
Passivation of the grain boundaries of CH3NH3PbI3 using carbon quantum dots for highly efficient perovskite solar cells with excellent environmental stability†
Qiang
Guo‡
a,
Fanglong
Yuan‡
b,
Bing
Zhang‡
a,
Shijie
Zhou
a,
Jin
Zhang
c,
Yiming
Bai
a,
Louzhen
Fan
*b,
Tasawar
Hayat
d,
Ahmed
Alsaedi
d and
Zhan'ao
Tan
 *ac
*ac
aState Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing 102206, China. E-mail: tanzhanao@mail.buct.edu.cn
bDepartment of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: lzfan@bnu.edu.cn
cBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China
dNAAM Research Group, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 20th November 2018
Abstract
Organic–inorganic hybrid perovskites are prone to defect formation due to iodine and methylamine ion/defect migration, leading to the formation of lots of defects at the perovskite surface and grain boundaries. Passivation of the defects is an effective method to improve the power conversion efficiency (PCE) and stability of perovskite solar cells (PSCs). To achieve stable passivation, the interaction between the perovskite and additive materials should be taken into consideration. In this work, we for the first time introduced carbon quantum dots (CQDs) as an additive for the stabilization of MAPbI3via passivation of the grain boundaries of the perovskite. Because the carboxylic groups, hydroxyl groups and amino-groups on the edge of CQDs can bond with the uncoordinated Pb in MAPbI3, strong and stable interactions between the perovskite and CQDs can be generated, inducing a lower trap-state density and better optoelectronic properties. The typical PCE of the PSCs based on CQD modified MAPbI3 films increases from 17.59% to 18.81% and the PCE of the optimized champion PSCs reaches 19.38%. Furthermore, the hydrophobic CQD molecules can block the contact between water and MAPbI3, and even if the CQD modified perovskite is kept under ambient atmosphere without controlling the humidity for 4 months, the MAPbI3 film still retained its original black color.
Introduction
Due to the superior optoelectronic properties of perovskite materials and unremitting efforts of researchers from the fields of materials, chemistry and physics, rapid progress has been made in organic–inorganic hybrid perovskite solar cells (PSCs). The certified best research-cell power conversion efficiency (PCE) reaches 23.3%,1 which is even higher than that of commercialized solar cells such as multicrystalline Si (22.3%), CdTe (22.1%), and CIGS (22.6%). Although the efficiency question has been answered, there still remain big issues in stability and cost that must be addressed before commercialization of the PSCs.A high-quality perovskite film with great crystallization and a low trap-state density is a primary requirement for high efficiency PSCs. Since the all-solid-state PSCs reported by Nam-Gyu Park et al. in 2012,2 a lot of methods such as solution spin-coating,3–5 vapor assisted deposition6–8 and an anti-solvent assisted process9–11 have been developed to improve the crystallization of the perovskite. However, organohalide perovskites are prone to defect formation, and lots of defects are found at the perovskite surface and grain boundaries, due to the iodine and methylamine ion/defect migration.12 Defects on the perovskite surface and at grain boundaries inevitably introduce localized trap states, which could act as a recombination center of the photogenerated carriers, leading to unsatisfactory photovoltaic performance. Therefore, passivation of the defect is an effective method to improve the PCE and stability of PSCs.
Excess PbI2 or CH3CH3I (MAI) has been used to passivate or heal the defects at the grain boundaries for suppressing nonradiative charge recombination.13–18 Besides the passivation by introducing intrinsic ions of perovskites, fullerene based electron acceptors deposited on the perovskites can simultaneously passivate the charge trap states and transport electrons.19 Recently, contact-passivation by using a chlorine-capped TiO2 nanocrystal film has also been performed to reduce the interfacial recombination and to enhance the interface binding for large dimensional PSCs.20 Lewis bases thiophene and pyridine are used to treat the perovskite crystal surfaces, and a significant reduction of the non-radiative electron–hole recombination can be observed due to the electronic passivation of the uncoordinated Pb atoms within the crystals.21 As a guidance for this approach, various semiconductor polymers containing electron-rich atoms, such as S, N, and O, have been chosen to passivate perovskite films, and this has been proven to be a promising strategy to improve the performance of PSCs.22–24
To achieve stable passivation, the interaction between the perovskite and additive materials should be taken into consideration. CQDs exhibit unique quantum confinement and surface and edge effects, which can be easily controlled by tailoring the size, shape and heteroatom doping and modifying the surface and edge of CQDs.25,26 Various functional groups such as carbonyl, hydroxyl, carboxyl, and epoxy groups can be found on the surfaces of CQDs due to the solution based chemical synthesis routes, which makes it easy to functionalize them with various organic, polymeric, or biological species.27,28 In PSCs, an ultrathin CQD layer or a graphene quantum dot layer has been inserted between the perovskite and TiO2.29,30 By adding a CQD layer, the electron mobility and the electron extraction ability of the electron transport layer can be greatly improved. CQDs have also been used as sensitizers for all-inorganic CsPbBr3 inverse opal PSCs, which enhance light absorption and facilitate charge transfer.31
In this work, we for the first time introduced CQDs as an additive for the stabilization of MAPbI3via passivating the grain boundaries of the perovskite. Strong and stable interactions between the uncoordinated Pb atoms in MAPbI3 and the carboxylic groups, hydroxyl groups and amino-groups on the edge of CQDs were detected, which could induce a lower trap-state density and better optoelectrical properties. Furthermore, the hydrophobic CQD molecules can attach on the MAPbI3 surface to block the contact between water and MAPbI3, thus enhancing the stability of the perovskite. The PSCs with an appropriate concentration of CQDs exhibit more superior photovoltaic performance and long-time stability.
Experimental section
Materials
Fluorine doped tin oxide (FTO) glass (15 Ω sq−1) was purchased from Wuhan Geao company (China). 75% titanium (diisopropoxide) bis(2,4-pentanedionate) (TIPD) isopropanol solution, dimethyl sulfoxide (DMSO, extra dry 99.7+%) and N,N-dimethylformamide (DMF, extra dry 99.7+%) were purchased from Alfa Aesar. Extra dry isopropyl alcohol, ethanol and terpineol were bought from Acros Organics. 30 nm particle paste of mesoporous TiO2 was bought from Dyesol. PbI2, CH3NH3I, 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9-spirobifluorene (spiro-OMeTAD), 4-tertbutylpyridine (TBP) and lithium bis(trifluoromethanesulfonyl)imide (Li-TFSI) for perovskite film and hole transport layer fabrication were purchased from Xi'an Polymer Light Technology Corp.Synthesis of CQDs
The CQDs were synthesized according to our previously reported solvothermal treatment method.25 Firstly, 100 mg of citric acid and 20 mg of 1,5-diaminonaphthalene (DAN) were dissolved in 15 mL ethanol, and the solution was then transferred into a 25 mL poly(tetrafluoroethylene) (Teflon)-lined autoclave and heated at 200 °C for 4 h for the formation of CQDs. After the reaction, the reactors were naturally cooled down to room temperature, the solution was diluted with deionized water, and the supernatant was collected by centrifugation. Finally, the solution was subjected to dialysis for two days in order to obtain the CQDs.Precursor preparation
The pure PbI2 precursor was obtained by dissolving 1.3 M PbI2 in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF mixed solvent for 5 hours. For the PbI2 precursor of the CQD modified MAPbI3 film, different amounts of CQDs were added to the PbI2 precursor to obtain solutions with different concentrations of CQDs. MAI was dissolved in isopropanol with a concentration of 50 mg mL−1.
:DMF mixed solvent for 5 hours. For the PbI2 precursor of the CQD modified MAPbI3 film, different amounts of CQDs were added to the PbI2 precursor to obtain solutions with different concentrations of CQDs. MAI was dissolved in isopropanol with a concentration of 50 mg mL−1.
Device fabrication
The patterned FTO glass was ultrasonically cleaned twice in succession with detergent, water, deionized water and ethanol. The cleaned FTO glass was blown dry with nitrogen and then heated to 450 °C. Then, a compact TiO2 (c-TiO2) layer was fabricated by spraying 10 mL of TIPD/ethanol (1:9) solution onto the heated FTO glass and sintered at 450 °C for 30 minutes. 100–150 nm mesoporous TiO2 (mass ratio of TiO2:ethanol:terpineol is 1:7.5:0.75) was spin-coated on c-TiO2 at 5000 rpm for 30 s, the substrate was dried at 125 °C for 10 min and then the organic part was removed at 500 °C for 30 min. The pure PbI2 precursor and different CQD added PbI2 precursors were spin-coated on the TiO2 substrate at 3000 rpm for 30 s after the substrate cooled down to room temperature. Subsequently, the MAI solution was spin-coated on the as-prepared PbI2 film in a glove-box and then annealed at 100 °C for 30 min in atmospheric environment with a RH less than 30% to form a MAPbI3 film. The spiro-OMeTAD solution (72.3 mg of spiro-OMeTAD, 29 μL of TBP and 17.5 μL of Li-TFSI (520 mg mL−1 in acetonitrile) dissolved in 1 mL of chlorobenzene) was spin-coated on the perovskite film at 3000 rpm for 30 s. Finally, 100 nm Ag was thermally evaporated on spiro-OMeTAD at a pressure of 3 × 10−4 Pa.
Instrumentation and characterization
TEM images were obtained by JEOL JEM 2100 transmission electron microscopy. SEM was conducted using a Hitachi S4800 at acceleration voltages of 2.5 kV and 5 kV. XRD was measured using a D8 Advance (Bruker) diffractometer with an X-ray tube for Cu Kα radiation (λ = 0.1542 nm). The PL of the MAPbI3 film was acquired using a FluoroLog-3 modular spectrofluorometer from Horiba Scientific and the samples for the PL test were excited using a 500 nm laser. A steady/transient state fluorescence spectrometer F900 (Edinburgh Instruments) was used for TRPL measurements. UV-vis absorption spectra of the perovskite films were recorded on a Shimadzu UV-2450. FTIR spectra were recorded using a Nicolet 380 spectrograph. The pristine MAPbI3 film, CQD modified MAPbI3 film and CQD added PbI2 films for FTIR test were deposited on the KBr substrate. J–V curves of the PSCs were obtained using a computer-controlled Keithley 2400 source meter under standard one sun AM1.5G (100 mW cm−2) illumination. The active area of the device is 4 mm2. The EQE of the PSCs was tested using QE-R systems (Enli Tech.).Density functional theory (DFT) calculations
The structure of the CQDs was firstly optimized using Gaussian 09 based on Hartree-Fock (HF) with basis sets 6-31G*. Then first-principles calculation was performed using a Vienna ab initio simulation package (VASP) based on DFT.32,33 The generalized-gradient approximation (GGA)-Perdew–Burke–Ernzerhof (PBE) functional of the augmented plane wave was applied. A cutoff energy of 500 eV for the plane-wave basis set and a Monkhorst–Pack mesh of 1 × 3 × 2 for the Brillouin zone integration were employed. A vacuum layer of 10 Å was adopted in the calculation. The structure is fully relaxed until the maximum Hellmann–Feynman forces acting on each atom is less than 0.01 eV Å−1. The binding energy Ebinding between CQDs and MAPbI3 is defined as Ebinding = E(MAPbI3+CQDs) − E(MAPbI3) − E(CQDs), where E(MAPbI3+CQDs) is the total energy of MAPbI3 with absorbed CQDs and E(MAPbI3) and E(CQDs) are the total energies of pristine MAPbI3 and isolated CQDs, respectively.Results and discussion
The CQDs are synthesized according to our previous literature report,25 and the structure of the CQDs is illustrated in Fig. 1a. The diameter of the CQDs is 3–5 nm according to the transmission electron microscopy (TEM) image (Fig. 1b). To investigate the effect of the CQDs on the morphology of the MAPbI3 film, scanning electron microscopy (SEM) of the MAPbI3 films with different concentrations of CQDs is performed. Fig. 1c shows the SEM image of the pristine MAPbI3 film, where big gaps between the perovskite grains can be observed. On adding 0.02 mg mL−1 of CQDs into the PbI2 precursor solution, the obtained MAPbI3 film (Fig. 1d) demonstrates much more compact crystal grains and the gaps between the grains become much smaller than that of the pristine MAPbI3 film. As the concentration of the CQDs increases to 0.05 mg mL−1 (Fig. 1e), the gaps between the perovskite grains disappear and the grain boundaries can be clearly identified, which can also be verified from the cross-section SEM image as shown in Fig. 1h. Pristine MAPbI3 and MAPbI3 with 0.02 mg mL−1 and 0.05 mg mL−1 CQDs show similar grain size distribution. On further increasing the concentration of CQDs to 0.1 (Fig. 1f) and 0.25 mg mL−1 (Fig. 1g), the grain size of MAPbI3 is sharply decreased. The crystallization of MAPbI3 is obviously damaged when the concentration of CQDs reaches 0.25 mg mL−1 (Fig. 1g). To investigate the reason for the bad morphology of the perovskite film with excess CQDs, UV-visible (UV-vis) absorption spectroscopy and X-ray diffraction (XRD) characterization of the PbI2 film with different concentrations of CQDs were conducted. As shown in Fig. S1,† the intensity of the absorption shoulder (around 490 nm) and XRD peaks (12.7°, 25.6°, 38.7°, and 52.4°) of PbI2 are significantly decreased as the concentration of CQDs increases to 0.1 and 0.25 mg mL−1, implying that the PbI2 crystal growth is greatly inhibited. The reduction of the XRD peak of PbI2 is caused by the interaction between CQDs and PbI2, which can be validated using the Fourier transform infrared spectroscopy (FTIR) spectra of CQDs and the PbI2 film with CQDs (10 mg mL−1). As shown in Fig. S2,† the CQDs show C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations at 1713 cm−1, while they shifts to 1700 cm−1 for the PbI2 film with the CQD additive. The shift of the CO stretching vibrations indicates that the PbI2 film interacts with CQDs. Besides, from the SEM images of PbI2 films with different concentrations of CQDs (Fig. S3†), we can see that the number of PbI2 particles increases and the size of the particles decreases with higher concentrations of CQDs (0.1 and 0.25 mg mL−1). These phenomena indicate that the addition of excessive CQDs in the PbI2 film would lead to greater and faster nucleation of PbI2 and inhibition of PbI2 crystal growth. It has been reported that PbI2 acts as a framework and provides “nucleation” centers for the subsequent crystallization of the perovskite film.6,34 Thus, the excess nuclei in the PbI2 film with 0.1 and 0.25 mg mL−1 CQDs accelerate the nucleation of the perovskite and hinder the growth of the perovskite grain, resulting in the bad morphology of the perovskite film.
O stretching vibrations at 1713 cm−1, while they shifts to 1700 cm−1 for the PbI2 film with the CQD additive. The shift of the CO stretching vibrations indicates that the PbI2 film interacts with CQDs. Besides, from the SEM images of PbI2 films with different concentrations of CQDs (Fig. S3†), we can see that the number of PbI2 particles increases and the size of the particles decreases with higher concentrations of CQDs (0.1 and 0.25 mg mL−1). These phenomena indicate that the addition of excessive CQDs in the PbI2 film would lead to greater and faster nucleation of PbI2 and inhibition of PbI2 crystal growth. It has been reported that PbI2 acts as a framework and provides “nucleation” centers for the subsequent crystallization of the perovskite film.6,34 Thus, the excess nuclei in the PbI2 film with 0.1 and 0.25 mg mL−1 CQDs accelerate the nucleation of the perovskite and hinder the growth of the perovskite grain, resulting in the bad morphology of the perovskite film.
 | ||
| Fig. 1 (a) Molecular structure and (b) TEM image of CQDs: top-view SEM images of (c) pristine MAPbI3 film and MAPbI3 films with (d) 0.02, (e) 0.05, (f) 0.1, and (g) 0.25 mg mL−1 CQDs deposited on the FTO/c-TiO2/mp-TiO2 substrate; (h) cross-section SEM images of the MAPbI3 films with 0.05 mg mL−1 CQDs deposited on the FTO/c-TiO2/mp-TiO2 substrate. | ||
To further study whether CQDs can affect the crystal structure of the MAPbI3 perovskite, XRD characterization was conducted. The XRD patterns of the MAPbI3 film and MAPbI3 films with different concentrations (<0.1 mg mL−1) of CQDs are presented in Fig. 2a. The strong diffraction peaks of the perovskite at 14.1°, 28.4°, 31.9° and 40.7° can be observed in the XRD patterns of the MAPbI3 films without CQDs.35,36 The same perovskite peaks can also be identified for MAPbI3 films with low concentrations (<0.1 mg mL−1) of CQDs, and there are no extra peaks in the XRD curves of the MAPbI3 films with CQDs, which means that the CQDs do not affect the perovskite crystal structure and the CQDs present in the MAPbI3 films are in the amorphous state. Fig. 2b illustrates the normalized XRD curves from 13.7° to 14.4°. We can see that the XRD patterns of the pristine MAPbI3 film and MAPbI3 films with 0.02 and 0.05 mg mL−1 CQDs exhibit almost equivalent full width at half maximum (FWHM) values, while the FWHM becomes broader in the XRD patterns of the MAPbI3 films with 0.1 mg mL−1 CQDs. The wider FWHM indicates that the grain size is smaller in the perovskite film with 0.1 mg mL−1 CQDs, which is in good agreement with the result of the SEM images.37,38
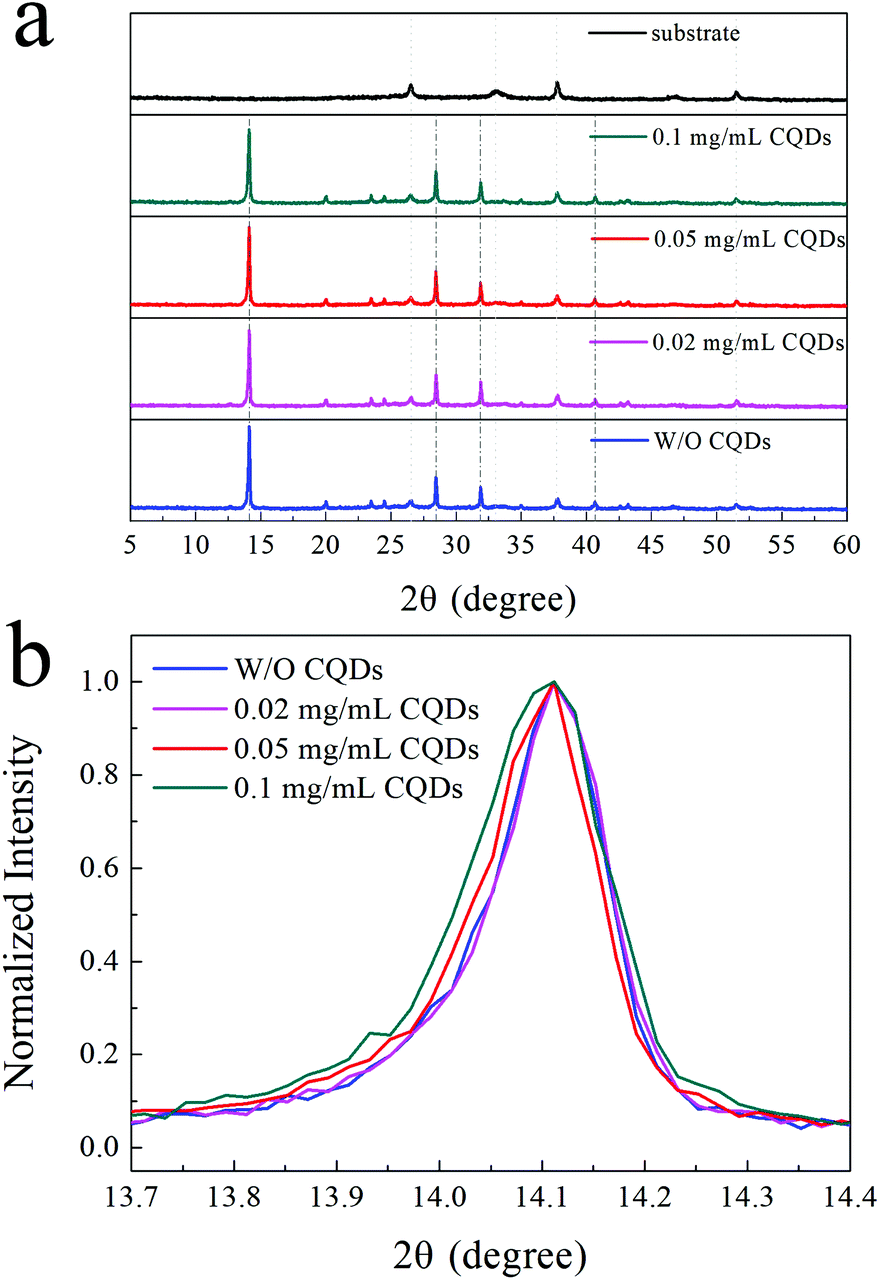 | ||
| Fig. 2 (a) XRD patterns of the pristine MAPbI3 film and MAPbI3 films with 0.02, 0.05 and 0.1 mg mL−1 CQDs; (b) normalized XRD patterns ((110) lattice planes) of the pristine MAPbI3 film and MAPbI3 films with 0.02, 0.05 and 0.1 mg mL−1 CQDs. | ||
According to the results of XRD and SEM, we can conclude that the CQD additive does not change the perovskite crystal structure but affects perovskite growth. Recently, Wang et al. proposed that a single-walled carbon nanotube with an octadecylamine functional group incorporated in perovskite film fabrication can restrict the evaporation of the solvent, thus enhancing the perovskite crystal and grain size.39 In our work, due to the interaction between the functional group of CQDs and the perovskite, the CQDs are absorbed on the perovskite grain boundaries and impede the evaporation of the solvent during film formation. Besides, the interaction between PbI2 and the functional group of CQDs can also retard the perovskite growth and improve crystallization.40 So, more compact and well-crystallized perovskite films are obtained with a small amount of the CQD additive (less than 0.05 mg mL−1). However, compared to the positive effect on perovskite crystallization induced by CQDs, when excess CQD additive was introduced in perovskite film fabrication, the negative effect induced by excess PbI2 “nuclei” is more pronounced. Therefore, an appropriate amount of the CQD additive is crucial to form a compact perovskite film with enhanced crystallization.
Besides the morphology and crystallization, the photophysical properties of the perovskite film are also critical for high-performance PSCs. To investigate the photophysical properties of the MAPbI3 films with CQDs, steady-state photoluminescence (PL) and time-resolved photoluminescence (TRPL) of the MAPbI3 films were measured. According to the SEM and XRD results, the optimized concentration of CQDs is fixed at 0.05 mg mL−1 to simplify the process of analysis. As shown in Fig. 3a and b, the CQD modified MAPbI3 film shows an increased PL intensity compared to the pristine MAPbI3 film. The increased PL intensity should be attributed to the decreased nonradiative recombination caused by the defect,41 indicating that the CQDs induce passivation of the perovskite and greatly reduce the defect in the CQD modified MAPbI3 film. The same information is also reflected by the TRPL curves (Fig. 3b), and the PL decay of the pristine MAPbI3 film is faster than that of the CQD modified MAPbI3 film. By fitting the decay curves with a three-component exponential, the average PL lifetime of pristine and CQD modified MAPbI3 films are calculated as 154.35 ns and 170.48 ns, respectively (detailed fitting parameters are listed in Table S1†). To compare the trap-state density of the pristine MAPbI3 film and the CQD modified MAPbI3 film, space-charge-limited current (SCLC) devices with the structure of FTO/TiO2/perovskite/PCBM/Ag were fabricated. The current density–voltage (J–V) characteristic curves of the SCLC devices were tested in a dark environment. The trap-state density (nt) can be calculated using the following equation:
| nt = (VTFLεε0)/(eL2) |
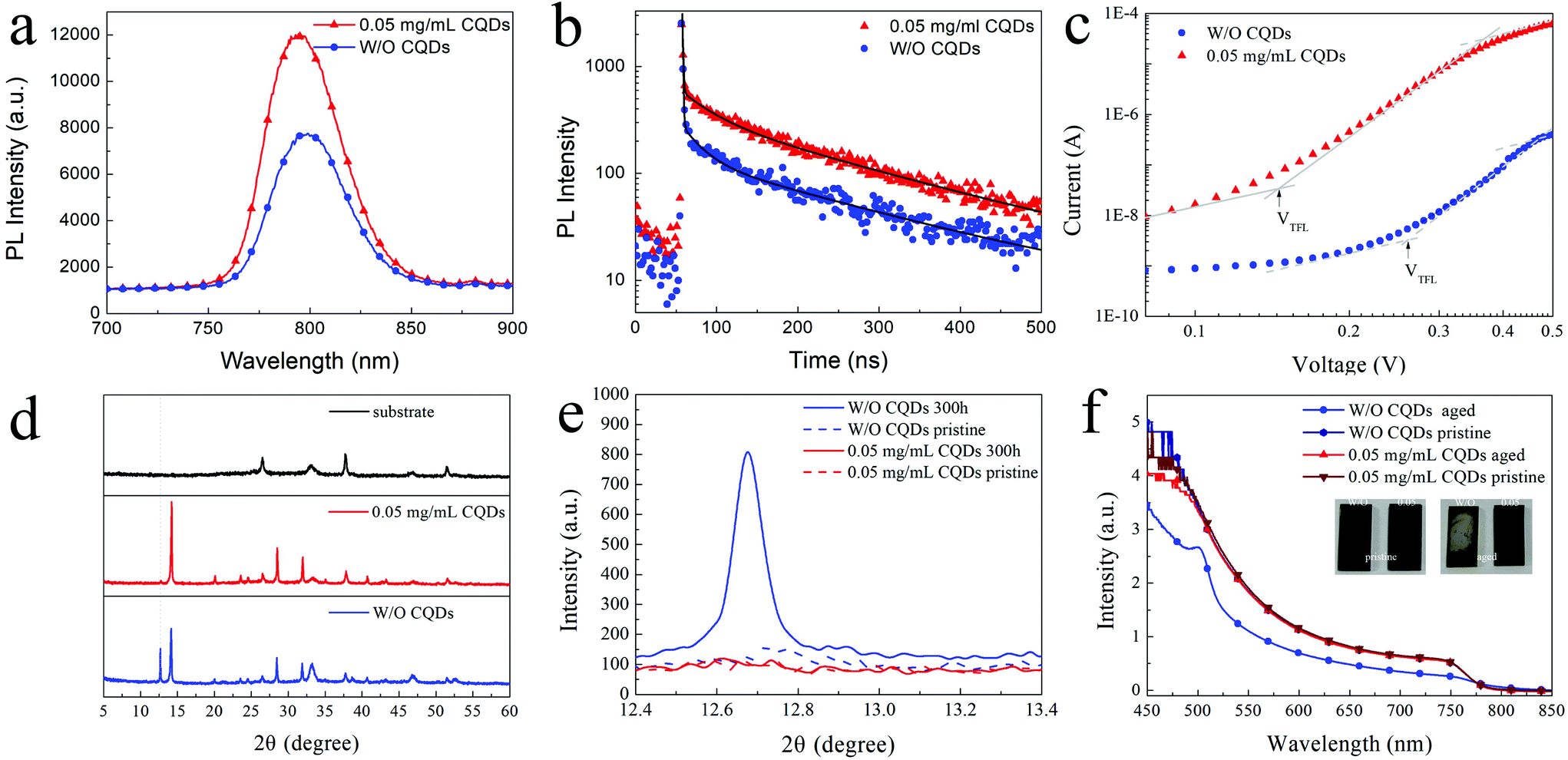 | ||
| Fig. 3 (a) PL and (b) TRPL curves of the pristine MAPbI3 film and the MAPbI3 film with 0.05 mg mL−1 CQDs deposited on the quartz substrate; (c) J–V curves of devices with the structure ITO/c-TiO2/perovskite (300 nm)/PCBM/Ag; (d) XRD patterns of the pristine MAPbI3 film and the CQD modified MAPbI3 film after exposure to ambient atmosphere for 300 hours (RH: 50%–60%); (e) XRD patterns of the initial and aged perovskite films (12.4° to 13.4°); (f) UV-vis absorption spectra of the pristine MAPbI3 film and the MAPbI3 film after exposing to ambient atmosphere without controlling the humidity for 4 months; the insets show the corresponding photographs of the films. | ||
In addition to the passivation of defects and improvement of photophysical properties, the addition of the CQDs can significantly improve the stability of the MAPbI3 film. After aging in ambient environment with a relative humidity (RH) of 50–60% for 300 hours, the CQD modified MAPbI3 film (0.05 mg mL−1) exhibits much less degradation compared to the pristine MAPbI3 film. The obvious difference appears in their XRD patterns (Fig. 3d). The strong peaks related to PbI2 (12.7°) can be observed in the XRD pattern for the aged pristine MAPbI3 film, while this signal of PbI2 cannot be detected in the XRD pattern for the aged MAPbI3 film with CQDs. Meanwhile, the intensities of the XRD peaks related to the MAPbI3 perovskite (14.1°, 28.4°, 31.9° and 40.7°) are obviously reduced for the aged pristine MAPbI3 film, revealing that many MAPbI3 crystals are decomposed into PbI2. Fig. 3e shows the XRD patterns of the initial and aged perovskite films with the 2θ range from 12.4° to 13.4°. In contrast to PbI2 significantly increasing in the pristine MAPbI3 film, the XRD curves of the initial and aged MAPbI3 films with 0.05 mg mL−1 CQDs show almost no difference. The UV-vis absorption spectra also indicate that the MAPbI3 film with CQDs shows superior ambient stability (Fig. 3f). Even when the sample is left under ambient atmosphere without controlling the humidity for 4 months, the MAPbI3 film with 0.05 mg mL−1 CQDs still retains its original black color and its UV-vis spectra show no obvious decrease in the intensity.
To explore the reasons for the passivation and improved stability of the CQD modified perovskite film, the interaction between the CQDs and MAPbI3 is detected by FTIR measurement. The FTIR spectra of CQDs, MAPbI3, and CQD modified MAPbI3 films are shown in Fig. 4a and b. The perovskite films for the FTIR test are deposited on the KBr substrate and the concentration of CQDs is increased to 10 mg mL−1 to enhance the signal. The characteristic peak of the CO bond stretching vibrations in CQDs locates at 1713 cm−1, while this characteristic peak shifts to 1705 cm−1 in the CQD modified perovskite film. The shift of this peak indicates a strong interaction between CQDs and MAPbI3, which should be caused by the lone pair electrons on the carboxylic group of CQDs delocalized to the empty orbit of Pb2+ in MAPbI3.41 In addition, the interaction between CQDs and MAPbI3 is also calculated by density functional theory (DFT). The optimized structures of CQDs and the MAPbI3 perovskite simulated with Gaussian 09 are shown in Fig. 4c. Because the ammonium groups co-exist in MAPbI3 and CQDs, it is difficult to distinguish the FTIR absorption features of the ammonium groups of MAPbI3 and CQDs. Hence, the interactions between uncoordinated Pb atoms and the amino groups of CQDs are also taken into consideration in DFT calculations. To study the effect of CQDs on MAPbI3, in the initial structure, the CQDs are put near the MAPbI3 surface, the distances between Pb1 of MAPbI3 and O1 (O of the carboxyl group) of CQDs (Fig. 4d), Pb3 of the MAPbI3 and O2 (O of the hydroxyl group) of CQDs (Fig. 4f), Pb2 of the MAPbI3 and N2 (N of the amino-group) of CQDs (Fig. 4h) and H1 of the MAPbI3 and N1 (N of the amino-group) of CQDs (Fig. 4j) are set to 3.3, 3.0, 2.9 and 2.1 Å, respectively. During the optimization processes, the CQDs are found to move closer to the perovskite surface (Fig. 4c); eventually, the distances between Pb1 and O1, Pb3 and O2, Pb2 and N2, and H1 and N1 are found to be 2.5, 2.6, 2.7 and 1.8 Å, respectively. According to the sum of van der Waals radii of ca. 3.05 Å of Pb and oxygen atoms,44 the distances of 2.5 and 2.6 Å are deemed to be close enough to form a stable Pb–O bond (Fig. 4d). The binding energies of Pb1 and O1, Pb3 and O2, Pb2 and N2, and H1 and N1 are calculated to be −2.04, −0.90, −1.16 and −1.45 eV, respectively. The first adsorption model (Pb1–O1) is the most stable adsorption pattern owing to its lowest binding energy among these four models. The negative adsorption energy indicates that the adsorption process is exothermic, i.e. the adsorption of the organic molecules on the perovskite surface is beneficial for the system stability. Because the uncoordinated Pb atoms always act as electronic trap states,45 the CQDs bonding with uncoordinated Pb atoms can passivate the trap states. Furthermore, according to Edoardo Mosconi's simulations,44,46 MAPbI3 undergoes a rapid solvation process in a liquid water environment; therefore, when multiple CQD molecules are attached on the MAPbI3 surface, they would form a barrier layer and that helps to block the contact between water and MAPbI3, which makes MAPbI3 more stable in the moisture environment.
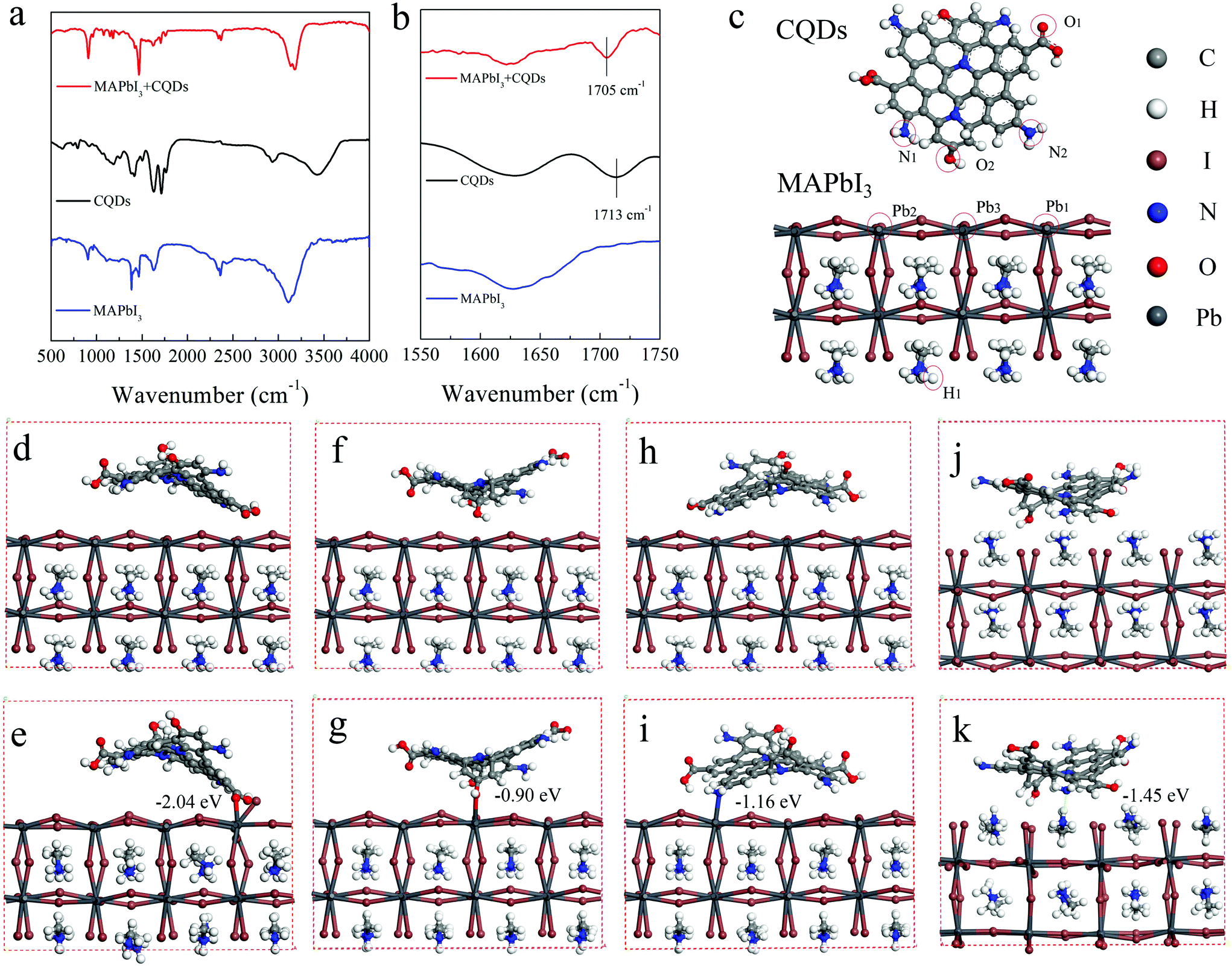 | ||
| Fig. 4 (a, b) FTIR spectra of CQDs, MAPbI3 and CQD modified MAPbI3 films; (c) the structures of CQDs and MAPbI3 optimized with Gaussian 09; initial and optimized structures of CQDs on MAPbI3 based on the (d, e) Pb1 and O1 model, (f, g) Pb3 and O2 model, (h, i) Pb2 and N2 model, and (j, k) H1 and N1 model. | ||
Due to the passivation and promoted optoelectronic properties of the CQD modified MAPbI3 film, the PSCs based on appropriate CQD modified MAPbI3 films exhibit better photovoltaic performance. The typical J–V curves of the PSCs based on the MAPbI3 film with different concentrations of CQDs are presented in Fig. 5a, and the detailed photovoltaic parameters are listed in Table 1. The photovoltaic performance distribution of 15 individual PSCs based on the pristine MAPbI3 film and 0.02, 0.05, and 0.1 mg mL−1 CQD modified MAPbI3 films are shown in Fig. S4.† The PSCs made of pristine MAPbI3 films show a PCE of 17.59% for reverse scan with a short-circuit current density (Jsc) of 21.88 mA cm−2, an open-circuit voltage (Voc) of 1.103 V and a fill factor (FF) of 72.90% (Table 1), while the PSCs based on CQD (0.02 mg mL−1) modified MAPbI3 films show an increased PCE of 18.22%. When the concentration of CQDs increases to 0.05 mg mL−1, the typical PCE is boosted to 18.81% with a Jsc of 22.19 mA cm−2, a Voc of 1.134 and an FF of 74.75% (reverse scan). The improved photovoltaic performance of devices (0.05 mg mL−1) can be attributed to the sufficient passivation of the perovskite. Finally, the PSCs with 0.05 mg mL−1 CQDs achieve a highest PCE of 19.38% (Jsc = 22.30 mA cm−2, Voc = 1.142 V and FF = 76.1%) as shown in Fig. 5b. However, on further increasing the concentration of CQDs to 0.1 mg mL−1, the device performance is obviously deteriorated, which can be ascribed to the bad morphology and crystallization of the perovskite film. Compared to the referenced device (16.31%), the typical forward scan PCE of the devices with 0.02 and 0.05 mg mL−1 CQDs increases to 16.99% and 17.66%, respectively, and then decreases to 15.21% for devices with 0.1 mg mL−1 CQD additive. The hysteresis for the 0.05 mg mL−1 CQD modified devices is the smallest among the devices. Because the defects in the perovskite layer could act as traps for charges and aggravate hysteresis,47 the reduced trap density of the 0.05 mg mL−1 CQD modified perovskite layer could be the reason for the decrease of hysteresis. Fig. 5c shows the external quantum efficiency (EQE) curves and integrated current density (Jint) from the EQE test. The Jint of PSCs without and with 0.02, 0.05 and 0.1 mg mL−1 CQDs are 20.89, 21.14, 21.82 and 20.79 mA cm−2, respectively, which are in good agreement with the Jsc from the J–V test (Fig. 5a). In addition, due to the lower trap state and more efficient charge extraction of the CQD passivated perovskite film, the photoresponse of 0.05 mg mL−1 CQD modified PSCs is much faster than that of the PSCs without CQDs, according to the steady-state output current density at the maximum power point shown in Fig. 5d. However, the PSCs with excess CQDs show worse photoresponse in that the excess CQDs increase the device series resistance, and thus hinder the charge extraction.
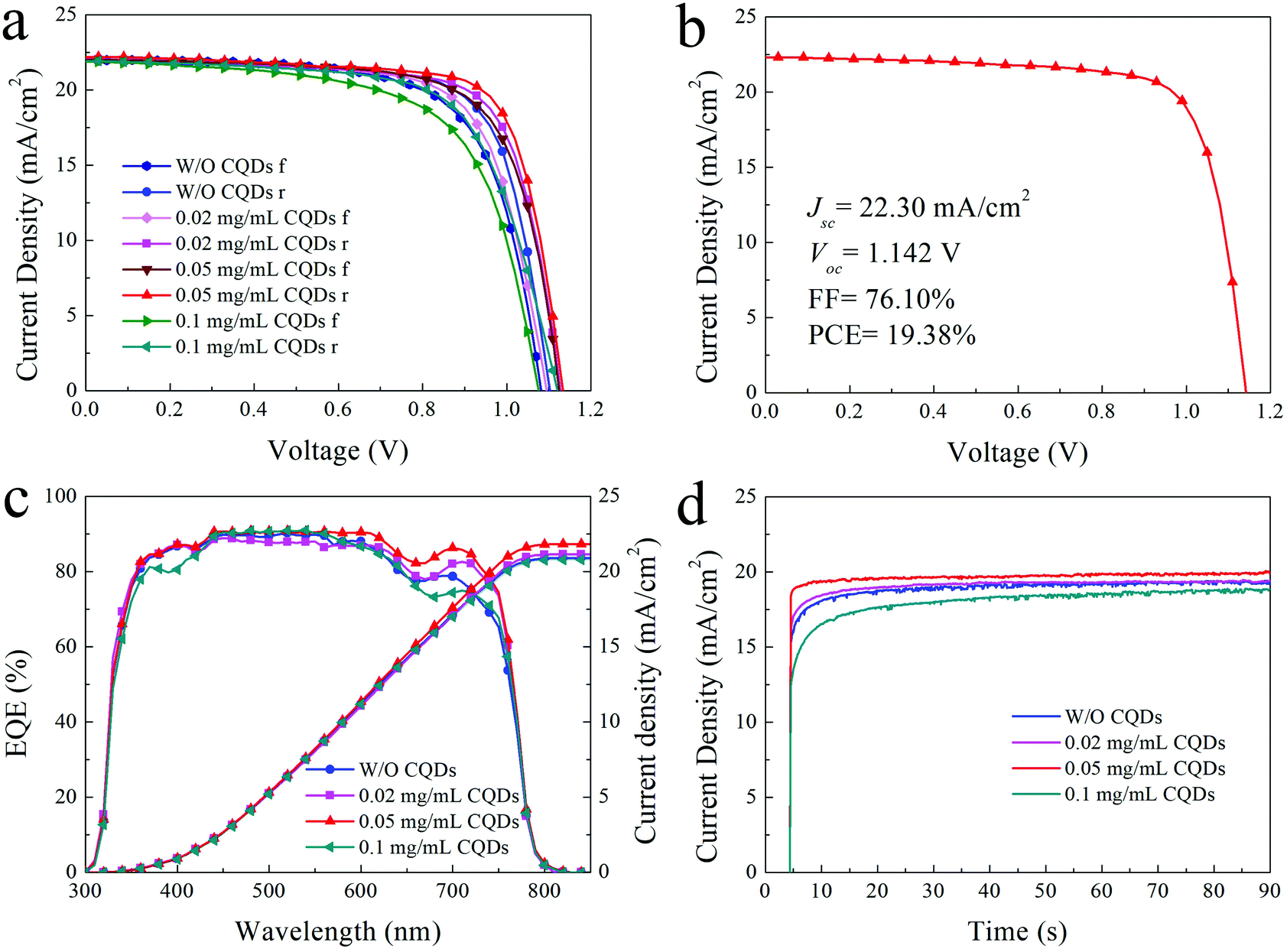 | ||
| Fig. 5 (a) Forward and reverse J–V curves of PSCs without and with 0.02, 0.05 and 0.1 mg mL−1 CQDs under AM 1.5G one sun (100 mW cm−2) illumination; (b) J–V curve of the champion device with 0.05 mg mL−1 CQDs; (c) EQE curves and Jint of PSCs without and with CQDs; (d) steady-state output current density at the maximum power point of PSCs without and with CQDs. | ||
| Scan | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | |
|---|---|---|---|---|---|
| W/O | Forward | 22.00 | 1.082 | 68.51 | 16.31 |
| Reverse | 21.88 | 1.103 | 72.90 | 17.59 | |
| 0.02 mg mL−1 CQDs | Forward | 22.08 | 1.095 | 70.26 | 16.99 |
| Reverse | 22.04 | 1.128 | 73.29 | 18.22 | |
| 0.05 mg mL−1 CQDs | Forward | 22.01 | 1.125 | 71.31 | 17.66 |
| Reverse | 22.19 | 1.134 | 74.75 | 18.81 | |
| 0.1 mg mL−1 CQDs | Forward | 21.88 | 1.076 | 64.55 | 15.21 |
| Reverse | 21.92 | 1.121 | 67.25 | 16.52 |
Finally, the long-term stability of PSCs based on the pristine MAPbI3 film and the 0.05 mg mL−1 CQD modified MAPbI3 film is investigated, and the PCE of samples stored in a N2-filled glove box and under ambient atmosphere (RH: 50%–60%) are determined. Fig. 6a and b exhibit the normalized PCE of the samples. After aging in the glove box for 1500 hours, the PCE of the PSCs based on the pristine MAPbI3 film retains 74.1% of the original value, while the PCE of PSCs fabricated with the 0.05 mg mL−1 CQD modified MAPbI3 film retains 90.1% of the original PCE. For samples stored under ambient atmosphere with a RH of 50–60% for 216 hours, the CQD modified PSCs are also more stable than the PSCs without CQDs. Since the hydrophilic salt in spiro-OMeTAD exposed to wet air would accelerate the destruction of the perovskite film, the PCE of PSCs without CQDs only retains 17.0% of the original PCE, while the CQD modified PSCs still retain 70.5% of the original PCE. Fig. 6b insets show the photographs of the corresponding devices before and after aging for 216 hours under ambient atmosphere. The yellowish PbI2 can be easily identified for the device based on the MAPbI3 film without CQDs, while there is almost no change for the device based on the CQD modified perovskite film.
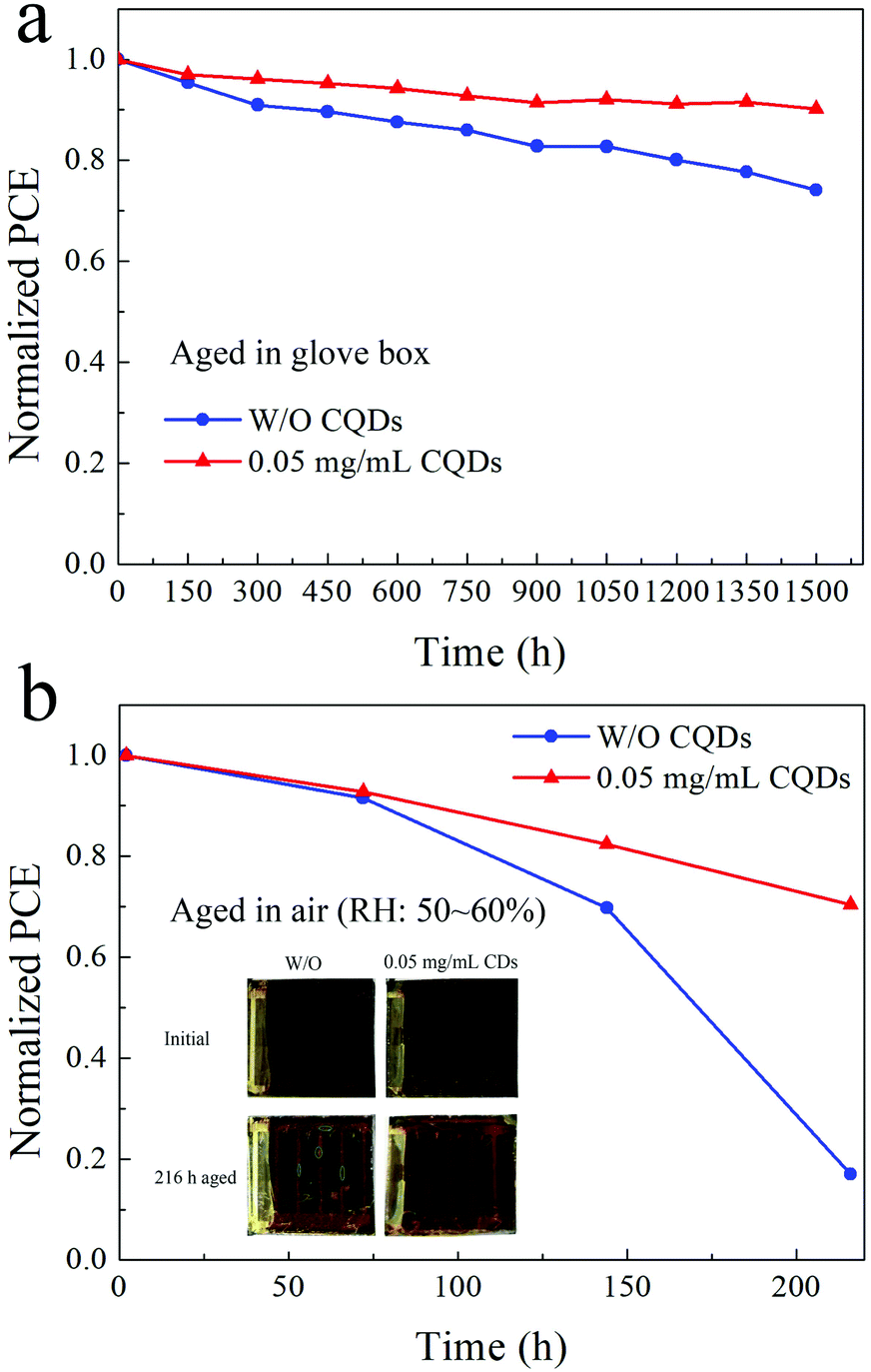 | ||
| Fig. 6 Normalized PCE of unencapsulated PSCs based on the pure MAPbI3 film and the 0.05 mg mL−1 CQD modified MAPbI3 film stored in (a) a N2-filled glove box and (b) under air atmosphere (RH: 50%–60%). | ||
Conclusion
In summary, efficient and stable PSCs are demonstrated by passivating the grain boundaries of CH3NH3PbI3via bonding with CQDs. The typical PCE of PSCs based on the CQD modified MAPbI3 film increases from 17.59% to 18.81% and the PCE of the optimized champion PSCs reaches 19.38%. An appropriate concentration of CQDs in the MAPbI3 film shows negligible influence on the MAPbI3 crystal structure but can passivate the grain boundaries and decrease the trap-state density of the MAPbI3 film. The passivation of CQDs should be attributed to the carboxylic groups, hydroxyl groups and amino-groups of CQDs bonding with the uncoordinated Pb ions in the perovskite crystal, which is proved by the FTIR and DFT results. Moreover, the bonding between CQDs and MAPbI3 leads to a stable absorption of CQDs on the MAPbI3 surface, forming a protective layer to prevent the perovskite from coming in contact with water, thereby enhancing the stability of PSCs. Our results indicate that the bonding between the additive and the perovskite crystal is crucial to achieve stable passivation for efficient and stable PSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51573042, 51873007, 21835006, and 51602102), and the Fundamental Research Funds for the Central Universities in China (2018MS032, 2016MS50, 2017MS027, 2018ZD07, and 2017XS084).References
- Best research-cell efficiencies, NREL. https://www.nrel.gov/pv/assets/images/efficiency-chart.png.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Grätzel and N. G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- J. H. Im, I. H. Jang, N. Pellet, M. Gratzel and N. G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- W. Nie, H. Tsai, R. Asadpour, J. C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H. L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- C. Li, Q. Guo, W. Qiao, Q. Chen, S. Ma, X. Pan, F. Wang, J. Yao, C. Zhang, M. Xiao, S. Dai and Z. A. Tan, Org. Electron., 2016, 33, 194–200 CrossRef CAS.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H. S. Duan, H. H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622–625 CrossRef CAS PubMed.
- Q. Guo, C. Li, W. Qiao, S. Ma, F. Wang, B. Zhang, L. Hu, S. Dai and Z. A. Tan, Energy Environ. Sci., 2016, 9, 1486–1494 RSC.
- C. Li, F. Wang, J. Xu, J. Yao, B. Zhang, C. Zhang, M. Xiao, S. Dai, Y. Li and Z. A. Tan, Nanoscale, 2015, 7, 9771–9778 RSC.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- N. Ahn, D. Y. Son, I. H. Jang, S. M. Kang, M. Choi and N. G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef CAS PubMed.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y.-B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 126, 10056–10061 CrossRef.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- C. Roldan-Carmona, P. Gratia, I. Zimmermann, G. Grancini, P. Gao, M. Graetzel and M. K. Nazeeruddin, Energy Environ. Sci., 2015, 8, 3550–3556 RSC.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J. P. Correa Baena, J. D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, 1501170 CrossRef PubMed.
- D. Y. Son, J. W. Lee, Y. J. Choi, I. H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N. G. Park, Nat. Energy, 2016, 1, 16081 CrossRef CAS.
- M. Yang, Y. Zhou, Y. Zeng, C. S. Jiang, N. P. Padture and K. Zhu, Adv. Mater., 2015, 27, 6363–6370 CrossRef CAS PubMed.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv. Mater., 2017, 29, 1703852 CrossRef PubMed.
- Q. Chen, H. Zhou, T. B. Song, S. Luo, Z. Hong, H.-S. Duan, L. Dou, Y. Liu and Y. Yang, Nano Lett., 2014, 14, 4158–4163 CrossRef CAS PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. García de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z.-H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722–726 CrossRef CAS PubMed.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815–9821 CrossRef CAS PubMed.
- C.-C. Zhang, M. Li, Z.-K. Wang, Y.-R. Jiang, H.-R. Liu, Y.-G. Yang, X.-Y. Gao and H. Ma, J. Mater. Chem. A, 2017, 5, 2572–2579 RSC.
- J. Jiang, Q. Wang, Z. Jin, X. Zhang, J. Lei, H. Bin, Z.-G. Zhang, Y. Li and S. F. Liu, Adv. Energy Mater., 2018, 8, 1701757 CrossRef.
- T. Niu, J. Lu, R. Munir, J. Li, D. Barrit, X. Zhang, H. Hu, Z. Yang, A. Amassian, K. Zhao and S. F. Liu, Adv. Mater., 2018, 30, 1706576 CrossRef PubMed.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. A. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436 CrossRef PubMed.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. A. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249 CrossRef PubMed.
- F. Yuan, L. Ding, Y. Li, X. Li, L. Fan, S. Zhou, D. Fang and S. Yang, Nanoscale, 2015, 7, 11727–11733 RSC.
- F. Yuan, S. Li, Z. Fan, X. Meng, L. Fan and S. Yang, Nano Today, 2016, 11, 565–586 CrossRef CAS.
- H. Li, W. Shi, W. Huang, E. P. Yao, J. Han, Z. Chen, S. Liu, Y. Shen, M. Wang and Y. Yang, Nano Lett., 2017, 17, 2328–2335 CrossRef CAS PubMed.
- Z. Zhu, J. Ma, Z. Wang, C. Mu, Z. Fan, L. Du, Y. Bai, L. Fan, H. Yan and D. L. Phillips, J. Am. Chem. Soc., 2014, 136, 3760–3763 CrossRef CAS PubMed.
- S. Zhou, R. Tang and L. Yin, Adv. Mater., 2017, 29, 1703682 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- L. Tanghao, H. Qin, W. Jiang, C. Ke, Z. Lichen, L. Feng, W. Cheng, L. Hong, J. Shuang, R. Thomas, Z. Rui and G. Qihuang, Adv. Energy Mater., 2016, 6, 1501890 CrossRef.
- S. R. Raga, M. C. Jung, M. V. Lee, M. R. Leyden, Y. Kato and Y. Qi, Chem. Mater., 2015, 27, 1597–1603 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- C. Li, Q. Guo, H. Zhang, Y. Bai, F. Wang, L. Liu, T. Hayat, A. Alsaedi and Z. A. Tan, Nano Energy, 2017, 40, 248–257 CrossRef CAS.
- Z. Wang, T. Cheng, F. Wang, S. Dai and Z. A. Tan, Small, 2016, 12, 4412–4420 CrossRef CAS PubMed.
- V. T. Tiong, N. D. Pham, T. Wang, T. Zhu, X. Zhao, Y. Zhang, Q. Shen, J. Bell, L. Hu, S. Dai and H. Wang, Adv. Funct. Mater., 2018, 28, 1705545 CrossRef.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- L. Zuo, H. Guo, D. W. deQuilettes, S. Jariwala, N. De Marco, S. Dong, R. DeBlock, D. S. Ginger, B. Dunn, M. Wang and Y. Yang, Sci. Adv., 2017, 3, 1700106 CrossRef PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- T. Bu, X. Liu, Y. Zhou, J. Yi, X. Huang, L. Luo, J. Xiao, Z. Ku, Y. Peng, F. Huang, Y. B. Cheng and J. Zhong, Energy Environ. Sci., 2017, 10, 2509–2515 RSC.
- J. M. Grevy, F. Tellez, S. Bernés, H. Nöth, R. Contreras and N. Barba-Behrens, Inorg. Chim. Acta, 2002, 339, 532–542 CrossRef CAS.
- W. J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104, 063903 CrossRef.
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS.
- H. J. Snaith, A. Abate, J. M. Ball, G. E. Eperon, T. Leijtens, N. K. Noel, S. D. Stranks, J. T. Wang, K. Wojciechowski and W. Zhang, J. Phys. Chem. Lett., 2014, 5, 1511–1515 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08295b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |