
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputer-aided re-engineering of nonribosomal peptide and polyketide biosynthetic assembly lines
Mohammad
Alanjary†
 a,
Carolina
Cano-Prieto†
b,
Harald
Gross
*b and
Marnix H.
Medema
*a
a,
Carolina
Cano-Prieto†
b,
Harald
Gross
*b and
Marnix H.
Medema
*a
aBioinformatics Group, Wageningen University, Wageningen, The Netherlands. E-mail: marnix.medema@wur.nl
bDepartment of Pharmaceutical Biology, Pharmaceutical Institute, Eberhard Karls Universität Tübingen, Tübingen, Germany. E-mail: harald.gross@uni-tuebingen.de
First published on 1st July 2019
Abstract
Covering: 2014 to 2019
Nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) have been the subject of engineering efforts for multiple decades. Their modular assembly line architecture potentially allows unlocking vast chemical space for biosynthesis. However, attempts thus far are often met with mixed success, due to limited molecular compatibility of the parts used for engineering. Now, new engineering strategies, increases in genomic data, and improved computational tools provide more opportunities for major progress. In this review we highlight some of the challenges and progressive strategies for the re-design of NRPSs & type I PKSs and survey useful computational tools and approaches to attain the ultimate goal of semi-automated and design-based engineering of novel peptide and polyketide products.
 Mohammad Alanjary | Dr Mohammad Alanjary obtained his PhD in bioinformatics at the University of Tübingen, Germany, with a focus on antibiotic resistance. Previously he aided in the launch of the first commercial semi-conductor gene-sequencing platform and developed several procedures for performance optimization while working at IonTorrent. He is currently working at Wageningen University to develop computational methods leveraging comparative genomics for natural product discovery and engineering. |
 Carolina Cano-Prieto | Carolina Cano Prieto was born in 1984. She obtained her bachelors degree in Biology at University of Granada (Spain) followed by a MSc in Microbiology under the Spanish Educational Minister fellowship. She completed her PhD studies in 2015 under the supervision of Prof. José Antonio Salas and Dr Carlos Olano at University of Oviedo where her work was focused in identification of the polyketide biosynthesis produced by streptomycetes. Since 2016 she is a post-doctoral researcher in the lab of Prof. Dr Harald Gross at the University of Tübingen. There she currently works on the characterization and modification of NRPS biosynthetic gene clusters in Pseudomonas and Bacillus strains and with the development of biosynthetic gene clusters using synthetic biology tools. |
 Harald Gross | Harald Gross is a pharmacist by training and obtained his PhD degree in 2004 from the University of Bonn, Germany, followed by 2 years of postdoctoral research at the Oregon State University in Corvallis/OR, USA and at the Scripps Institution of Oceanography in La Jolla/CA, USA. Back at the University of Bonn, he established in 2006 his own independent research group where he was mainly working on the genomics and secondary metabolism of Pseudomonas bacteria. Since 2012 he is Full Professor at the University of Tübingen. His research interests are in the field of genome-driven discovery of microbial secondary metabolites, biosynthesis research and genetic engineering. |
 Marnix Medema | Marnix Medema is an Assistant Professor of Bioinformatics at Wageningen University, The Netherlands. In 2013, he completed his PhD in the groups of Eriko Takano and Rainer Breitling, at the University of Groningen. During his PhD he spent time in the lab of Michael Fischbach at the University of California, San Francisco, as a visiting research fellow. Following a postdoctoral fellowship in the group of Frank Oliver Glöckner at the Max Planck Institute for Marine Microbiology in Bremen, Germany, he joined Wageningen University in 2015. His group develops and applies computational tools to understand bacterial, fungal and plant natural product biosynthesis from a genomic perspective. |
1. NRPSs and PKSs: enzymatic factories for compound production
Using biology to produce high-value compounds has been a boon to humanity; cell factories are an integral production method to supply antibiotics and other useful natural products to the world, with a market value estimated at several hundreds of billions of USD per year.1 For example, complex structures such as vancomycin require over 40 steps for total synthesis,2 while production in Amycolatopsis orientalis can be made in continuous batch cultures with relative ease.3 Along with optimization efforts,4,5 cell factories can be a more economic strategy for mass production or for the generation of new leads, especially for the variety of natural products with intricate structures.Non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) can be described as the factory ‘machinery’ for many natural products. Engineering these multi-modular systems has been a “holy grail” for synthetic biology due to their modular nature and the endless combinatorial design possibilities available through module deletions, insertions, duplications or exchanges. Leveraging these systems can thus accelerate discovery of high-value compounds, especially to help refill the waning antimicrobial pipeline.6 However, ever since the earliest attempts in the 1990s, modifying PKSs and NRPSs on demand has turned out to be much more challenging than first expected: due to the complex molecular interactions among modules and domains, their engineering requires deep understanding of protein–protein interactions and domain specificities. For this reason, many engineering successes have led to low yields impractical for industrial production. Now, new breakthroughs are finally providing solutions to leverage these systems, opening up countless new opportunities that can be exploited more effectively with the help of computer-aided design tools. Here, we review state-of-the-art strategies for engineering of modular PKSs and NRPSs, as well as the computational tools that can be used to support and accelerate this process.
Modular PKSs as well as NRPSs constitute complex enzymatic assembly lines comprised of multiple enzymatic components that are responsible for initiation, elongation and termination of polyketide or peptide chains to produce the core scaffolds of natural products. In the NRPS context, each module catalyzes the addition and modification of a specific amino acid and successively extends the peptide in an assembly line manner. Each module consists of at least three domains that define a minimal module: the adenylation (A) domain, the thiolation (T) domain and the condensation (C) domain. The A domain selects, activates and transfers a specific amino acid to the T domain. Subsequently, the C domain catalyzes the peptide bond formation of the amino acid tethered to the T-domain of the same module and one of the preceding module. The integrated amino acids can be further modified concerning their absolute configuration at the Cα atom by epimerization domains (E) and the degree of C- or N-methylation by methyltransferases (MT). Furthermore, heterocyclization can be performed by cyclization domains (Cy), while redox-active domains (Ox, Red) determine their redox state. A loading module typically comprises an A and a T domain, an elongation module a C–A–T set, while a termination module contains a C–A–T and thioesterase domain (TE). The latter is responsible for the release, or optionally, macrocyclization of the peptide product. Thus, a vast variety of compounds can be generated based on different composition and arrangements of these elements. With only a few exceptions,7–9 the organization and order of modules corresponds to the amino acid sequence of the peptide product (co-linearity rule). Thanks to the pioneering work of the Lipmann10,11 and the Laland12 groups in the 1960s and later on by Stachelhaus and coworkers, who discovered the specificity conferring code of the A domains,13 the logic of NRPSs is reasonably understood and set the stage for engineering of the NRPS assembly lines.
In a similar fashion to NRPSs, modular PKSs synthesize polyketides through the stepwise elongation of the starter unit of 2-, 3- or 4-carbon units molecule such as acetyl-CoA, propionyl-CoA, butyryl-CoA, and their activated derivatives, malonyl-, methylmalonyl-, and ethylmalonyl-CoA extender units.14 Three main types of PKSs have been described: type I PKSs, type II PKSs and type III PKSs.15 In this review, we will focus on modular type I PKSs. The minimal module of these is also formed by three core domains: the acetyltransferase (AT) domain which selects for the extender unit and transfers it to the acyl carrier protein (ACP) domain. Finally, a ketosynthase (KS) domain catalyzes the condensation reaction between two modules – analogous to the C-domain in NRPSs. Elongation modules comprise all three core domains, while the loading module lacks a KS domain and the terminal module contains a TE domain, which is responsible for the release of the linear polyketide chain or of the release with macrocyclization. The sole use of KS–AT–ACP-modules leads to β-keto chains. The additional integration of a ketoreductase (KR) domain converts the keto-functionality into a β-OH group, which can be eliminated by a dehydratase (DH) domain to give an alpha-beta unsaturated alkene, which in turn can be reduced to a single bond with the help of an enoyl-reductase (ER) domain. Thus, the final PKS assembly line can be defined as KS–AT–(DH–ER–KR)–ACP. For more detailed information on NRPS and PKSs, we refer the reader to several excellent reviews.16–21
2. Strategies for re-engineering PKS/NRPS systems
Various strategies have been employed to leverage NRPS/PKS systems, ranging from combinatorial design to direct refactoring of processing steps. The following is a brief overview of these tactics with some of their successes and challenges (Fig. 1). | ||
| Fig. 1 Various strategies for re-engineering PKS/NRPS systems including: (a) precursor-directed biosynthesis to leverage domain promiscuity. (b) Domain editing to re-program monomer specificities. (c) Domain exchanges to replace partial or whole modules. (d) Multi-module exchanges to produce chimeric clusters. (e) Insertion or deletion of domains. (f) Post processing enzyme addition or deletion. | ||
2.1 Precursor directed biosynthesis
Precursor directed biosynthesis (PDB) and mutasynthesis were among the first re-engineering attempts applied on PKSs and NRPSs. PDB is based on feeding of alternative monomers to change the final product by leveraging domain promiscuity. This strategy is based on the frequent observation of major and minor products.22,23 In an NRPS context, the reason is that the A domains possess a natural relaxed substrate flexibility, e.g. a Glx-specific domain recognizes, and also to a minor extent activates, Asx. Likewise the Leu/Val/Ile-specific domains recognize their respective closely related member(s) of the branched amino acid family. Occasionally, A-domains display an intriguing promiscuity: e.g., the putatively Pro-specific A domain of the pyreudione NRPS accepts not only proline derivatives, but also ring-extended residues.24 This has proven to be an effective strategy to diversify NRPS products, for example with the addition of a fluorinated non-proteinogenic amino acid to the lipopeptide iturin.25 However, empirically the yields of the obtained new compounds are commonly low because the synthetic amino acids compete with the natural amino acids for cellular uptake as well as A-domain recognition. To overcome this problem, the mutasynthesis concept can be integrated,26,27 where the microorganism is deficient in the synthesis of the natural precursor and relies on the fed synthetic precursor to complete the secondary metabolite.28 Successful examples include e.g. the alteration of the regular dihydroxyphenylglycine and β-OH–Tyr units in the glycopeptide antibiotic balhimycin with methoxylated and fluoro-derivatives, respectively.29,30In the same way, PDB has been applied to PKSs. One of the first such experiments conducted with the 6-deoxyerythronolide B biosynthesis pathway led to the new analogue 15-fluoroethyl-6 deoxyerythronolide B.31 Similarly, mutasynthesis approaches have also been applied to PKSs. An example of this is the work on the antitumoral polyketide geldanamycin by Eichner and coworkers,32 in which variants of the natural 3-amino-5-hydroxybenzoic acid starter unit were incorporated into the polyketide product.
2.2 Domain exchanges
Considering the modular structure of the NRPS, it is most appealing to simply exchange a module with another. Such exchanges have been demonstrated at the level of a single A domain,33 combined C–A or A–T domains34 and finally of complete C–A–T modules.35 However, with a few exceptions, the success of these experiments remained modest due to reduced product yields. These swaps worked best if domains are exchanged with those from the same biosynthetic gene cluster (BGC) or from closely related BGCs that encode the production of molecules within the same compound family. The most extensive and successful example of this approach represents the work from Baltz and coworkers, who engineered the NRPS assembly line of the clinically applied lipopeptide antibiotic daptomycin in 2006. They replaced single or multiple C–A–T modules with modules from the same BGC or from the closely related NRPSs for the calcium-dependent antibiotics (CDA) and A54145.36 The authors demonstrated that 8 of the 13 AA positions in daptomycin can be modified by module exchange, which led to a combinatorial library of 40 daptomycin derivatives.37 In a recent review, Baltz exposed the failure and success in daptomycin NRPS engineering and suggested that C–A linkers are not flexible while T–C linkers and A–T are.38 In summary, these impressive results showed that while domain swapping is possible, there are also considerable limitations due to the lack of understanding of inter-modular communications and downstream specificity filters.A recent breakthrough in the understanding of the NRPS logic was achieved by contributions of the Bode group. During investigations of the flexibility of inter-domain linkers, they observed that C–A linkers are indeed more flexible than previously suggested. Two separate structural parts form these linkers, and 22 N-terminal amino acids appear to mediate the interactions between C and A domains. Based on these findings, they defined a new concept of an exchange unit (XU). An XU consists of sets of A–T–C (or A–T–C/E) domains instead of canonical C–A–T modules. The consequent application of these rules resulted in the successful de novo design of the NRPS of xenotetrapeptides.39 However, the yields decreased drastically, maintaining the common problem encountered in previous NRPS engineering attempts. In a follow-up work, Bode and coworkers refined and expanded their model and recognized the importance of the involved C-domains. In general, C domains catalyze the condensation of the downstream T-domain-bound amino acid (donor substrate) with the activated upstream T-domain-bound amino acid or peptide (acceptor substrate). For the fusion, donor and acceptor substrates have to be coordinated in their respective Cacceptor- and Cdonor-subdomains. The authors established that particularly the acceptor site is very selective for the nature of the side chain and chirality. However, the A domains have been considered to act as the primary specificity determinants in NRPSs. Bode and coworkers picked up the hypothesis that both subdomains display specificity for the corresponding amino acids and act as gatekeepers.40 Thus, an exchange that respects involved Cacceptor- and Cdonor-subdomains, as well as native protein–protein interactions adjacent to an A–T-bidomain, yielded a redefined exchange unit (referred as XUC, see Fig. 2a). During their experiment series, the Bode group located the exact dissection position in the linker region of the C-domain, so that it can be complemented with the respected counterpart for combinatorial biosynthesis and stays fully functional. The application of these rules led to the production of the target compounds without a loss in yields, a breakthrough for NRPS engineering.41
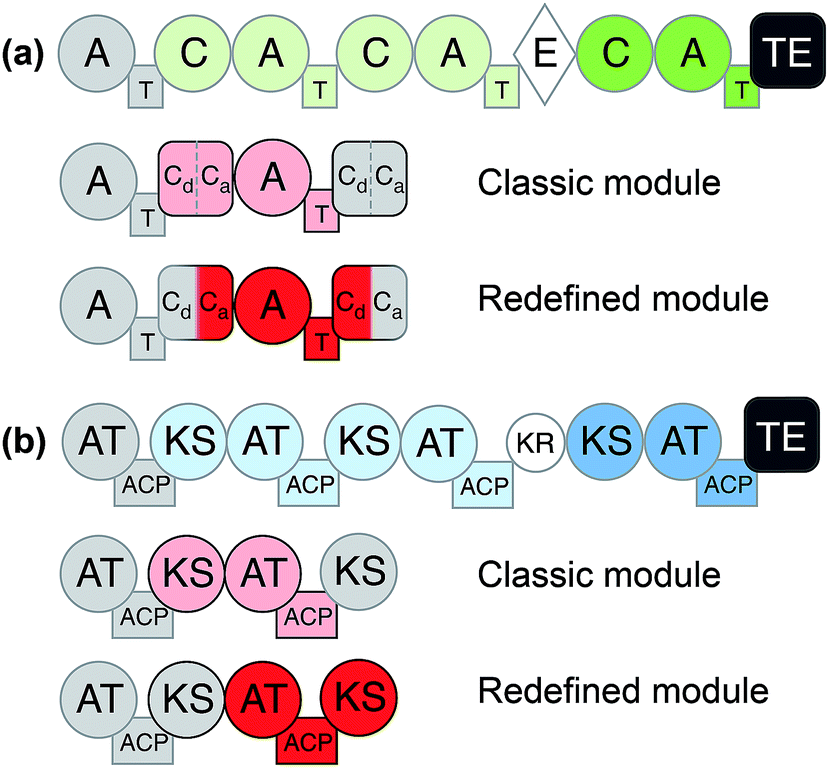 | ||
| Fig. 2 General domain and module organization of (a) non-ribosomal peptide synthetase (NRPS) and (b) polyketide synthase (PKS).42 Classic module definitions are highlighted in pink and redefined modules, such as XUC, are shown in red. | ||
Similar to the exchange of domains and modules in NRPSs, many engineering attempts for PKSs have been based on swapping or insertion of AT domains. For the erythromycin assembly line, new derivatives have been obtained by such domain insertions.43 DEBS-PKS engineering has been performed in the native producer (Saccharopolyspora erythraea), but also in heterologous hosts (Streptomyces coelicolor CH1999 and S. lividans K4-114). For example, Oliynyk and co- workers exchanged the AT of the module 1 of erythromycin PKS by the AT of module 2 of the rapamycin PKS, resulting in an extension unit exchange from methylmalonyl-CoA to malonyl-CoA and obtaining a novel triketide lactone instead of a lactone without methylation in the C-4.44 Similar approaches have recently also been used for the engineering of polyketides related to biofuel production.45 However these single domain exchanges have been known for some time to introduce complications in downstream compatibility46 and result in lower product yields. Yuzawa and coworkers (2016) related this problem with the sequence of the linkers among domains, specifically, between AT and KR domains and KS and AT domains. The AT–KR linker region is denominated by the authors as post AT-linker (PAL) region and can be further subdivided in PAL1 and PAL2, while the KS–AT linker region was designated as KAL. In-depth analysis of the linker sequences led to the identification of significant consensus sequences between the linker regions of DEBS-PKS and epothilone (EPO) PKS. The authors conducted diverse swapping experiments using different variants of swapped regions and finally resolved that the best region for the swapping is the region formed by KAL–AT–PAL1, while PAL2 was kept native. The production yield showed a minimal decrease compared to wild type. Subsequently, the group confirmed these rules using the lipomycin PKS assembly line and showed successful exchanges using modules from 5 different PKSs. With yields on par or greater than wild-type production, this illustrates that this approach is both robust and effective.47
Like for NRPSs, recent advances in PKS engineering have led to the redefinition of the domain organisation of PKS modules. PKS modules were previously organised into KS–AT–(DH–ER–KR)–ACP modules, but recent phylogenetic analysis by Abe et al. has highlighted that processing enzymes co-migrate during the assembly line with the KS domain downstream of the ACP.42,48,49 Thus, new PKS modules would be termed AT + (DH + ER + KR) + ACP + KS. These updated boundary definitions have already shown to have a beneficial effect in the rational design of a PKS to produce homoaureothin.50 These findings are a milestone in engineering NRPS/PKS systems and open the possibility for realizing total de novo design of clusters.
2.3 Specificity code mutations
Another approach to maintain inter-domain compatibility is to simply alter the specificity-coding regions of a domain. Structural biology investigations with the initial A domain of the gramicidin NRPS with L-Phe and AMP revealed the eight key amino acid residues in the active site of the A-domain13 which allows the substrate specificity prediction straight from the DNA level. This 8 AA-containing code, also referred as “Stachelhaus-code” was later on refined.51 While it needs to be adapted when applied to NRPS BGCs originating from certain bacterial genera or from fungi,52 it still forms the basis for today's bioinformatics-driven prediction of the products of NRPS gene clusters. This specificity code can also be modified to alter the chemical outcome of an assembly line. A successful example for A-domain code mutations is provided by the Micklefield group, who reprogrammed the 10th calcium-dependent antibiotic (CDA)-synthetase module to recognize Gln and Me–Gln instead of Glu.53 This has also recently been shown to be effective in altering non-promiscuous domains to accept unnatural extender units.54 Furthermore, besides rationally guided specificity code mutagenesis, it is also conceivable to perform directed evolution experiments, i.e. mutant libraries are generated and screened for enhanced properties such as an increased bioactivity of the final metabolite or an increased or different A-domain-selectivity. For details about the progress in this field, the reader is referred to other reviews.55Regarding PKS systems, it is also established to change the specificity of the AT-domains concerning natural substrates by mutagenesis. However, recently, some studies are taking a step forward trying to incorporate non-natural extender units. Based on the mutations which were made in module 6 of DEBS-PKS54 the Williams group repeated the same mutations in the pikromycin synthase cluster.56 Initially, they did not obtain satisfactory results with the mutation of Try–Arg in the corresponding AT domains but with the site-specific mutation of Try to Val at position 755, they achieved the desired substrate shift; they were also able to produce derivatives with the non-natural units propargyl-, ethyl- and allylmalonyl-CoA in robust yields.54 The incorporation of above mentioned non-natural units enables a further derivatization through semisynthetic chemistry, especially by click chemistry. In this way, the polyketide of interest can be conjugated with other molecules such as a further pharmacophore-containing moiety or dyes and would expand in this way the possibilities for drug discovery and diverse chemical-biological applications with the polyketidic compound.
2.4 Starter units and tailoring modifications
Other factors to take into account are the engineering of the starter domains or tailoring domains. The swapping of loading modules (LM) in PKSs has proven successful. For example, Leadlay and coworkers exchanged the 6-deoxyerythonolide LM for the avermectin LM, and Long et al. exchanged the DEBS LM for the oleandomycin LM.57,58 NRPS initiation modules have also been modified with increasingly positive results. These starting modules are attractive areas for redesign because they omit upstream restraints that confound similar extender module redesigns.59 Challenges still remain however with downstream compatibility. For example, several failed attempts at reprogramming tyrocidine initiation modules were shown to result from incompatibilities between PCP and E-domain interfaces.60Further successful domain engineering was achieved regarding internal tailoring domains, for example KR domain replacements with a tri-domain to form a new PKS.61 Various studies have shown positive swapping between two different KR domains in isolation, however the engineering of DH domains depends on the KR domains that precede them because DH domains are sensitive to the stereochemical configuration of the substrate.62 Tailoring domains can also be embedded as subdomains within functional NRPS domains, as seen with methylation interrupted A-domains. These proved to be a boon to NRPS design by allowing site-specific and selective methylation of monomer side chains (O- or S-) or at N termini.63 Additionally, exogenous reactions can also be incorporated to diversify compounds; examples include halogenation, glycosylation, acylation and, sulfation tailoring enzymes.28
2.5 Multi-module exchange
To govern the protein–protein interactions between modules that are part of separate polypeptides, not only the interactions between ACP–KS domain pairs are important, but also sets of small specialized N- and C-terminal regions called docking domains (DDs) for PKSs and communication (COM) domains for NRPSs. These interactions have been shown to be a dominant cause for lowered product turnover rates, overshadowing the impact of substrate recognition in chimeric clusters.64 By manipulation of these regions, examples of combinatorial clusters have been generated65,66 or re-designed to modulate products of single-module NRPS systems.67 DDs ensure the correct PKS assembly into a functional enzyme. Studies on virginiamycin biosynthesis show that the DDs are essential for the correct assembly of the enzymatic complex and finally for the communication between the KS and ACP domains. For successful engineering of PKSs, the identification of the interaction regions and the elucidation of the mechanism involved in the communication between the domains or modules is required.68For PKS engineering, the studies have been based on two research lines: modification of AT domains and docking domains (DDs). Principally, the engineering of ATs has concentrated on the swapping of solely AT domains or the complete module. Furthermore, direct mutagenesis to change the precursors' specificity of the targeted AT has been conducted. All of these concepts were applied extensively to the erythromycin PKS (syn. 6-deoxyerythronolide B synthase = DEBS). One example is the exchange of module 1 and 2 (load methylmalonyl-CoA) of DEBS by specific malonyl-CoA modules of Streptomyces hygroscopicus ATCC 29253 and the rapamycin module 14 PKS.68,69 New derivatives were obtained, however, again with strongly decreased yield. It is noteworthy to mention that the drop in production was highly dependent on the swapping position of the PKS assembly line. It appears that the linker region is of considerable importance to mediate communication between heterologous modules. A recent study showed that the yields of a chimeric DEBS system can be improved by introducing non-native docking domains between the native ones; this apparently added flexibility between processing and condensing domains70, which led to improved yields to near-native amounts.
3. Computational tools for re-engineering biosynthetic assembly lines
3.1 Search and collection of parts
Extensive genetic and biosynthetic parts catalogs have been growing in recent years, such as the iGEM Registry of Standard Biological Parts.71 In addition, databases of BGCs are helping to provide valued comparative context for design efforts. Some of the most successful computational approaches to NRPS/PKS re-design have relied on comparative analysis of related BGCs, which can help decipher optimal fusion/recombination points, identify new biosynthetic parts, and improve our understanding of module specificities.39,41,72 Furthermore, large-scale comparative analyses have shed light on natural evolutionary patterns that can guide future efforts and approaches.73 For instance, conglomerate BGCs show significant acquisition and refactoring of cluster fragments that encode functionally independent elements. These sub-clusters, responsible for similar moieties in the final product (including, e.g., the biosynthesis of nonproteinogenic amino acids or the synthesis and transfer of modified sugar units), are shared across a variety of distinct BGCs and can potentially be leveraged for combinatorial re-design of new chimeric clusters.73 Similar lessons in BGC evolution have been shown on laboratory time scales, as seen with sequencing comparisons of PKSs that underwent deletion and recombination events.74 These experiments showed analogs that produced near-native product yields, further underscoring the benefit of re-defined PKS modules as building blocks for engineering. Comparative approaches have also aided traditional combinatorial design; a classic example is the generation of 154 bimodular PKSs based on raw material from eight related PKS assembly lines.75 To take advantage of this perspective, cataloging and rapid search methods are required. Fortunately, progress in recent years has made this process easier and more automated through the use of computational tools, which are increasingly necessary as genomic data continues to grow.With maturing methods for BGC detection76 and increasing amounts of genomic data, there is a growing need to effectively navigate available BGC sequences. Several databases and tools facilitate the cataloging and comparison of known and putative BGCs. The recently updated antiSMASH database contains predicted gene clusters for ∼25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 representative complete and high-quality draft bacterial genomes, whereas the IMG Atlas of Biosynthetic gene Clusters contains a higher number of BGCs by also including lower-quality genomes as well as metagenomes.77,78 Both databases provide various search options to quickly identify biosynthetic parts of interest; for example, one can quickly query the antiSMASH database to download all adenylation domains specific for hydroxyphenylglycine in BGCs with a high similarity to the vancomycin BGC. The MiBIG repository provides data on >1800 BGCs of known function, with annotations and evidence codes that document functions of gene clusters, enzymes and domains; these are all downloadable in a computer-readable JSON format, which makes it possible to quickly screen for enzymes or domains of interest.77–79 The NORINE database is yet another resource for cluster mining, which contains extensive catalogs of NRPS/PKS monomers.80 This resource has served as an aid for retro-biosynthetic structure matching, which has recently become an automated process using the rBAN server.81 While much of the putative BGC data (over 95%)82 have yet to be associated with a known product, they can be leveraged through gene and structure similarity methods to aid with collection of similar clusters. Besides sequence identity-based methods such as clusterblast and MultiGeneBlast,83,84 tools that allow large-scale comparison of BGCs based on architectural similarity are recently coming to light. BiG-SCAPE85 is one such tool, which incorporates Pfam domain similarity as demonstrated previously.86 Multi-locus phylogenetic analysis of gene cluster families (GCFs) using CORASON85 can be used to provide even higher-resolution analysis of the relationships between groups of similar BGCs. Biocompass87 is another tool to perform BGC networking, with a focus on identifying syntenic blocks of sub-clusters. These approaches allow for a rapid collection of related BGCs, or GCFs with similar subunits, even for understudied or novel BGCs (Fig. 3).
000 representative complete and high-quality draft bacterial genomes, whereas the IMG Atlas of Biosynthetic gene Clusters contains a higher number of BGCs by also including lower-quality genomes as well as metagenomes.77,78 Both databases provide various search options to quickly identify biosynthetic parts of interest; for example, one can quickly query the antiSMASH database to download all adenylation domains specific for hydroxyphenylglycine in BGCs with a high similarity to the vancomycin BGC. The MiBIG repository provides data on >1800 BGCs of known function, with annotations and evidence codes that document functions of gene clusters, enzymes and domains; these are all downloadable in a computer-readable JSON format, which makes it possible to quickly screen for enzymes or domains of interest.77–79 The NORINE database is yet another resource for cluster mining, which contains extensive catalogs of NRPS/PKS monomers.80 This resource has served as an aid for retro-biosynthetic structure matching, which has recently become an automated process using the rBAN server.81 While much of the putative BGC data (over 95%)82 have yet to be associated with a known product, they can be leveraged through gene and structure similarity methods to aid with collection of similar clusters. Besides sequence identity-based methods such as clusterblast and MultiGeneBlast,83,84 tools that allow large-scale comparison of BGCs based on architectural similarity are recently coming to light. BiG-SCAPE85 is one such tool, which incorporates Pfam domain similarity as demonstrated previously.86 Multi-locus phylogenetic analysis of gene cluster families (GCFs) using CORASON85 can be used to provide even higher-resolution analysis of the relationships between groups of similar BGCs. Biocompass87 is another tool to perform BGC networking, with a focus on identifying syntenic blocks of sub-clusters. These approaches allow for a rapid collection of related BGCs, or GCFs with similar subunits, even for understudied or novel BGCs (Fig. 3).
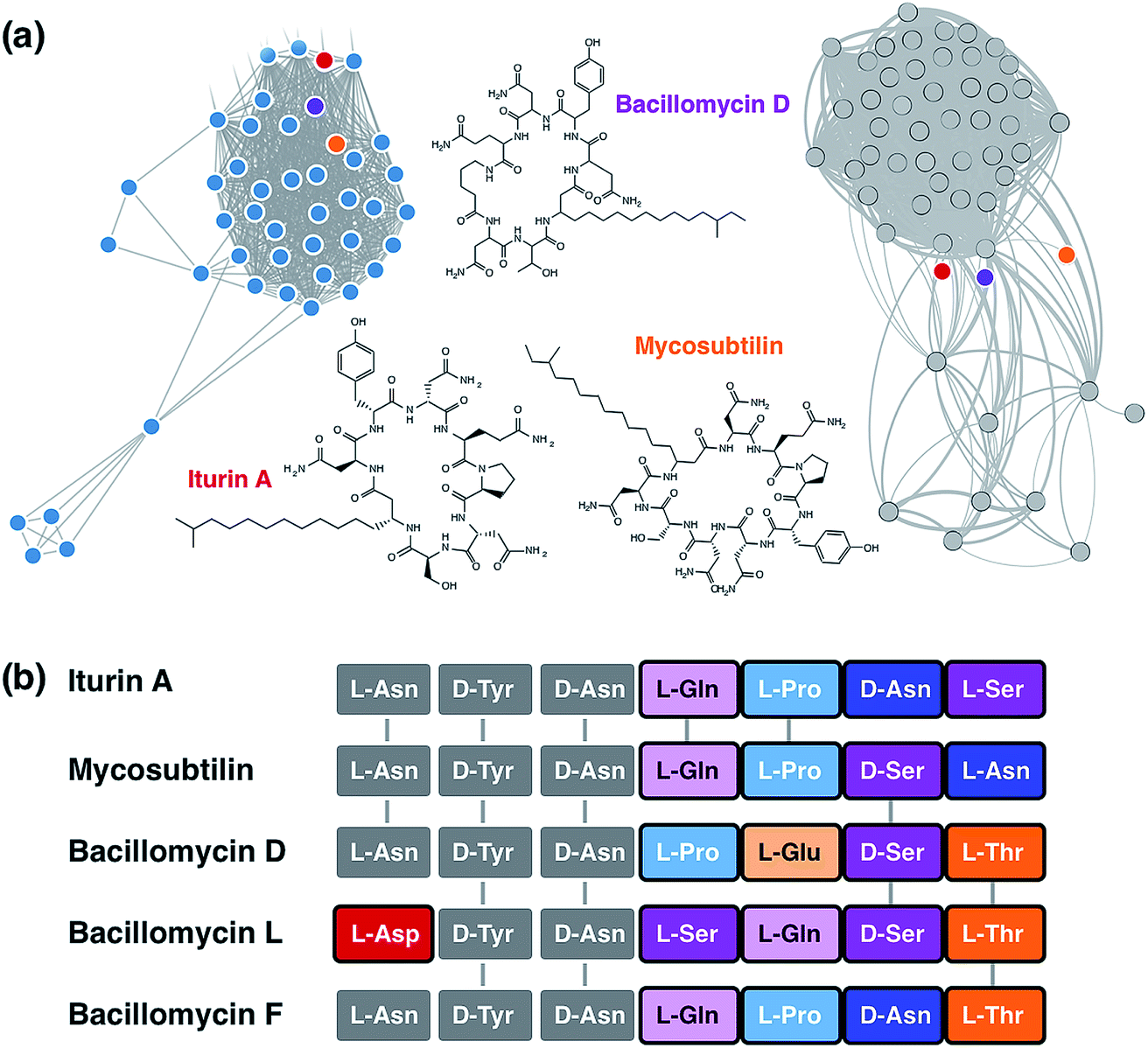 | ||
| Fig. 3 (a) Example chemical network using NP Atlas (left), BiG-SCAPE network (right), and structures using the lipopeptide iturin as a query. (b) Modules of iturin-like clusters illustrate parts mining for different substrate specificities. | ||
In addition to genomic comparisons, structure prediction and search methods can help with collecting similar known compounds or screen for desired products. One interesting result of structure comparison is a virtual library and generator of polyketides, PKS enumerator, which is based on several biologically active macrolides.88 While current structure prediction methods still require advancements in specialized cases (e.g. iterative PKS domains, external tailoring, macro-cyclization), they are capable of accurately predicting core chemical scaffolds quite well for NRPS/PKS systems. Detection tools such as antiSMASH and PRISM include recent advancements in predicting structures from BGCs, such as enhanced NRPS specificity detection using SANDPUMA and improved exploration of product permutations, respectively.72,89,90 In addition to defining module and domain specificities, these predictions can be used to help corroborate putative GCFs or find a starting cluster for engineering a desired compound. Intuitive web servers such as ChemSpider,91 SIMCOMP,92 and the NP Atlas93 provide user-friendly search interfaces for global and sub-structure searches; results are also enhanced with similarity scoring, moiety highlighting, and exploration in a chemical network. For any compounds of interest, the MIBiG repository can then be queried to identify whether a BGC has been characterized for it. Based on these connections, a larger set of similar clusters can be compared to ultimately help identify useful parts for constructing new clusters.
Another useful product of comparative analysis is the recent NRPS-linker database.94 This resource houses a catalog of inter-module linkers (IMLs) taken from NRPS clusters found in all publicly available genomes and a parser to extract IMLs from query sequences. Protein–protein linkers have been shown to play an important role in multi-domain dynamics and flexibility,95 and they potentially encode for important module compatibility elements. The database analyses showed IMLs have significant structured elements and were consistently associated to A-domain specificity pairs, suggesting they are crucial to module compatibility.94 Thus, conservation of IMLs may aid in module swapping. Furthermore, the authors noticed consistent phylogenetic conservation of IMLs, which implies host optimization of engineered NRPSs might benefit by using IMLs taken from related phyla. This is an interesting unexploited resource, as it expands parts mining to also include a library of “mortar” that can help ‘glue together’ desired combinations of BioBricks.
3.2 Optimization and refactoring
Concerted evolution (the homogenization of domain sequences within assembly lines through recombination) and family-specific adaptions provide other important evolutionary lessons for BGC refactoring.73 This underscores a need to custom-tailor re-engineered components, which includes codon optimization for host expression or protein level changes via directed evolution to maximize compatibility. Prioritizing the exchange of modules or domains for ones with closer phylogenetic proximity and respecting integral interfaces can potentially increase the success of redesigning native clusters. While phylogenetic proximity is not an absolute requirement, as interkingdom hybrid BGCs have been observed,96 it is likely to increase the chances of success. Improvements via codon optimization, reviewed elsewhere,97 illustrate this point. Many web services can perform this optimization as well as account for mRNA secondary structure and GC content;97 these functions are also included in DNA supplier workflows, such as IDT's codon optimization tool (https://www.idtdna.com/codonopt). One recent study on the effect of codon context revealed that the influence of neighboring codons can limit the rate of translation,98 and may be an additional measure to consider. CCtool (http://algo.tcnj.edu/cctool/) is a recent server that aims to address this consideration, albeit with limited host selection.99Optimizing amino acid sequence via directed evolution experiments is another level of refinement that can have major benefits, as protein–protein interactions or substrate specificities can be enhanced, by e.g. increasing their specificity. One example shows a drastic 500 fold improvement of enterobactin production after only 3 rounds of mutations.100 As the number of generated variants and testing of engineered BGCs can be quite time consuming, it is important to use the most educated mutations as possible. Using computational methods to reduce this search space is a valuable solution, as illustrated with the redesign of an NRPS A domain using the K* algorithm, implemented in the OSPREY application,101 which evaluates protein variants that improve or maintain protein flexibility and ligand binding via energy minimization methods.102 Many tools have been developed, and covered thoroughly in previous reviews,103–105 that aim to predict the impact of variants using different criteria: structure consistency, protein stability, evolutionary consistency, and coverage of mutant library space (Table 1).
| Description | URL | Year (latest) | Ref. | ||
|---|---|---|---|---|---|
| Databases & comparative tools | |||||
| antismashDB | Antismash-detected BGCs from public genomes | http://antismash-db.secondarymetabolites.org | 2018 | 77 | |
| MiBIG | Experimentally verified BGCs | http://mibig.secondarymetabolites.org | 2015 | 79 | |
| IMG-ABC | Clusterfinder-detected BGCs from all deposited genomes | http://img.jgi.doe.gov/abc | 2015 | 78 | |
| NRPS-linker | Parser and database of Inter Module Linkers (IMLs) for NRPSs | http://nrps-linker.unc.edu | 2019 | 94 | |
| NPAtlas | Collection of known natural products with structural similarity searches | http://www.npatlas.org | 2018 | 93 | |
| SIMCOMP | Structural similarity search tool | http://www.genome.jp/tools/simcomp | 2010 | 92 | |
| BiG-SCAPE | Calculates similarity between BGCs and groups into gene cluster families | http://bigscape-corason.secondarymetabolites.org | 2018 | 85 | |
| CORASON | Groups gene clusters using multi-locus phylogeny methods | http://bigscape-corason.secondarymetabolites.org | 2018 | 85 | |
| Biocompass | Calculates similarity between BGCs with a focus on shared syntenic sub-clusters | http://np-omix.github.io/BioCompass | 2017 | 87 | |
| Norine | Database and analysis tools for NRPS clusters and monomers | http://bioinfo.lifl.fr/norine | 2016 | 80 | |
| rBAN | Retro-biosynthetic analysis of nonribosomal peptides | http://bioinfo.cristal.univ-lille.fr/rban | 2019 | 81 | |
| PKS Enumerator | Virtual database and generator of macrolide polyketides | http://www.fourches-laboratory.com/single-post/2018/09/04/PKS-Enumerator | 2018 | 88 | |
|
|||||
| Directed evolution tools | |||||
| SCHEMA | Minimization of disruptions to local amino acids | http://github.com/mattasmith/SCHEMA-RASPP | 2002 | 106 | |
| HotSpot Wizard 3.0 | Energy minimization scoring from homology modeling results | http://loschmidt.chemi.muni.cz/hotspotwizard | 2018 | 107 | |
| OSPREY/K* | Substrate binding approximation method | http://www.cs.duke.edu/donaldlab/osprey.php | 2013/2009 | 101 and 102 | |
| CADEE | Reactivity approximation using empirical valence-bond theory | http://github.com/kamerlinlab/cadee | 2017 | 108 | |
|
|||||
| Homology search/modeling tools | |||||
| IntFOLD | 3D modeling with additional built in features: quality assessment, ligand binding, and disorder prediction | http://www.reading.ac.uk/bioinf/IntFOLD | 2015 | 109 | |
| RaptorX | Uses closely related homologs and structural data to create context specific models. Ranked 1st in CASP12 for contact point prediction | http://raptorx.uchicago.edu | 2015 | 110 | |
| HHpred | Remote homology search with alignment | http://toolkit.tuebingen.mpg.de/#/tools/hhpred | 2005 | 111 | |
| MODELLER | Search, model, and evaluation of models in one pipeline | http://salilab.org/modeller | 2003 | 112 | |
| PHYRE2 | Homology search and additional applications such as loop refinement and variant analysis | http://www.sbg.bio.ic.ac.uk/phyre2 | 2015 | 113 | |
| ROBETTA | Server suite with de novo prediction and template based 3d model generation | http://robetta.bakerlab.org | 2011 | 114 | |
| SWISS-MODEL | Server suite with variety of model building applications including quartinary structure prediction | http://swissmodel.expasy.org | 2018 | 115 | |
| I-TASSER | Apart of suite of software that offers function prediction and contact mapping with consistent high rankings in CASP competitions | http://zhanglab.ccmb.med.umich.edu/I-TASSER | 2018 | 116 | |
|
|||||
| Automated parts/pathway mining tools | |||||
| ClusterCAD | Automated PKS parts discovery engine for de novo design of polyketides | http://clustercad.jbei.org | 2018 | 117 | |
| RetroPATH | Automated pathway design using known and permiscious enzymes | http://www.jfaulon.com/category/retropath | 2014 | 118 | |
| genoCAD | Designer for DNA/vectors | http://www.genocad.com | 2009 | 119 | |
Unfortunately some of these, such as quantum mechanics/molecular mechanics (QM/MM) methods, can be impractical for high-throughput screening due to the lengthy compute times required. A recent addition, CADEE, aims to bridge this gap between fast heuristic methods and higher accuracy QM/MM methods using an empirical valence-bond (EVB) approach.108 Although these methods still struggle to accurately predict multi-site influence on structure, they can help to avoid severely deleterious mutations and save valuable experimental time.
Rational design constraints, such as focusing on certain hotspot regions, can further complement these methods. One way to highlight these areas is to interrogate the protein structure. Utilization of the growing number of available structures in the Protein Data Bank120 for comparative structural analysis can help indicate areas that might be tolerant to change. For example, the T domain central to NRPSs is now known to maintain a relatively consistent conformation throughout its different catalytic states.121 This was shown through comparative analysis of 18 structures at various catalytic states and further supports that tight interaction of T and surrounding domains and linkers are less amenable to change. Structural context not only informs subdomain lobes and flexible regions, but can also identify binding pocket residues, domain interface locations, and contact points. For example, homology modeling helped to confirm a linker fusion point that enabled the generation of several chimeric variants of antimycin.122 These structural homology approaches also led to the identification of a successful recombination site in the GameXPeptide-producing NRPS GxpS of Photorhabdus luminescens. This involved altering the alpha-5 helix into a consistent consensus sequence of all XUs involved, which resulted in the production of several peptide variants.41Fig. 4 illustrates how these regions can be identified using homology modeling with tools like MODELLER.112
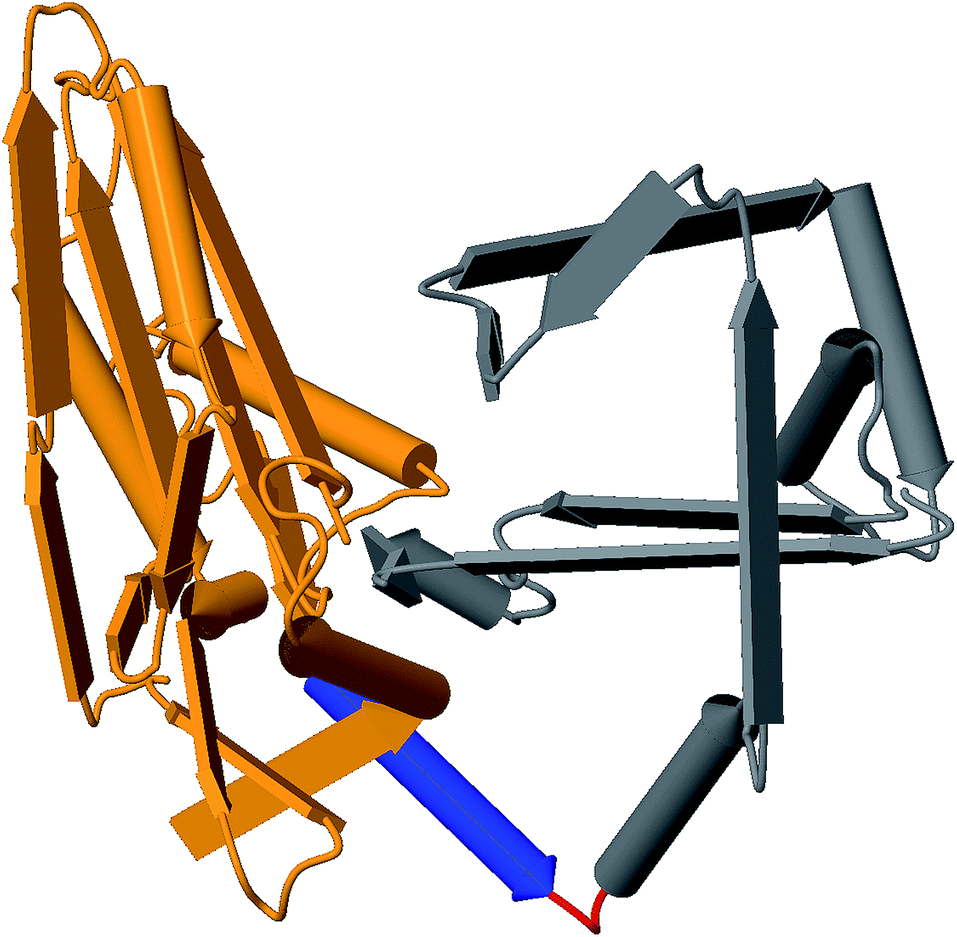 | ||
| Fig. 4 Example 3D structure generated from the C-domain sequence of module 2 of Gramicidin synthetase using MODELLER with PDB:2JGP as the template. Identified regions: C-donor (orange), alpha-5 helix (blue), linker (red), and C-acceptor (grey). | ||
With more structures coming to light, for example the first methyltransferase-interrupted A-domain,123 more accurate template-based sequence-to-structure modeling can be leveraged for design efforts. For example, using this analysis, a mutation of a key residue in TioS from the thiocoraline biosynthetic pathway was shown to abolish methylation activity while retaining yields nearly identical to the wild-type.63
Homology modeling can be performed with a variety of web servers and standalone tools (Table 1). The bi-annual Critical Assessment of techniques for protein Structure Prediction (CASP) competitions has helped to mature these methods, showing a significant increase in model accuracy, particularly for template-based methods;124 additionally, refinement and template-free modeling have also seen improvements. While much work is still required to further predict protein–protein docking, and ultimately the impact of mutations, these models have helped to identify fruitful areas for directed evolution.125
3.3 Toward automated cluster design
Collection of optimal parts for individual modules, domains or enzymes is an important step for successful re-factoring experiments. Currently, homology tools such as BLAST126 and HMMer127 are largely employed to extract these elements, but steps toward automated identification of useful elements based on a (desired) chemical structure are being employed. ClusterCAD117 is one such tool that focuses on cataloging and highlighting of PKS elements (collected from MIBiG), with the ultimate goal of de novo design of clusters. In addition to identifying the best starting BGC most similar to a desired structure, ClusterCAD aims to identify a parsimonious number of domain swaps, deletions and additions based on sequence homology to known clusters. This was shown to automatically select similar components for a previously validated re-engineering of an adipic acid producing cluster.128A more generalized tool, RetroPATH,129 aims to identify reactions beyond PKS and NRPS systems. This can be useful for PKS/NRPS engineering by identifying tailoring enzymes or improving the precursor supply of a pathway. The webserver genoCAD130 is another application that aims to leverage known context-free parts for designing DNA and vectors; this also has been shown fruitful for the design of plant expression systems.131 Despite requiring manual curation and optimization of these automated pipelines, these applications provide a foundation for automated design and ultimately can be matured to account for other constraints such as compatibility of compound intermediates.
4. Conclusion and future outlook
Traditional approaches for cell-factory design have largely focused on optimization of native production or expression in model hosts. Beyond these goals, synthetic biology aims to provide the plans and parts for de novo pathway construction, “BioBricks”, that can produce any desired compound. Some examples are the construction of synthetic pathways from plant polyketides: olivetolic acid in Escherichia coli132 and the biosynthesis of opioids, cannabinoids, and biofuels in yeast.133–135 The field is gradually progressing toward the ability to biosynthesize valued compounds “from scratch” in a design-based fashion.136 Considering the complexity of genomic regulation, multiple catalytic pathways, and environmental interactions of a cell, it is clear this goal will not be met overnight. Detailed reviews on synthetic biology have illustrated this long, and worthwhile, road for the design, build, and test hurdles toward this end.137–139 Although examples of successful computer-aided redesign efforts are discouragingly few, it is promising to see that many barriers for employing these methods are reducing. As accessible web interfaces and automated methods are coming to light, we can expect a greater number of experiments leveraging these approaches and improved generations of these tools from these results. While these methods still have room to mature, and account for more complex design restraints, they have already demonstrated great utility in aiding the rational design of biological machinery.Here, we highlight another longstanding goal within the general scope of cell factory design: manipulation of modular NRPS and PKS enzyme factories. Although many attempts to design these systems have lead to mere proof-of-concept levels of production, a new understanding of inter-module connections, new computational tools, and increasing comparative data analysis provide exciting new opportunities. With the re-definition of module boundaries, it has been shown that combinatorial design can lead to novel products with near-native yields; fully leveraging catalogs of such redefined modules is currently still an underexplored space, but will be made easier with new computational tools. Several applications can help with this step of parts mining, including gene cluster and structural similarity networking. While much of this process still remains manual and can benefit greatly from optimization and custom-tailoring efforts, tools to automate collection and assembly of components are already underway. These applications, such as ClusterCAD for PKSs, currently do not take into account potential incompatibilities via protein–protein interactions or substrate specificities but provide the groundwork for searching and structuring a de novo BGC in an automated pipeline. Future addition of NRPSs as well as the updated module definitions (exchange units) to ClusterCAD or similar tools, as well as integration with refactoring tools for inserting regulatory elements and optimizing codon usage, will enable even more full-fledged computer-aided design of PKS and NRPS assembly lines, especially if this can be supported by e.g. drag-and-drop functionality to assemble new polypeptides in silico. As parts collections continues to expand, we are confident that these tools can eventually help solve remaining challenges and thus facilitate the dreams of combinatorial design of these wonderful modular systems to allow accessing a vast chemical space through biosynthesis.
5. Conflicts of interest
MHM is a member of the Scientific Advisory Board of Hexagon Bio.6. References
- A. M. Davy, H. F. Kildegaard and M. R. Andersen, Cell Syst., 2017, 4, 262–275 CrossRef CAS PubMed.
- D. A. Evans, M. R. Wood, B. W. Trotter, T. I. Richardson, J. C. Barrow and J. L. Katz, Angew. Chem., Int. Ed., 1998, 37, 2700–2704 CrossRef CAS PubMed.
- J. J. McIntyre, A. T. Bull and A. W. Bunch, Biotechnol. Bioeng., 1996, 49, 412–420 CrossRef CAS PubMed.
- K. S. Lee, B. M. Lee, J. H. Ryu, D. H. Kim, Y. H. Kim and S.-K. Lim, Lett. Appl. Microbiol., 2016, 63, 222–228 CrossRef CAS PubMed.
- J. Thykaer, J. Nielsen, W. Wohlleben, T. Weber, M. Gutknecht, A. E. Lantz and E. Stegmann, Metab. Eng., 2010, 12, 455–461 CrossRef CAS PubMed.
- E. Kalkreuter and G. J. Williams, Curr. Opin. Microbiol., 2018, 45, 140–148 CrossRef CAS PubMed.
- S. Lautru, R. J. Deeth, L. M. Bailey and G. L. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS PubMed.
- D. Reimer, K. N. Cowles, A. Proschak, F. I. Nollmann, A. J. Dowling, M. Kaiser, R. ffrench-Constant, H. Goodrich-Blair and H. B. Bode, ChemBioChem, 2013, 14, 1991–1997 CrossRef CAS PubMed.
- A. C. Ross, Y. Xu, L. Lu, R. D. Kersten, Z. Shao, A. M. Al-Suwailem, P. C. Dorrestein, P.-Y. Qian and B. S. Moore, J. Am. Chem. Soc., 2013, 135, 1155–1162 CrossRef CAS PubMed.
- F. Lipmann, Science, 1971, 173, 875–884 CrossRef CAS PubMed.
- F. Lipmann, W. Gevers, H. Kleinkauf and R. J. Roskoski, Adv. Enzymol. Relat. Areas Mol. Biol., 1971, 35, 1–34 CAS.
- T. L. Berg, L. O. Froholm and S. G. Laland, Biochem. J., 1965, 96, 43–52 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- J. Lau, H. Fu, D. E. Cane and C. Khosla, Biochemistry, 1999, 38, 1643–1651 CrossRef CAS PubMed.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS PubMed.
- A. Koglin and C. T. Walsh, Nat. Prod. Rep., 2009, 26, 987–1000 RSC.
- G. H. Hur, C. R. Vickery and M. D. Burkart, Nat. Prod. Rep., 2012, 29, 1074–1098 RSC.
- R. D. Sussmuth and A. Mainz, Angew. Chem., Int. Ed. Engl., 2017, 56, 3770–3821 CrossRef PubMed.
- M. Klaus and M. Grininger, Nat. Prod. Rep., 2018, 35, 1070–1081 RSC.
- T. Robbins, Y.-C. Liu, D. E. Cane and C. Khosla, Curr. Opin. Struct. Biol., 2016, 41, 10–18 CrossRef CAS PubMed.
- K. J. Weissman, Nat. Prod. Rep., 2016, 33, 203–230 RSC.
- J. M. Crawford, C. Portmann, R. Kontnik, C. T. Walsh and J. Clardy, Org. Lett., 2011, 13, 5144–5147 CrossRef CAS PubMed.
- Y. Xie, Q. Cai, H. Ren, L. Wang, H. Xu, B. Hong, L. Wu and R. Chen, J. Nat. Prod., 2014, 77, 1744–1748 CrossRef CAS PubMed.
- M. Klapper, D. Braga, G. Lackner, R. Herbst and P. Stallforth, Cell Chem. Biol., 2018, 25, 659–665 CrossRef CAS PubMed.
- S. Moran, D. K. Rai, B. R. Clark and C. D. Murphy, Org. Biomol. Chem., 2009, 7, 644–646 RSC.
- A. Kirschning and F. Hahn, Angew. Chem., Int. Ed. Engl., 2012, 51, 4012–4022 CrossRef CAS PubMed.
- R. J. M. Goss, S. Shankar and A. A. Fayad, Nat. Prod. Rep., 2012, 29, 870–889 RSC.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Sussmuth, Angew. Chem., Int. Ed. Engl., 2002, 41, 3383–3385 CrossRef CAS PubMed.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Sussmuth, J. Am. Chem. Soc., 2004, 126, 5942–5943 CrossRef CAS PubMed.
- S. L. Ward, R. P. Desai, Z. Hu, H. Gramajo and L. Katz, J. Ind. Microbiol. Biotechnol., 2007, 34, 9–15 CrossRef CAS PubMed.
- S. Eichner, H. G. Floss, F. Sasse and A. Kirschning, ChemBioChem, 2009, 10, 1801–1805 CrossRef CAS PubMed.
- M. Crüsemann, C. Kohlhaas and J. Piel, Chem. Sci., 2013, 4, 1041–1045 RSC.
- M. J. Calcott, J. G. Owen, I. L. Lamont and D. F. Ackerley, Appl. Environ. Microbiol., 2014, 80, 5723–5731 CrossRef PubMed.
- S. Zobel, S. Boecker, D. Kulke, D. Heimbach, V. Meyer and R. D. Sussmuth, ChemBioChem, 2016, 17, 283–287 CrossRef CAS PubMed.
- K. T. Nguyen, D. Kau, J.-Q. Gu, P. Brian, S. K. Wrigley, R. H. Baltz and V. Miao, Mol. Microbiol., 2006, 61, 1294–1307 CrossRef CAS PubMed.
- R. H. Baltz, ACS Synth. Biol., 2014, 3, 748–758 CrossRef CAS PubMed.
- R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2018, 45, 1003–1006 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2017, 10, 275 CrossRef PubMed.
- K. Bloudoff and T. M. Schmeing, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 1587–1604 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y. N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 1755–4349, DOI:10.1038/s41557-019-0276-z.
- A. T. Keatinge-Clay, Angew. Chem., Int. Ed., 2017, 56, 4658–4660 CrossRef CAS PubMed.
- C. J. Rowe, I. U. Böhm, I. P. Thomas, B. Wilkinson, B. A. M. Rudd, G. Foster, A. P. Blackaby, P. J. Sidebottom, Y. Roddis, A. D. Buss, J. Staunton and P. F. Leadlay, Chem. Biol., 2001, 8, 475–485 CrossRef CAS PubMed.
- M. Oliynyk, M. J. Brown, J. Cortes, J. Staunton and P. F. Leadlay, Chem. Biol., 1996, 3, 833–839 CrossRef CAS PubMed.
- W. Cai and W. Zhang, Curr. Opin. Biotechnol., 2018, 50, 32–38 CrossRef CAS PubMed.
- M. Hans, A. Hornung, A. Dziarnowski, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 2003, 125, 5366–5374 CrossRef CAS PubMed.
- S. Yuzawa, K. Deng, G. Wang, E. E. K. Baidoo, T. R. Northen, P. D. Adams, L. Katz and J. D. Keasling, ACS Synth. Biol., 2016, 7–10 Search PubMed.
- D. A. Vander Wood and A. T. Keatinge-Clay, Proteins: Struct., Funct., Bioinf., 2018, 86, 664–675 CrossRef CAS PubMed.
- L. Zhang, T. Hashimoto, B. Qin, J. Hashimoto, I. Kozone, T. Kawahara, M. Okada, T. Awakawa, T. Ito, Y. Asakawa, M. Ueki, S. Takahashi, H. Osada, T. Wakimoto, H. Ikeda, K. Shin-Ya and I. Abe, Angew. Chem., Int. Ed. Engl., 2017, 56, 1740–1745 CrossRef CAS PubMed.
- Y. Sugimoto, K. Ishida, N. Traitcheva, B. Busch, H.-M. Dahse and C. Hertweck, Chem. Biol., 2015, 22, 229–240 CrossRef CAS PubMed.
- M. Rottig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- D. Kalb, G. Lackner and D. Hoffmeister, Appl. Environ. Microbiol., 2014, 80, 6175–6183 CrossRef PubMed.
- M. Styles, J. Thirlway, M. Al Nakeeb, C. P. Smith, R. Lewis, J. Micklefield, L. Nunns and A.-W. Struck, Angew. Chem., Int. Ed., 2012, 51, 7181–7184 CrossRef PubMed.
- E. Kalkreuter, J. M. CroweTipton, A. N. Lowell, D. H. Sherman and G. J. Williams, J. Am. Chem. Soc., 2019, 141, 1961–1969 CrossRef CAS PubMed.
- H. Kries and A. Stanišić, ChemBioChem, 2019, 20, 1347, DOI:10.1002/cbic.201800750.
- B. Vögeli, K. Geyer, P. D. Gerlinger, S. Benkstein, N. S. Cortina and T. J. Erb, Cell Chem. Biol., 2018, 25, 833–839.e4 CrossRef PubMed.
- A. F. Marsden, B. Wilkinson, J. Cortes, N. J. Dunster, J. Staunton and P. F. Leadlay, Science, 1998, 279, 199–202 CrossRef CAS PubMed.
- P. F. Long, C. J. Wilkinson, C. P. Bisang, J. Cortes, N. Dunster, M. Oliynyk, E. McCormick, H. McArthur, C. Mendez, J. A. Salas, J. Staunton and P. F. Leadlay, Mol. Microbiol., 2002, 43, 1215–1225 CrossRef CAS PubMed.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- U. Linne, S. Doekel and M. A. Marahiel, Biochemistry, 2001, 40, 15824–15834 CrossRef CAS PubMed.
- S. Yuzawa, J. D. Keasling and L. Katz, J. Antibiot., 2016, 69, 494–499 CrossRef CAS PubMed.
- J. F. Barajas, J. M. Blake-Hedges, C. B. Bailey, S. Curran and J. D. Keasling, Synth. Syst. Biotechnol., 2017, 2, 147–166 CrossRef PubMed.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, ACS Synth. Biol., 2018, 7, 399–404 CrossRef CAS PubMed.
- M. Klaus, M. P. Ostrowski, J. Austerjost, T. Robbins, B. Lowry, D. E. Cane and C. Khosla, J. Biol. Chem., 2016, 291, 16404–16415 CrossRef CAS PubMed.
- M. Hahn and T. Stachelhaus, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 275–280 CrossRef CAS PubMed.
- C. Chiocchini, U. Linne and T. Stachelhaus, Chem. Biol., 2006, 13, 899–908 CrossRef CAS PubMed.
- C. Hacker, X. Cai, C. Kegler, L. Zhao, A. K. Weickhmann, J. P. Wurm, H. B. Bode and J. Wöhnert, Nat. Commun., 2018, 9, 4366 CrossRef PubMed.
- E. Musiol-kroll and W. Wohlleben, Antibiotics, 2018, 7, 62, DOI:10.3390/antibiotics7030062.
- X. Ruan, A. Pereda, D. L. Stassi, D. Zeidner, R. G. Summers, M. Jackson, A. Shivakumar, S. Kakavas, M. J. Staver, S. Donadio and L. Katz, J. Bacteriol., 1997, 179, 6416–6425 CrossRef CAS PubMed.
- M. Klaus, A. D. D'Souza, A. Nivina, C. Khosla and M. Grininger, ACS Chem. Biol., 2017, 6, 139–147, DOI:10.1021/acschembio.8b01060.
- R. Kelwick, L. Bowater, K. H. Yeoman and R. P. Bowater, FEMS Microbiol. Lett., 2015, 362, 16, DOI:10.1093/femsle/fnv129.
- M. G. Chevrette, F. Aicheler, O. Kohlbacher, C. R. Currie and M. H. Medema, Bioinformatics, 2017, 33, 3202–3210 CrossRef CAS PubMed.
- M. H. Medema, P. Cimermancic, A. Sali, E. Takano and M. A. Fischbach, PLoS Comput. Biol., 2014, 10, 12, DOI:10.1371/journal.pcbi.1004016.
- A. Wlodek, S. G. Kendrew, N. J. Coates, A. Hold, J. Pogwizd, S. Rudder, L. S. Sheehan, S. J. Higginbotham, A. E. Stanley-Smith, T. Warneck, M. Nur-E-Alam, M. Radzom, C. J. Martin, L. Overvoorde, M. Samborskyy, S. Alt, D. Heine, G. T. Carter, E. I. Graziani, F. E. Koehn, L. McDonald, A. Alanine, R. M. Rodríguez Sarmiento, S. K. Chao, H. Ratni, L. Steward, I. H. Norville, M. Sarkar-Tyson, S. J. Moss, P. F. Leadlay, B. Wilkinson and M. A. Gregory, Nat. Commun., 2017, 8, 1206 CrossRef PubMed.
- H. G. Menzella, R. Reid, J. R. Carney, S. S. Chandran, S. J. Reisinger, K. G. Patel, D. A. Hopwood and D. V. Santi, Nat. Biotechnol., 2005, 23, 1171 CrossRef CAS PubMed.
- T. Weber and H. U. Kim, Synth. Syst. Biotechnol., 2016, 1, 69–79 CrossRef PubMed.
- K. Blin, V. Pascal Andreu, E. L. C. de los Santos, F. Del Carratore, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2018, 47, 625–630 CrossRef PubMed.
- M. Hadjithomas, I.-M. A. Chen, K. Chu, A. Ratner, K. Palaniappan, E. Szeto, J. Huang, T. B. K. Reddy, P. Cimermancic, M. A. Fischbach, N. N. Ivanova, V. M. Markowitz, N. C. Kyrpides and A. Pati, mBio, 2015, 6, e00932 CrossRef CAS PubMed.
- M. H. Medema, R. Kottmann, P. Yilmaz, M. Cummings, J. B. Biggins, K. Blin, I. de Bruijn, Y. H. Chooi, J. Claesen, R. C. Coates, P. Cruz-Morales, S. Duddela, S. Dusterhus, D. J. Edwards, D. P. Fewer, N. Garg, C. Geiger, J. P. Gomez-Escribano, A. Greule, M. Hadjithomas, A. S. Haines, E. J. N. Helfrich, M. L. Hillwig, K. Ishida, A. C. Jones, C. S. Jones, K. Jungmann, C. Kegler, H. U. Kim, P. Kotter, D. Krug, J. Masschelein, A. V. Melnik, S. M. Mantovani, E. A. Monroe, M. Moore, N. Moss, H.-W. Nutzmann, G. Pan, A. Pati, D. Petras, F. J. Reen, F. Rosconi, Z. Rui, Z. Tian, N. J. Tobias, Y. Tsunematsu, P. Wiemann, E. Wyckoff, X. Yan, G. Yim, F. Yu, Y. Xie, B. Aigle, A. K. Apel, C. J. Balibar, E. P. Balskus, F. Barona-Gomez, A. Bechthold, H. B. Bode, R. Borriss, S. F. Brady, A. A. Brakhage, P. Caffrey, Y.-Q. Cheng, J. Clardy, R. J. Cox, R. De Mot, S. Donadio, M. S. Donia, W. A. van der Donk, P. C. Dorrestein, S. Doyle, A. J. M. Driessen, M. Ehling-Schulz, K.-D. Entian, M. A. Fischbach, L. Gerwick, W. H. Gerwick, H. Gross, B. Gust, C. Hertweck, M. Hofte, S. E. Jensen, J. Ju, L. Katz, L. Kaysser, J. L. Klassen, N. P. Keller, J. Kormanec, O. P. Kuipers, T. Kuzuyama, N. C. Kyrpides, H.-J. Kwon, S. Lautru, R. Lavigne, C. Y. Lee, B. Linquan, X. Liu, W. Liu, A. Luzhetskyy, T. Mahmud, Y. Mast, C. Mendez, M. Metsa-Ketela, J. Micklefield, D. A. Mitchell, B. S. Moore, L. M. Moreira, R. Muller, B. A. Neilan, M. Nett, J. Nielsen, F. O'Gara, H. Oikawa, A. Osbourn, M. S. Osburne, B. Ostash, S. M. Payne, J.-L. Pernodet, M. Petricek, J. Piel, O. Ploux, J. M. Raaijmakers, J. A. Salas, E. K. Schmitt, B. Scott, R. F. Seipke, B. Shen, D. H. Sherman, K. Sivonen, M. J. Smanski, M. Sosio, E. Stegmann, R. D. Sussmuth, K. Tahlan, C. M. Thomas, Y. Tang, A. W. Truman, M. Viaud, J. D. Walton, C. T. Walsh, T. Weber, G. P. van Wezel, B. Wilkinson, J. M. Willey, W. Wohlleben, G. D. Wright, N. Ziemert, C. Zhang, S. B. Zotchev, R. Breitling, E. Takano and F. O. Glockner, Nat. Chem. Biol., 2015, 11, 625–631 CrossRef CAS PubMed.
- M. Pupin, Q. Esmaeel, A. Flissi, Y. Dufresne, P. Jacques and V. Leclere, Synth. Syst. Biotechnol., 2016, 1, 89–94 CrossRef CAS PubMed.
- E. Ricart, V. Leclère, A. Flissi, M. Mueller, M. Pupin and F. Lisacek, J. Cheminf., 2019, 11, 13 Search PubMed.
- J. J. J. van der Hooft, J. Wandy, M. P. Barrett, K. E. V. Burgess and S. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13738–13743 CrossRef PubMed.
- M. H. Medema, E. Takano and R. Breitling, Mol. Biol. Evol., 2013, 30, 1218–1223 CrossRef CAS PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef CAS PubMed.
- J. Navarro-Muñoz, N. Selem-Mojica, M. Mullowney, S. Kautsar, J. Tryon, E. Parkinson, E. De Los Santos, M. Yeong, P. Cruz-Morales, S. Abubucker, A. Roeters, W. Lokhorst, A. Fernandez-Guerra, L. Teresa Dias Cappelini, R. Thomson, W. Metcalf, N. Kelleher, F. Barona-Gomez and M. H. Medema, bioRxiv, 2018, 445270, DOI:10.1101/445270.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. a. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. a. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
- T. Leao, G. Castelão, A. Korobeynikov, E. A. Monroe, S. Podell, E. Glukhov, E. E. Allen, W. H. Gerwick and L. Gerwick, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3198–3203 CrossRef CAS PubMed.
- P. P. K. Zin, G. Williams and D. Fourches, J. Cheminf., 2018, 10, 53 Search PubMed.
- K. Blin, T. Wolf, M. G. Chevrette, X. Lu, C. J. Schwalen, S. A. Kautsar, H. G. Suarez Duran, E. L. C. de Los Santos, H. U. Kim, M. Nave, J. S. Dickschat, D. A. Mitchell, E. Shelest, R. Breitling, E. Takano, S. Y. Lee, T. Weber and M. H. Medema, Nucleic Acids Res., 2017, 45, W36–W41 CrossRef CAS PubMed.
- C. W. Johnston, M. A. Skinnider, N. J. Merwin and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef PubMed.
- H. E. Pence and A. Williams, J. Chem. Educ., 2010, 87, 1123–1124 CrossRef CAS.
- M. Hattori, N. Tanaka, M. Kanehisa and S. Goto, Nucleic Acids Res., 2010, 38, W652–W656 CrossRef CAS PubMed.
- R. G. Linington, NP Atlas, https://www.npatlas.org Search PubMed.
- S. Farag, R. M. Bleich, E. A. Shank, O. Isayev, A. A. Bowers and A. Tropsha, Bioinformatics, 2019, btz127, DOI:10.1093/bioinformatics/btz127.
- W. Wriggers, S. Chakravarty and P. A. Jennings, Biopolymers, 2005, 80, 736–746 CrossRef CAS PubMed.
- D. P. Lawrence, S. Kroken, B. M. Pryor and A. E. Arnold, PLoS One, 2011, 6, e28231 CrossRef CAS PubMed.
- C. Elena, P. Ravasi, M. E. Castelli, S. Peirú and H. G. Menzella, Front. Microbiol., 2014, 5, 21 CrossRef PubMed.
- F. F. V. Chevance, S. Le Guyon and K. T. Hughes, PLoS Genet., 2014, 10, e1004392 CrossRef PubMed.
- D. Papamichail, H. Liu, V. Machado, N. Gould, J. R. Coleman and G. Papamichail, IEEE/ACM Trans. Comput. Biol. Bioinf., 2018, 15, 452–459 CAS.
- Z. Zhou, J. R. Lai and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11621–11626 CrossRef CAS PubMed.
- P. Gainza, K. E. Roberts, I. Georgiev, R. H. Lilien, D. A. Keedy, C.-Y. Chen, F. Reza, A. C. Anderson, D. C. Richardson, J. S. Richardson and B. R. Donald, Methods Enzymol., 2013, 523, 87–107 CAS.
- C.-Y. Chen, I. Georgiev, A. C. Anderson and B. R. Donald, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3764–3769 CrossRef CAS PubMed.
- R. Verma, U. Schwaneberg and D. Roccatano, Comput. Struct. Biotechnol. J., 2012, 2, e201209008 CrossRef PubMed.
- M. S. Packer and D. R. Liu, Nat. Rev. Genet., 2015, 16, 379 CrossRef CAS PubMed.
- R. E. Cobb, R. Chao and H. Zhao, AIChE J., 2013, 59, 1432–1440 CrossRef CAS PubMed.
- C. A. Voigt, C. Martinez, Z.-G. Wang, S. L. Mayo and F. H. Arnold, Nat. Struct. Biol., 2002, 9, 553 CAS.
- L. Sumbalova, J. Stourac, T. Martinek, D. Bednar and J. Damborsky, Nucleic Acids Res., 2018, 46, W356–W362 CrossRef CAS PubMed.
- B. A. Amrein, F. Steffen-Munsberg, I. Szeler, M. Purg, Y. Kulkarni and S. C. L. Kamerlin, IUCrJ, 2017, 4, 50–64 CrossRef CAS PubMed.
- L. J. McGuffin, J. D. Atkins, B. R. Salehe, A. N. Shuid and D. B. Roche, Nucleic Acids Res., 2015, 43, W169–W173 CrossRef CAS PubMed.
- M. Kallberg, H. Wang, S. Wang, J. Peng, Z. Wang, H. Lu and J. Xu, Nat. Protoc., 2012, 7, 1511–1522 CrossRef CAS PubMed.
- J. Söding, A. Biegert and A. N. Lupas, Nucleic Acids Res., 2005, 33, W244–W248 CrossRef PubMed.
- A. Fiser and A. B. T.-M. in E. Šali, in Macromolecular Crystallography, Part D, Academic Press, 2003, vol. 374, pp. 461–491 Search PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- D. E. Kim, D. Chivian and D. Baker, Nucleic Acids Res., 2004, 32, W526–W531 CrossRef CAS PubMed.
- A. Waterhouse, C. Rempfer, F. T. Heer, G. Studer, G. Tauriello, L. Bordoli, M. Bertoni, R. Gumienny, R. Lepore, S. Bienert, T. A. P. de Beer and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- C. Zhang, S. M. Mortuza, B. He, Y. Wang and Y. Zhang, Proteins, 2018, 86(Suppl 1), 136–151 CrossRef CAS PubMed.
- C. H. Eng, T. W. H. Backman, C. B. Bailey, C. Magnan, H. García Martín, L. Katz, P. Baldi and J. D. Keasling, Nucleic Acids Res., 2018, 46, D509–D515 CrossRef CAS PubMed.
- P. Carbonell, P. Parutto, C. Baudier, C. Junot and J.-L. Faulon, ACS Synth. Biol., 2014, 3, 565–577 CrossRef CAS PubMed.
- M. J. Czar, Y. Cai and J. Peccoud, Nucleic Acids Res., 2009, 37, W40–W47 CrossRef CAS PubMed.
- S. K. Burley, H. M. Berman, G. J. Kleywegt, J. L. Markley, H. Nakamura and S. Velankar, Methods Mol. Biol., 2017, 1607, 627–641 CrossRef CAS PubMed.
- T. Izoré and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 1120–1139 RSC.
- T. Awakawa, T. Fujioka, L. Zhang, S. Hoshino, Z. Hu, J. Hashimoto, I. Kozone, H. Ikeda, K. Shin-Ya, W. Liu and I. Abe, Nat. Commun., 2018, 9, 3534 CrossRef PubMed.
- S. Mori, A. H. Pang, T. A. Lundy, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Nat. Chem. Biol., 2018, 14, 428–430 CrossRef CAS PubMed.
- J. Moult, K. Fidelis, A. Kryshtafovych, T. Schwede and A. Tramontano, Proteins, 2018, 86(Suppl 1), 7–15 CrossRef CAS PubMed.
- F. Goedegebuur, L. Dankmeyer, P. Gualfetti, S. Karkehabadi, H. Hansson, S. Jana, V. Huynh, B. R. Kelemen, P. Kruithof, E. A. Larenas, P. J. M. Teunissen, J. Ståhlberg, C. M. Payne, C. Mitchinson and M. Sandgren, J. Biol. Chem., 2017, 292, 17418–17430 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed.
- R. D. Finn, J. Clements and S. R. Eddy, Nucleic Acids Res., 2011, 39, W29–W37 CrossRef CAS PubMed.
- A. Hagen, S. Poust, T. de Rond, J. L. Fortman, L. Katz, C. J. Petzold and J. D. Keasling, ACS Synth. Biol., 2016, 5, 21–27 CrossRef CAS PubMed.
- B. Delépine, T. Duigou, P. Carbonell and J. Faulon, Metab. Eng., 2018, 45, 158–170 CrossRef PubMed.
- B. Hartnett, C. Gustafsson, J. Peccoud and Y. Cai, Bioinformatics, 2007, 23, 2760–2767 CrossRef PubMed.
- A. Coll, M. L. Wilson, K. Gruden and J. Peccoud, Methods Mol. Biol., 2016, 1482, 219–232 CrossRef CAS PubMed.
- Z. Tan, J. M. Clomburg and R. Gonzalez, ACS Synth. Biol., 2018, 7, 1886–1896 CrossRef CAS PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. Filsinger Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS PubMed.
- X. Luo, M. A. Reiter, L. d'Espaux, J. Wong, C. M. Denby, A. Lechner, Y. Zhang, A. T. Grzybowski, S. Harth, W. Lin, H. Lee, C. Yu, J. Shin, K. Deng, V. T. Benites, G. Wang, E. E. K. Baidoo, Y. Chen, I. Dev, C. J. Petzold and J. D. Keasling, Nature, 2019, 567, 123–126 CrossRef CAS PubMed.
- P. P. Peralta-Yahya, F. Zhang, S. B. del Cardayre and J. D. Keasling, Nature, 2012, 488, 320 CrossRef CAS PubMed.
- D. E. Cameron, C. J. Bashor and J. J. Collins, Nat. Rev. Microbiol., 2014, 12, 381 CrossRef CAS PubMed.
- J. C. Blain and J. W. Szostak, Annu. Rev. Biochem., 2014, 83, 615–640 CrossRef CAS PubMed.
- P. Carbonell, A. Currin, A. J. Jervis, N. J. W. Rattray, N. Swainston, C. Yan, E. Takano and R. Breitling, Nat. Prod. Rep., 2016, 33, 925–932 RSC.
- E. Kim, B. S. Moore and Y. J. Yoon, Nat. Chem. Biol., 2015, 11, 649 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |