, it is important to understand the cellular and molecular mechanisms by which complex mixtures exert their effects. To ascertain the molecular targets of mixtures, direct and indirect approaches can be taken.166 The direct approach utilizes targeted biological assays to identify molecules that affect specific molecular targets while indirect approaches aim to identify mechanisms of action through the evaluation of changes in gene, protein, and/or metabolite profiles in an untargeted manner.166 While these technologies show great promise, their effectiveness for identifying mechanisms of synergy and antagonism remains to determined.6.1 Targeted assays evaluating specific mechanisms of action (direct approaches)
Targeted approaches to identify mechanisms of action rely on appropriate in vitro and in vivo models. One important example involves identifying compounds that synergize with existing antibiotics through the inhibition of bacterial efflux pumps.18,141,167 A popular method for evaluating efflux pump inhibition involves the use of an efflux pump substrate (such as ethidium bromide or Nile Red) that fluoresces upon contact with cellular DNA.167–169 When efflux pumps are inhibited, fluorescence of the substrate increases due to increased cellular accumulation. This approach has been successfully utilized in numerous studies to identify efflux pump inhibitors from complex botanical mixtures.18,141 While often successful, these fluorescence-based methods are subject to false results due to matrix quenching effects, particularly when screening complex natural product mixtures.167 Fluorescence quenching is so common in the biological evaluation of drug candidates that fluorescence quenchers have been tagged as one type of “PAINS” (pan-assay interference compounds).170,171 However, the ability to absorb UV-vis light (and quench fluorescence) is a common feature of druggable small molecules (for example, tetracycline antibiotics) and only constitutes a problem with fluorescence assays. To overcome this limitation, mass spectrometric assays have been developed to monitor efflux pump inhibition or cellular accumulation in Staphylococcus aureus,167Escherichia coli,172,173Bacillus subtilis,172 and Mycobacterium smegmatis.172 These assays also offer the distinct advantage of being able to monitor drug accumulation of molecules that do not fluoresce.Efflux pump inhibition assays, like many other assays used in classical drug discovery approaches, test compounds or mixtures one at a time to identify compounds with promising biological activity. To improve efficiency of these methods, mixtures of drugs can be simultaneously evaluated, but identifying which molecules in these mixtures exert biological effects can be challenging.174 To overcome this limitation, pulsed ultrafiltration mass spectrometry (PUF-MS) was developed, which enables screening of mixtures such as natural products and synthetic combinatorial libraries.174 PUF-MS involves the incubation of small molecule mixtures with a target protein in solution. Those molecules with affinity for the target will bind to the protein, and compounds that are not bound can be washed away using an ultrafiltration membrane.174 This approach, though effective, is slowed by the ultrafiltration step. To improve the speed of screening, a Magnetic Microbead Affinity Selection Screening (MagMASS) protocol was developed, in which the protein target of interest is not free in solution, but rather is bound to magnetic beads.175 To separate compounds with and without affinity for the given target, the receptor-bound fraction can be held in solution using a magnet.175 In a recent study, PUF-MS and MagMASS were compared, and both screening methods were found to reliably identify ligands of a specific molecular target from complex botanical matrices.175 MagMASS showed a 6-fold faster separation of bound and unbound compounds when compared to PUF-MS and is compatible with a 96-well plate format.175 Notably, these methods do not require molecules to bind to a particular active site on the target of interest, and can identify ligands that bind to active or allosteric sites. In this way, the assay could be modified to identify combination effects in which the protein's activity is changed through allosteric activation or inhibition.175 However, given that this approach utilizes protein targets rather than whole cells, the combination effects discovered may not translate to intact biological systems. Additionally, promiscuous inhibitors may cause false positive results using this method, requiring orthogonal approaches to confirm validity of results. Furthermore, these approaches require access to purified material of the protein target of interest. Therefore, while these assays provide target-specific information that can generate valuable hypotheses on mechanisms of action, methods based on PUF-MS and its iterations are not applicable for situations where the target of the active compound is either not known or not available.
6.2 Indirect approaches to identify multiple targets
While targeted approaches may be useful for identifying compounds that act upon specific molecular targets, assays involving single targets only are not capable of identifying combination effects that involve multiple targets. To identify these multi-target effects, whether it be for a single compound or a combination of multiple constituents, indirect approaches are particularly useful. As discussed in recent review articles, synergistic drug combinations and their modes of action have been explored using molecular interaction profiles,76,112 and investigation of herbal ingredients using molecular interaction profiles may enable detection of synergistic mechanisms of action. As stated in the 2009 review by Ma et al.,76 over 1800 active ingredients from more than 1200 herbs had been subjected to molecular interaction profiling and found to interact with nearly 1000 proteins, many of which were therapeutic targets.76 Although these connections can be utilized to detect potential synergies, the efficacy of complex natural product mixtures and their impact on molecular targets can be influenced by variations in genetics, environment, host behaviour, and timing and dosage of treatment.76 These tools should, therefore, be considered hypothesis-generating, providing a framework for more comprehensive assessment. Lewis et al.176 recently produced a new visualization technique termed “Synergy Maps” which integrates Bliss independence-based combination data of individual compound combinations with their chemical properties into a single visualization. By identifying relationships between individual compound properties and their combination effects, insight into mechanism of action may be provided. Importantly, this tool is only applicable when individual constituents acting in combination are known.176The use of DNA and RNA microarrays is another popular tool for probing combination effects within complex mixtures, enabling identification of genes that are up- or down-regulated by natural product extracts alone and in combination. In a recent study, an RNA microarray of neuroglia cells was utilized to compare the number of genes impacted by treatment with Andrographis paniculata (Burm.f.) Nees (Acanthaceae), Eleutherococcus senticosus (Rupr. & Maxim.) Maxim. (Araliaceae), and their fixed combination Kan Jang.177 Results illustrated that A. paniculata and E. senticosus deregulated 211 and 207 genes, respectively, 36 of which were common to cells treated with each extract. Using this information, researchers expected that 382 genes would be deregulated in cells treated with the fixed combination Kan Jang. However, only 250 genes were deregulated in Kan Jang treated cells, 111 of which were unique to the Kan Jang combination, potentially due to synergistic interactions between A. paniculata and E. senticosus. Alternatively, 170 genes were only affected by treatments with A. paniculata or E. senticosus and not by the Kan Jang mixture, possibly due to antagonistic interactions between the plant species when applied in combination.177 Importantly, microarray analyses do not provide infallible evidence that genes induced by treatments are responsible for physiological effects or mechanisms of synergy, but provide a framework for future research.
Because of the material- and time-consuming nature of biological testing, in silico approaches have been developed that enable prediction of activity and mechanism of action without the need for direct biological testing. Existing experimental activity data can be used to mine ligand–target relationships and reveal potential biological activities of diverse molecules.178 Key to the success of this approach for identifying putative mechanisms of action is the availability of compound databases that will facilitate sharing of data and innovation in drug discovery research with both single-target and multi-target approaches.178 Similarly, computational approaches including molecular docking, pharmacophore modelling, and similarity searching can be used as so-called “virtual screening” techniques to identify candidate compounds for follow-up testing.178 Of course, these techniques are subject to error and may not provide accurate representation of the biological system in question, particularly if the model datasets are based on incorrect literature-based annotations of compound activities and/or incomplete understanding of molecular processes of disease.
A systems biology-based approach, network pharmacology, predicts the complex interactions between small molecules and proteins in a biological system, and shows potential as a way to evaluate pharmacological effects of natural product mixtures.178 Unlike the classic “silver bullet” approach where single-target mechanisms are identified for single drugs, network pharmacology focuses on multiple constituents with multiple targets. Several studies have successfully utilized network pharmacology to putatively identify active constituents with both known and unknown molecular targets.166,176,178,179 Networks can be built using existing literature data, computationally-derived data, or experimental data. The predictive accuracy of the resulting networks relies on the completeness of databases, the robustness of the computational models, the understanding of the underlying mechanisms of disease, and/or the chosen biological assay.178
Recently, a broad-scale approach was developed in which functional signature ontology (FUSION) maps are utilized to classify putative mechanisms of action of natural products.180 With this method, cellular responses to natural product treatment can be tracked by measuring gene expression of a small, representative subset of genes that provide insight into the physiological state of the cell. The resulting data can then be combined into FUSION maps capable of linking putative bioactive molecules to the proteins and biological pathways that they target in cells.180 This approach has been successfully utilized to link natural products to their mechanisms of action180 and to identify a marine-derived natural product that inhibits AMPK kinase activity in colon tumor cells.181
A similar approach, the Connectivity Map, or CMap, was developed in which genes, drugs, and disease states are connected based on the gene expression fingerprints that they share.182 Originally produced using 164 drugs and mRNA expression profiling, the CMap has since been expanded more than 1000-fold, and now contains over 1.3 million publicly available profiles. This scale-up was achieved using a high-throughput, reduced representation in which only 1000 landmarks are assessed rather than the full transcriptome. This approach, termed L1000, is sufficient to recover 81% of the information contained in the full transcriptome. The L1000 approach offers advantages over popular approaches such as gene expression microarrays and RNA sequencing because of its low cost and hybridization-based nature, making detection of low-abundant transcripts possible without the need for deep sequencing.182 Preliminary testing has illustrated the potential of the expanded L1000 CMap to determine the mechanisms of action of small molecules based on the similarities of their genetic perturbations to those of compounds with known activities. This approach can also be utilized to identify potential off-target effects of a drug or drug combinations.182
During a pilot study, the L1000 CMap was successfully utilized to recover known mechanisms of action from 63% of existing drugs under analysis, to identify the mechanism of action of a previously uncharacterized compound, and to identify compounds with a particular activity of interest. Importantly, the L1000 CMap is not infallible, and 37% of small molecules with known mechanisms of action were not linked to their expected targets during this study. The authors suggest six reasons for this failure: (1) incomplete inhibition of the target by the compound, (2) off target effects, (3) incomplete information in the L1000 data, (4) incorrect data in the literature, (5) biological differences between complete loss of function and loss of a specific protein function, and (6) existence of previously unrecognized connections with stronger connections than expected ones.182 Despite these limitations, the preliminary results of this study emphasize the potential of the L1000 CMap as a launching point for both target- and ligand-based drug discovery.182 Although they have not been explicitly applied to identify mechanisms of synergy or antagonism, the utilization of FUSION maps and the L1000 CMap platform may represent useful tools to enable identification of genes and pathways impacted by a synergistic/antagonistic combination, providing insight into potential mechanisms of action in complex natural product mixtures.
7. Conclusions and future directions
In recent years, the concept of synergy in natural product mixtures has gained attention, and the importance of multi-target combination therapies has come to the forefront. However, the classification of combination effects within complex mixtures and the identification of contributing constituents remains a challenging task, particularly when the majority of established tools have been designed to reduce complexity of natural product mixtures. Additionally, there remains a lack of consensus in the field about which reference models are best for defining combination effects, making interpretation of studies challenging. Recent models using the explicit mean equation55 and the zero interaction potency model47 represent newly developed and robust reference models that may permit improved identification combination effects. These models have yet to be employed for real world applications in studying natural product mixtures, and future studies will reveal their applicability for this approach.Metabolomics and biochemometric approaches are promising tools for studying synergy, and have just begun to be applied to identifying constituents that participate in combination effects.22 While useful, biochemometric models are subject to limitations based on the biological assays and reference models used to define biological activity. Similarly, the linear regression models used to predict active constituents are inherently limited given that true linear relationships rarely exist, particularly when assessing complex mixtures with numerous unknown combination effects. The application of statistical tools capable of identifying non-linear relationships will be helpful for future research initiatives. In addition, untargeted approaches to identify molecular targets of synergy and unravel synergistic (or antagonistic) mechanisms of action have just begun to be explored, and continued studies on this topic are of the utmost importance. Advancements in Big Data approaches show great promise for identifying active mixture constituents, characterizing the nature of their interactions, and elucidating their potential mechanisms of action. Integrated technologies capable of completing all of these tasks simultaneously remain to be developed. The production of such integrated techniques will become increasingly important in our continued pursuit to understand the biological activities of complex mixtures.
8. Conflicts of interest
There are no conflicts to declare.9. Acknowledgements
This research was supported by the National Center for Complementary and Integrative Health of the National Institutes of Health under grant numbers 5 T32 AT008938 and 1 R15 AT010191. Ashley Scott is also acknowledged for her help preparing figures for this manuscript.10. References
- B. B. Petrovska, Pharmacogn. Rev., 2012, 6, 1–5 CrossRef PubMed.
- K. Kelly, The history of medicine, Facts on file, 2009 Search PubMed.
- W. M. Bandaranayake, in Modern Phytomedicine: Turning Medicinal Plants into Drugs, ed. I. Ahmad, F. Aqil and M. Owais, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006, pp. 25–57 Search PubMed.
- C.-K. Ong, G. Bodeker, C. Grundy and K. Shein, WHO Global Atlas of Traditional, Complementary and Alternative Medicine, World Health Organization, Kobe, Japan, 2005 Search PubMed.
- M. Ekor, Front. Pharmacol., 2014, 4, 177 CrossRef PubMed.
- L. I. Black, T. C. Clarke, P. M. Barnes, B. J. Stussman and R. L. Nahin, Natl. Health Stat. Report, 2015, 1 Search PubMed.
- T. Efferth and E. Koch, Curr. Drug Targets, 2011, 12, 122–132 CrossRef CAS PubMed.
- S. van Vuuren and A. Viljoen, Planta Med., 2011, 77, 1168–1182 CrossRef CAS PubMed.
- H. Wagner and G. Ulrich-Merzenich, Phytomedicine, 2009, 16, 97–110 CrossRef CAS PubMed.
- T. Burfield and S.-L. Reekie, Int. J. Aromather., 2005, 15, 30–41 CrossRef CAS.
- I. Raskin and C. Ripoll, Curr. Pharm. Des., 2004, 10, 3419–3429 CrossRef CAS PubMed.
- T. Pemovska, J. W. Bigenzahn and G. Superti-Furga, Curr. Opin. Pharmacol., 2018, 42, 102–110 CrossRef CAS PubMed.
- H. Wagner, Pure Appl. Chem., 2005, 77, 1–6 CAS.
- H. Wagner, Fitoterapia, 2011, 82, 34–37 CrossRef PubMed.
- Y. Liu, Q. Wei, G. Yu, W. Gai, Y. Li and X. Chen, Database, Oxford, 2014, vol. 2014, p. bau124 Search PubMed.
- C. G. Enke and L. J. Nagels, Anal. Chem., 2011, 83, 2539–2546 CrossRef CAS PubMed.
- H. A. Junio, A. A. Sy-Cordero, K. A. Ettefagh, J. T. Burns, K. T. Micko, T. N. Graf, S. J. Richter, R. E. Cannon, N. H. Oberlies and N. B. Cech, J. Nat. Prod., 2011, 74, 1621–1629 CrossRef CAS PubMed.
- F. R. Stermitz, P. Lorenz, J. N. Tawara, L. A. Zenewicz and K. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1433–1437 CrossRef CAS PubMed.
- F. R. Stermitz, L. N. Scriven, G. Tegos and K. Lewis, Planta Med., 2002, 68, 1140–1141 CrossRef CAS PubMed.
- G. Ulrich-Merzenich, D. Panek, H. Zeitler, H. Vetter and H. Wagner, Indian J. Exp. Biol., 2010, 48, 208–219 CAS.
- F. Qiu, G. Cai, B. U. Jaki, D. C. Lankin, S. G. Franzblau and G. F. Pauli, J. Nat. Prod., 2013, 76, 413–419 CrossRef CAS PubMed.
- E. R. Britton, J. J. Kellogg, O. M. Kvalheim and N. B. Cech, J. Nat. Prod., 2017, 81, 484–493 CrossRef PubMed.
- B. Bunterngsook, L. Eurwilaichitr, A. Thamchaipenet and V. Champreda, Bioresour. Technol., 2015, 176, 129–135 CrossRef CAS PubMed.
- G. Chevereau and T. Bollenbach, Mol. Syst. Biol., 2015, 11, 807 CrossRef PubMed.
- J. J. Piggott, C. R. Townsend and C. D. Matthaei, Ecol. Evol., 2015, 5, 1538–1547 CrossRef PubMed.
- J. Tang, K. Wennerberg and T. Aittokallio, Front. Pharmacol., 2015, 6, 181 CrossRef PubMed.
- M. C. Berenbaum, Pharmacol. Rev., 1989, 41, 93–141 CAS.
- M. A. Rather, B. A. Bhat and M. A. Qurishi, Phytomedicine, 2013, 21, 1–14 CrossRef PubMed.
- E. M. Williamson, Phytomedicine, 2001, 8, 401–409 CrossRef CAS PubMed.
- J. L. Medina-Franco, M. A. Giulianotti, G. S. Welmaker and R. A. Houghten, Drug Discovery Today, 2013, 18, 495–501 CrossRef PubMed.
- A. C. Palmer and P. K. Sorger, Cell, 2017, 171, 1678–1691 CrossRef CAS PubMed.
- G. R. Zimmermann, J. Lehar and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS PubMed.
- A. A. Borisy, P. J. Elliott, N. W. Hurst, M. S. Lee, J. Lehár, E. R. Price, G. Serbedzija, G. R. Zimmermann, M. A. Foley and B. R. Stockwell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7977–7982 CrossRef CAS PubMed.
- J. Vande Voorde, T. Ackermann, N. Pfetzer, D. Sumpton, G. Mackay, G. Kalna, C. Nixon, K. Blyth, E. Gottlieb and S. Tardito, Sci. Adv., 2019, 5, eaau7314 CrossRef PubMed.
- J. Bisson, J. B. McAlpine, J. B. Friesen, S.-N. Chen, J. Graham and G. F. Pauli, J. Med. Chem., 2016, 59, 1671–1690 CrossRef CAS PubMed.
- B. Y. Feng and B. K. Shoichet, J. Med. Chem., 2006, 49, 2151–2154 CrossRef CAS PubMed.
- M. R. Senger, C. A. Fraga, R. F. Dantas and F. P. Silva Jr, Drug Discovery Today, 2016, 21, 868–872 CrossRef CAS PubMed.
- C. R. Bonapace, R. L. White, L. V. Friedrich and J. A. Bosso, Diagn. Microbiol. Infect. Dis., 2000, 38, 43–50 CrossRef CAS.
- R. Lewis, D. Diekema, S. Messer, M. Pfaller and M. Klepser, J. Antimicrob. Chemother., 2002, 49, 345–351 CrossRef CAS PubMed.
- R. L. White, D. S. Burgess, M. Manduru and J. A. Bosso, Antimicrob. Agents Chemother., 1996, 40, 1914–1918 CrossRef CAS PubMed.
- M. Wu and R. F. Woolson, Commun. Stat. Simulat. Comput., 1998, 27, 303–327 CrossRef.
- A. N. Shikov, O. N. Pozharitskaya and V. G. Makarov, Synergy, 2018, 7, 36–38 CrossRef.
- A. Ocana, E. Amir, C. Yeung, B. Seruga and I. F. Tannock, Ann. Oncol., 2012, 23, 2161–2166 CrossRef CAS PubMed.
- V. H. Tam, A. N. Schilling and M. Nikolaou, J. Antimicrob. Chemother., 2005, 55, 699–706 CrossRef CAS PubMed.
- J. An, G. Zuo, X. Hao, G. Wang and Z. Li, Phytomedicine, 2011, 18, 990–993 CrossRef CAS PubMed.
- S. Lederer, T. M. Dijkstra and T. Heskes, bioRxiv, 2018, 480608 Search PubMed.
- B. Yadav, K. Wennerberg, T. Aittokallio and J. Tang, Comput. Struct. Biotechnol. J., 2015, 13, 504–513 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- W. R. Greco, G. Bravo and J. C. Parsons, Pharmacol. Rev., 1995, 47, 331–385 CAS.
- S.-i. Lee, Korean J. Anesthesiol., 2010, 58, 421–434 CrossRef CAS PubMed.
- L. Zhao, J. L.-S. Au and M. G. Wientjes, Front. Biosci., Elite Ed., 2010, 2, 241 Search PubMed.
- C. I. Bliss, Ann. Appl. Biol., 1939, 26, 585–615 CrossRef CAS.
- S. Loewe, Arzneimittelforschung, 1953, 3, 285–290 CAS.
- N. Geary, Am. J. Physiol.: Endocrinol. Metab., 2012, 304, E237–E253 CrossRef PubMed.
- S. Lederer, T. M. Dijkstra and T. Heskes, Front. Pharmacol., 2018, 9, 31 CrossRef PubMed.
- R. J. Tallarida, J. Pharmacol. Exp. Ther., 2006, 319, 1–7 CrossRef CAS PubMed.
- M. Cokol, H. N. Chua, M. Tasan, B. Mutlu, Z. B. Weinstein, Y. Suzuki, M. E. Nergiz, M. Costanzo, A. Baryshnikova and G. Giaever, Mol. Syst. Biol., 2011, 7, 544 CrossRef PubMed.
- L. A. Mathews Griner, R. Guha, P. Shinn, R. M. Young, J. M. Keller, D. Liu, I. S. Goldlust, A. Yasgar, C. McKnight and M. B. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2349–2354 CrossRef CAS PubMed.
- T.-C. Chou, Cancer Res., 2010, 72, 440–446 CrossRef PubMed.
- J. B. Fitzgerald, B. Schoeberl, U. B. Nielsen and P. K. Sorger, Nat. Chem. Biol., 2006, 2, 458 CrossRef CAS PubMed.
- M. Kong and J. J. Lee, Biometrics, 2006, 62, 986–995 CrossRef PubMed.
- J. J. Lee, M. Kong, G. D. Ayers and R. Lotan, J. Biopharm. Stat., 2007, 17, 461–480 CrossRef CAS PubMed.
- W. Zhao, K. Sachsenmeier, L. Zhang, E. Sult, R. E. Hollingsworth and H. Yang, J. Biomol. Screening, 2014, 19, 817–821 CrossRef PubMed.
- W. Greco, H. Unkelbach, G. Pöch, J. Sühnel, M. Kundi and W. Bödeker, Arch. Complex Environ. Stud., 1992, 4, 65–69 Search PubMed.
- D. Russ and R. Kishony, Nat. Microbiol., 2018, 3, 1339 CrossRef CAS PubMed.
- K. Chandrasekaran, Z. Mehrabian, B. Spinnewyn, C. Chinopoulos, K. Drieu and G. Fiskum, Cell. Mol. Biol., 2002, 48, 663–669 CAS.
- T.-C. Chou and P. Talalay, Trends Pharmacol. Sci., 1983, 4, 450–454 CrossRef CAS.
- F. C. Odds, J. Antimicrob. Chemother., 2003, 52, 1 CrossRef CAS PubMed.
- G. Y. Di Veroli, C. Fornari, D. Wang, S. Mollard, J. L. Bramhall, F. M. Richards and D. I. Jodrell, Bioinformatics, 2016, 32, 2866–2868 CrossRef CAS PubMed.
- B. Gilbert and L. Alves, Curr. Med. Chem., 2003, 10, 13–20 CrossRef CAS PubMed.
- P. Rasoanaivo, C. W. Wright, M. L. Willcox and B. Gilbert, Malar. J., 2011, 10(Suppl. 1), S4 CrossRef PubMed.
- B. L. Beyerstein, Acad. Med., 2001, 76, 230–237 CrossRef CAS PubMed.
- T. J. Kaptchuk, Ann. Intern. Med., 2002, 136, 817–825 CrossRef PubMed.
- G. T. Lewith, M. E. Hyland and S. Shaw, Am. J. Public Health, 2002, 92, 1604–1606 CrossRef PubMed.
- F. A. Tausk, Arch. Dermatol., 1998, 134, 1422–1425 CAS.
- X. Ma, C. Zheng, L. Han, B. Xie, J. Jia, Z. Cao, Y. Li and Y. Chen, Drug Discovery Today, 2009, 14, 579–588 CrossRef CAS PubMed.
- T. Efferth, S. Zacchino, M. I. Georgiev, L. Liu, H. Wagner and A. Panossian, Phytomedicine, 2015, 22, 1–4 CrossRef PubMed.
- Y. Y. Tu, M. Y. Ni, Y. Zhong and L. N. Li, Zhongguo Zhongyao Zazhi, 1981, 6, 31–32 CAS.
- N. J. White, Science, 2008, 320, 330–334 CrossRef CAS PubMed.
- World Health Organization, WHO Guidelines for the Safe Use of Wastewater, Excreta and Greywater, World Health Organization, Geneva, Switzerland, 2006 Search PubMed.
- K. I. Barnes, D. N. Durrheim, F. Little, A. Jackson, U. Mehta, E. Allen, S. S. Dlamini, J. Tsoka, B. Bredenkamp and D. J. Mthembu, PLoS Med., 2005, 2, e330 CrossRef PubMed.
- A. Bhattarai, A. S. Ali, S. P. Kachur, A. Mårtensson, A. K. Abbas, R. Khatib, A.-w. Al-Mafazy, M. Ramsan, G. Rotllant and J. F. Gerstenmaier, PLoS Med., 2007, 4, e309 CrossRef PubMed.
- V. I. Carrara, S. Sirilak, J. Thonglairuam, C. Rojanawatsirivet, S. Proux, V. Gilbos, A. Brockman, E. A. Ashley, R. McGready and S. Krudsood, PLoS Med., 2006, 3, e183 CrossRef PubMed.
- C. W. Jefford, Curr. Med. Chem., 2001, 8, 1803–1826 CrossRef CAS PubMed.
- A. V. Pandey, B. L. Tekwani, R. L. Singh and V. S. Chauhan, J. Biol. Chem., 1999, 274, 19383–19388 CrossRef CAS PubMed.
- M. Wilcox, G. Bodeker, G. Bourdy, V. Dhingra, J. Falquet, J. F. S. Ferreira, B. Graz, H. Hans-Martin, E. Hsu, P. Melillo de Magalhaes, D. Provendier and C. W. Wright, in Traditional Medicinal Plants and Malaria, ed. M. Willcox, G. Bodeker and P. Rosaoanaivo, CRC Press, Boca Raton, FL, 2004, pp. 43–60 Search PubMed.
- F. H. Jansen, Trans. R. Soc. Trop. Med. Hyg., 2006, 100, 285–286 CrossRef CAS.
- K. Räth, K. Taxis, G. Walz, C. H. Gleiter, S.-M. Li and L. Heide, Am. J. Trop. Med. Hyg., 2004, 70, 128–132 CrossRef.
- R. K. Haynes, Curr. Top. Med. Chem., 2006, 6, 509–537 CrossRef CAS PubMed.
- P. J. Weina, Parassitologia, 2008, 50, 25 CAS.
- J. O. Suberu, A. P. Gorka, L. Jacobs, P. D. Roepe, N. Sullivan, G. C. Barker and A. A. Lapkin, PLoS One, 2013, 8, e80790 CrossRef PubMed.
- B. C. Elford, M. F. Roberts, J. D. Phillipson and R. J. Wilson, Trans. R. Soc. Trop. Med. Hyg., 1987, 81, 434–436 CrossRef CAS PubMed.
- K. C. Liu, S.-L. Yang, M. Roberts, B. Elford and J. Phillipson, Plant Cell Rep., 1992, 11, 637–640 CrossRef CAS PubMed.
- A. Nemeikaitė-Čėnienė, A. Imbrasaitė, E. Sergedienė and N. Čėnas, Arch. Biochem. Biophys., 2005, 441, 182–190 CrossRef PubMed.
- E. G. Yordi, E. M. Pérez, M. J. Matos and E. U. Villares, in Nutrition, Well-being, and Health, ed. J. B. Bohn, InTech, Rijeka, Croatia, 2012, pp. 24–48 Search PubMed.
- B. Barrett, R. Brown, D. Rakel, M. Mundt, K. Bone, S. Barlow and T. Ewers, Ann. Intern. Med., 2010, 153, 769–777 CrossRef.
- M. Jawad, R. Schoop, A. Suter, P. Klein and R. Eccles, J. Evidence-Based Complementary Altern. Med., 2012, 2012, 841315 CAS.
- T. Smith, M. Lynch, J. Johnson, K. Kawa, H. Bauman and M. Blumenthal, HerbalGram, 2015, 107, 52–59 Search PubMed.
- M. Rotblatt, Ann. Intern. Med., 2000, 133, 487 CrossRef.
- A. Proksch and H. Wagner, Phytochemistry, 1987, 26, 1989–1993 CrossRef CAS.
- N. D. Pugh, H. Tamta, P. Balachandran, X. Wu, J. L. Howell, F. E. Dayan and D. S. Pasco, Int. Immunopharmacol., 2008, 8, 1023–1032 CrossRef CAS PubMed.
- H. Tamta, N. D. Pugh, P. Balachandran, R. Moraes, J. Sumiyanto and D. S. Pasco, J. Agric. Food Chem., 2008, 56, 10552–10556 CrossRef CAS PubMed.
- M. H. Haron, H. L. Tyler, N. D. Pugh, R. M. Moraes, V. L. Maddox, C. R. Jackson and D. S. Pasco, Planta Med., 2016, 82, 1258 CrossRef CAS PubMed.
- D. A. Todd, T. V. Gulledge, E. R. Britton, M. Oberhofer, M. Leyte-Lugo, A. N. Moody, T. Shymanovich, L. F. Grubbs, M. Juzumaite and T. N. Graf, PLoS One, 2015, 10, e0124276 CrossRef.
- N. B. Cech, V. Kandhi, J. M. Davis, A. Hamilton, D. Eads and S. M. Laster, Int. Immunopharmacol., 2010, 10, 1268–1278 CrossRef CAS.
- J. Gertsch, R. Schoop, U. Kuenzle and A. Suter, FEBS Lett., 2004, 577, 563–569 CrossRef CAS.
- S. Raduner, A. Majewska, J.-Z. Chen, X.-Q. Xie, J. Hamon, B. Faller, K.-H. Altmann and J. Gertsch, J. Biol. Chem., 2006, 281, 14192–14206 CrossRef CAS.
- L. Stevenson, A. Matthias, L. Banbury, K. Penman, K. Bone, D. Leach and R. Lehmann, Molecules, 2005, 10, 1279–1285 CrossRef CAS.
- X. Zhou, S. W. Seto, D. Chang, H. Kiat, V. Razmovski-Naumovski, K. Chan and A. Bensoussan, Front. Pharmacol., 2016, 7, 201 Search PubMed.
- D. Wing-Shing Cheung, C. M. Koon, C. F. Ng, P. C. Leung, K. P. Fung, S. Kar-Sing Poon and C. Bik-San Lau, J. Ethnopharmacol., 2012, 143, 859–866 CrossRef PubMed.
- S. Hemaiswarya, A. K. Kruthiventi and M. Doble, Phytomedicine, 2008, 15, 639–652 CrossRef CAS PubMed.
- J. Jia, F. Zhu, X. Ma, Z. W. Cao, Y. X. Li and Y. Z. Chen, Nat. Rev. Drug Discovery, 2009, 8, 111 CrossRef CAS PubMed.
- X. Gong and N. J. Sucher, Trends Pharmacol. Sci., 1999, 20, 191–196 CrossRef CAS PubMed.
- B. D. Brooks and A. E. Brooks, Adv. Drug Delivery Rev., 2014, 78, 14–27 CrossRef CAS PubMed.
- C.-M. J. Hu and L. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS PubMed.
- F. Carmona and A. M. S. Pereira, Rev. Bras. Farmacogn., 2013, 23, 379–385 CrossRef.
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206 CrossRef CAS PubMed.
- M. A. Lila and I. Raskin, J. Food Sci., 2005, 70, R20–R27 CrossRef CAS.
- P. Imming, C. Sinning and A. Meyer, Nat. Rev. Drug Discovery, 2006, 5, 821 CrossRef CAS.
- P. Montes, E. Ruiz-Sanchez, C. Rojas and P. Rojas, CNS Neurol. Disord.: Drug Targets, 2015, 14, 132–149 CrossRef CAS.
- M. Spinella, Altern. Med. Rev., 2002, 7, 130–137 Search PubMed.
- V. Butterweck, U. Liefländer-Wulf, H. Winterhoff and A. Nahrstedt, Planta Med., 2003, 69, 189–192 CrossRef CAS.
- Y. H. Choi, J. van Spronsen, Y. Dai, M. Verberne, F. Hollmann, I. W. C. E. Arends, G.-J. Witkamp and R. Verpoorte, Plant Physiol., 2011, 156, 1701–1705 CrossRef CAS PubMed.
- M. Adams, A. Mahringer, R. Bauer, G. Fricker and T. Efferth, Planta Med., 2007, 73, 1475–1478 CrossRef CAS PubMed.
- M. Adams, A. Mahringer, O. Kunert, G. Fricker, T. Efferth and R. Bauer, Planta Med., 2007, 73, 1554–1557 CrossRef CAS PubMed.
- X.-L. Liang, Z.-G. Liao, J.-Y. Zhu, G.-W. Zhao, M. Yang, R.-L. Yin, Y.-C. Cao, J. Zhang and L.-J. Zhao, J. Ethnopharmacol., 2012, 139, 52–57 CrossRef.
- S. Wang, F. Zhu and M. F. Marcone, J. Med. Food, 2015, 18, 938–940 CrossRef CAS PubMed.
- L. M. McCune and T. Johns, J. Ethnopharmacol., 2002, 82, 197–205 CrossRef PubMed.
- R. Müller, J. Cancer Res. Clin. Oncol., 2004, 130, 429–444 CrossRef PubMed.
- W. Kassouf, C. P. Dinney, G. Brown, D. J. McConkey, A. J. Diehl, M. Bar-Eli and L. Adam, Cancer Res., 2005, 65, 10524–10535 CrossRef CAS PubMed.
- N. V. Sergina, M. Rausch, D. Wang, J. Blair, B. Hann, K. M. Shokat and M. M. Moasser, Nature, 2007, 445, 437–441 CrossRef CAS PubMed.
- Q. Hu, W. Sun, C. Wang and Z. Gu, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS.
- D. Sanglard, Front. Med., 2016, 3, 11 Search PubMed.
- L. Strasfeld and S. Chou, Infect. Dis. Clin. N. Am., 2010, 24, 413–437 CrossRef.
- J. O'Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations-The Review on Antimicrobial Resistance, 2016 Search PubMed.
- L. Mundy, B. Pendry and M. Rahman, J. Herb. Med., 2016, 6, 53–58 CrossRef.
- G. A. Jacoby and L. Sutton, Antimicrob. Agents Chemother., 1985, 28, 703–705 CrossRef CAS PubMed.
- L. Catteau, J. Olson, F. Van Bambeke, J. Leclercq and V. Nizet, FASEB J., 2017, 31, 1000.1005 Search PubMed.
- M. E. Falagas and I. A. Bliziotis, Int. J. Antimicrob. Agents, 2007, 29, 630–636 CrossRef CAS PubMed.
- L. J. Piddock, Clin. Microbiol. Rev., 2006, 19, 382–402 CrossRef CAS PubMed.
- K. A. Ettefagh, J. T. Burns, H. A. Junio, G. W. Kaatz and N. B. Cech, Planta Med., 2011, 77, 835 CrossRef CAS PubMed.
- M. Leyte-Lugo, E. R. Britton, D. H. Foil, A. R. Brown, D. A. Todd, J. Rivera-Chávez, N. H. Oberlies and N. B. Cech, Phytochem. Lett., 2017, 20, 54–60 CrossRef CAS PubMed.
- I. A. Khan, Life Sci., 2006, 78, 2033–2038 CrossRef CAS PubMed.
- F. Bucar, A. Wube and M. Schmid, Nat. Prod. Rep., 2013, 30, 525–545 RSC.
- T. Inui, Y. Wang, S. M. Pro, S. G. Franzblau and G. F. Pauli, Fitoterapia, 2012, 83, 1218–1225 CrossRef CAS PubMed.
- J. J. Kellogg, D. A. Todd, J. M. Egan, H. A. Raja, N. H. Oberlies, O. M. Kvalheim and N. B. Cech, J. Nat. Prod., 2016, 79, 376–386 CrossRef CAS PubMed.
- K. L. Kurita, E. Glassey and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11999–12004 CrossRef CAS PubMed.
- L. F. Nothias, M. Nothias-Esposito, R. da Silva, M. Wang, I. Protsyuk, Z. Zhang, A. Sarvepalli, P. Leyssen, D. Touboul, J. Costa, J. Paolini, T. Alexandrov, M. Litaudon and P. C. Dorrestein, J. Nat. Prod., 2018, 81, 758–767 CrossRef CAS PubMed.
- B. M. Dietz, S. N. Chen, R. F. R. Alvarenga, H. Dong, D. Nikolic, M. Biendl, R. B. van Breemen, J. L. Bolton and G. F. Pauli, J. Nat. Prod., 2017, 80, 2284–2294 CrossRef CAS PubMed.
- N. H. Oberlies and D. J. Kroll, J. Nat. Prod., 2004, 67, 129–135 CrossRef CAS PubMed.
- T. El-Elimat, M. Figueroa, B. M. Ehrmann, N. B. Cech, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2013, 76, 1709–1716 CrossRef CAS PubMed.
- S. P. Gaudencio and F. Pereira, Nat. Prod. Rep., 2015, 32, 779–810 RSC.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Crusemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C.A. Boya P., D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrov, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- B. C. Covington, J. A. McLean and B. O. Bachmann, Nat. Prod. Rep., 2017, 34, 6–24 RSC.
- M. T. Henke and N. L. Kelleher, Nat. Prod. Rep., 2016, 33, 942–950 RSC.
- T. Kind and O. Fiehn, Phytochem. Lett., 2017, 21, 313–319 CrossRef CAS PubMed.
- R. B. Williams, M. O'Neil-Johnson, A. J. Williams, P. Wheeler, R. Pol and A. Moser, Org. Biomol. Chem., 2015, 13, 9957–9962 RSC.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS PubMed.
- L. K. Caesar, J. J. Kellogg, O. M. Kvalheim and N. B. Cech, J. Nat. Prod., 2019, 82, 469–484 CrossRef CAS.
- L. K. Caesar, J. J. Kellogg, O. M. Kvalheim, R. A. Cech and N. B. Cech, Planta Med., 2018, 84, 721–728 CrossRef CAS PubMed.
- K. M. Chan, G. G. Yue, P. Li, E. C. Wong, J. K. Lee, E. J. Kennelly and C. B. Lau, J. Chromatogr. A, 2017, 1487, 162–167 CrossRef CAS PubMed.
- P. Li, H. AnandhiSenthilkumar, S. B. Wu, B. Liu, Z. Y. Guo, J. E. Fata, E. J. Kennelly and C. L. Long, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1011, 179–195 CrossRef CAS PubMed.
- H. Abdi, in The SAGE Encyclopedia of Social Science Research Methods, SAGE Publications, Inc., Thousand Oaks, CA, 2004, pp. 792–795 Search PubMed.
- T. Rajalahti, R. Arneberg, A. C. Kroksveen, M. Berle, K.-M. Myhr and O. M. Kvalheim, Anal. Chem., 2009, 81, 2581–2590 CrossRef CAS PubMed.
- T. Rajalahti and O. M. Kvalheim, Int. J. Pharm., 2011, 417, 280–290 CrossRef CAS PubMed.
- S. Chen, H. Jiang, Y. Cao, Y. Wang, Z. Hu, Z. Zhu and Y. Chai, Sci. Rep., 2016, 6, 24245 CrossRef PubMed.
- A. R. Brown, K. A. Ettefagh, D. Todd, P. S. Cole, J. M. Egan, D. H. Foil, T. N. Graf, B. D. Schindler, G. W. Kaatz and N. B. Cech, PLoS One, 2015, 10, e0124814 CrossRef PubMed.
- J. M. A. Blair and L. J. V. Piddock, mBio, 2016, 7, e00840 CrossRef CAS PubMed.
- J. A. Bohnert, B. Karamian and H. Nikaido, Antimicrob. Agents Chemother., 2010, 54, 3770–3775 CrossRef CAS PubMed.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- T. D. Davis, C. J. Gerry and D. S. Tan, ACS Chem. Biol., 2014, 9, 2535–2544 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- R. B. van Breemen, C.-R. Huang, D. Nikolic, C. P. Woodbury, Y.-Z. Zhao and D. L. Venton, Anal. Chem., 1997, 69, 2159–2164 CrossRef CAS.
- M. D. Rush, E. M. Walker, G. Prehna, T. Burton and R. B. van Breemen, J. Am. Soc. Mass Spectrom., 2017, 28, 479–485 CrossRef CAS PubMed.
- R. Lewis, R. Guha, T. Korcsmaros and A. Bender, J. Cheminf., 2015, 7, 36 Search PubMed.
- A. Panossian, E. J. Seo, G. Wikman and T. Efferth, Phytomedicine, 2015, 22, 981–992 CrossRef CAS PubMed.
- Y. Xiong, Y. Hu, L. Cheng, Z. Zhang, Y. Zhang, M. Niu and Z. Cui, Front. Pharmacol., 2019, 9, 1514 CrossRef PubMed.
- J. Liu, J. Liu, F. Shen, Z. Qin, M. Jiang, J. Zhu, Z. Wang, J. Zhou, Y. Fu, X. Chen, C. Huang, W. Xiao, C. Zheng and Y. Wang, Sci. Rep., 2018, 8, 380 CrossRef PubMed.
- M. B. Potts, H. S. Kim, K. W. Fisher, Y. Hu, Y. P. Carrasco, G. B. Bulut, Y.-H. Ou, M. L. Herrera-Herrera, F. Cubillos, S. Mendiratta, G. Xiao, M. Hofree, T. Ideker, Y. Xie, L. J.-s. Huang, R. E. Lewis, J. B. MacMillan and M. A. White, Sci. Signaling, 2013, 6, ra90 CrossRef PubMed.
- B. Das, B. K. Neilsen, K. W. Fisher, D. Gehring, Y. Hu, D. J. Volle, H. S. Kim, J. L. McCall, D. L. Kelly, J. B. MacMillan, M. A. White and R. E. Lewis, Sci. Rep., 2018, 8, 3770 CrossRef PubMed.
- A. Subramanian, R. Narayan, S. M. Corsello, D. D. Peck, T. E. Natoli, X. Lu, J. Gould, J. F. Davis, A. A. Tubelli, J. K. Asiedu, D. L. Lahr, J. E. Hirschman, Z. Liu, M. Donahue, B. Julian, M. Khan, D. Wadden, I. C. Smith, D. Lam, A. Liberzon, C. Toder, M. Bagul, M. Orzechowski, O. M. Enache, F. Piccioni, S. A. Johnson, N. J. Lyons, A. H. Berger, A. F. Shamji, A. N. Brooks, A. Vrcic, C. Flynn, J. Rosains, D. Y. Takeda, R. Hu, D. Davison, J. Lamb, K. Ardlie, L. Hogstrom, P. Greenside, N. S. Gray, P. A. Clemons, S. Silver, X. Wu, W. N. Zhao, W. Read-Button, X. Wu, S. J. Haggarty, L. V. Ronco, J. S. Boehm, S. L. Schreiber, J. G. Doench, J. A. Bittker, D. E. Root, B. Wong and T. R. Golub, Cell, 2017, 171, 1437–1452 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |