
In situ electron microscopy of the self-assembly of single-stranded DNA-functionalized Au nanoparticles in aqueous solution†
Eli
Sutter
 *a,
Bo
Zhang
a,
Stephan
Sutter
b and
Peter
Sutter
b
*a,
Bo
Zhang
a,
Stephan
Sutter
b and
Peter
Sutter
b
aDepartment of Mechanical & Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE 68588, USA
bDepartment of Electrical & Computer Engineering, University of Nebraska-Lincoln, Lincoln, NE 68588, USA. E-mail: esutter@unl.edu
First published on 3rd December 2018
Abstract
Solution-phase self-assembly of DNA-functionalized nanoparticles into mesoscale structures is a promising strategy for creating functional materials from nanocrystal building blocks. The predominant approach has been the use of Watson–Crick base pairing between complementary bases in designated ‘sticky ends’ to trigger programmable self-assembly into ordered superlattices. Here we demonstrate the ordered self-assembly of Au nanoparticles conjugated with single-stranded (ss) DNA in acidic solutions. Au nanoparticles functionalized with thiolated ssDNA are protected against coalescence and the DNA conformation undergoes significant modifications at low pH, which can be associated with the protonation of adenine bases and the formation of a parallel poly-adenine duplex, which govern the interaction between ssDNA–Au nanoparticle conjugates. In situ liquid cell electron microscopy enables real-time imaging of the self-assembly process and the identification of key characteristics, such as the preferred structural motifs and interparticle separations in the native solution environment. Our results highlight alternatives to conventional base-pairing interactions for building DNA-directed nanoparticle superlattices.
1. Introduction
The interaction of gold nanoparticles mediated by DNA strands is of interest since oligonucleotide-nanoparticle conjugates are suitable for a variety of applications. Watson–Crick base-pairing interactions between Au nanoparticles conjugated with DNA1 carry special promise for programmable self-assembly2,3 and the formation of ordered and reconfigurable superlattices.4–7 Non-specific interactions between gold nanoparticles functionalized with single-stranded DNA (ssDNA) are of importance for biosensing, e.g., DNA detection with high sensitivity and selectivity,8 screening for DNA mutations with single nucleotide sensitivity,9 development of rapid biomolecular assays for detection of oligonucleotides,10 enzymes, small ions, proteins, human gene mutations,11 bacterial analysis,12etc. Au nanoparticles can be functionalized with ssDNA that is thiolated (i.e., is terminated by a thiol group at either the 3′ or 5′ end) or non-thiolated (i.e., lacking such a functional group). While both thiolated and non-thiolated ssDNA can attach to gold surfaces, the attachment mechanisms are different, as are their capabilities to hybridize with added complementary strands.13 In the case of non-thiolated ssDNA, the oligonucleotide strands adsorb on the unmodified negatively charged, citrate-stabilized Au nanoparticle surfaces10,14 since the ssDNA is sufficiently flexible to partially uncoil and expose its bases, most of which have significant affinity for interaction with gold.10,14,15 By adsorbing on the surface of the Au nanoparticles, the ssDNA can provide protection against coalescence of the particles due to salt addition10 or lowering of the pH,11 and can enable the reversible assembly of Au nanoparticles.11 Thiolated ssDNA is commonly used for Au nanoparticle functionalization because the formation of a covalent S–Au bond upon displacement of citrate ligands strongly anchors the oligonucleotides to the nanoparticle surface.1,16–18 For both non-thiolated and thiolated ssDNA, the strands can achieve different conformations through interaction of the nitrogenous bases with the Au surface.19,20 For non-thiolated ssDNA, this base-surface contact provides the primary interaction between the oligonucleotides and the gold surface. Thiolated ssDNA conjugated to the Au nanoparticle surface is not expected to wrap around the particles but to remain extended in the solution, in particular when the loading is high. In both cases, the ssDNA-functionalized nanoparticles remain suspended in solution at neutral pH for extended time periods. Whereas the DNA bases are essentially charge-neutral in the pH range of 4.0–9.0,15 the sugar-phosphate backbone carries net negative charge that causes an electrostatic repulsion with (negatively charged) Au nanoparticles. Cytosine (C) and adenine (A) bases become protonated at pH < 4.21 The protonation of the bases promotes DNA adsorption, presumably by reducing the repulsion between DNA and the Au surface. The mutual interaction between protonated adenine bases (i.e., AH+–H+A) in acidic conditions has been shown to lead to the formation of a parallel duplex of poly-A DNA strands (A-motif).22,23 Among other possible implications, the ssDNA interaction and duplexation via protonated adenine bases has been used for pH sensing.24 Similarly, other pH sensitive interactions were proposed to detect pH changes in vivo and in vitro, among them the formation of cytosine–guanine-protonated cytosine (C–G–H+C) triplex DNA under acidic conditions,25 the association of thymine–adenine–thymine (T–A–T) triplex DNA at neutral pH values,25 and the stabilization of hemi-protonated cytosine–cytosine (C–H+C) base pairs under acidic conditions.26 Besides sensing applications, the formation of A-motif parallel duplex DNA has been shown to enable the conjugation of Au nanoparticles with thiol-functionalized, poly-A terminated ssDNA extremely rapidly and to very high surface coverages,17 thus providing an alternative to the conventional salt-ageing process for producing DNA–Au nanoparticle conjugates.27,28Here we use a combination of ex situ and in situ imaging by conventional and liquid-cell transmission electron microscopy and associated complementary measurements to explore the behavior of Au nanoparticles conjugated with thiolated ssDNA at low pH. A dense shell of covalently bound ssDNA effectively protects the Au particles against coalescence, similar to the protection shown for particles wrapped by non-thiolated ssDNA.11 Importantly, lowering of the pH allows the DNA–Au nanoparticle conjugates to approach and interact, thus enabling aggregation and self-assembly processes that give rise to ordered structures with well-defined local particle arrangements. Liquid cell electron microscopy is used to observe this self-assembly of ssDNA conjugated nanoparticles in real time in their native solution environment. Instead of adjusting the pH prior to loading of the liquid cell and its introduction into the microscope, which is problematic since this approach would likely miss the early stages of self-assembly, we use electron-beam effects on the solution to our advantage. Interacting with aqueous solutions, the high-energy electrons used for imaging are well known to inject different radicals such as aqueous electrons (eaq), OH˙ radicals, hydronium (H3O+) ions, etc., due to radiolysis of water.29–31 A specific by-product, local ‘acid spikes’ induced by electron exposure, can be used to create a low pH environment ‘on demand’ during imaging in the liquid-cell. In this way, stable suspensions of well-dispersed DNA–Au nanoparticle conjugates in an initially pH neutral solution can be loaded and the effects of an acidic environment triggered locally in the field of view during real-time microscopy to study the self-assembly processes in acidic solutions.
2. Results and discussion
2.a DNA-shell conformation and self-assembly of ssDNA–Au nanoparticle conjugates at low pH
Au nanoparticles with narrow size distribution (mean diameter ∼17.5 nm, determined from TEM image analysis (see below)) were functionalized with thiolated ssDNA according to the procedure described in the Methods. The base sequence of the ssDNA is given in Fig. 1(a). A thiol functional group at the 5′ end is separated by a poly-A (here A12) sequence from a terminal base sequence, as shown. At neutral pH (∼7.0–7.5), the ssDNA–Au nanoparticle conjugates remain suspended in (0.1 M phosphate buffer, 0.5 M NaCl (PBS)) aqueous solution for extended time periods, during which the solution retains its characteristic red color and UV-VIS spectra show a dominant peak at 525.8 nm that does not change over time (Fig. 1(b)).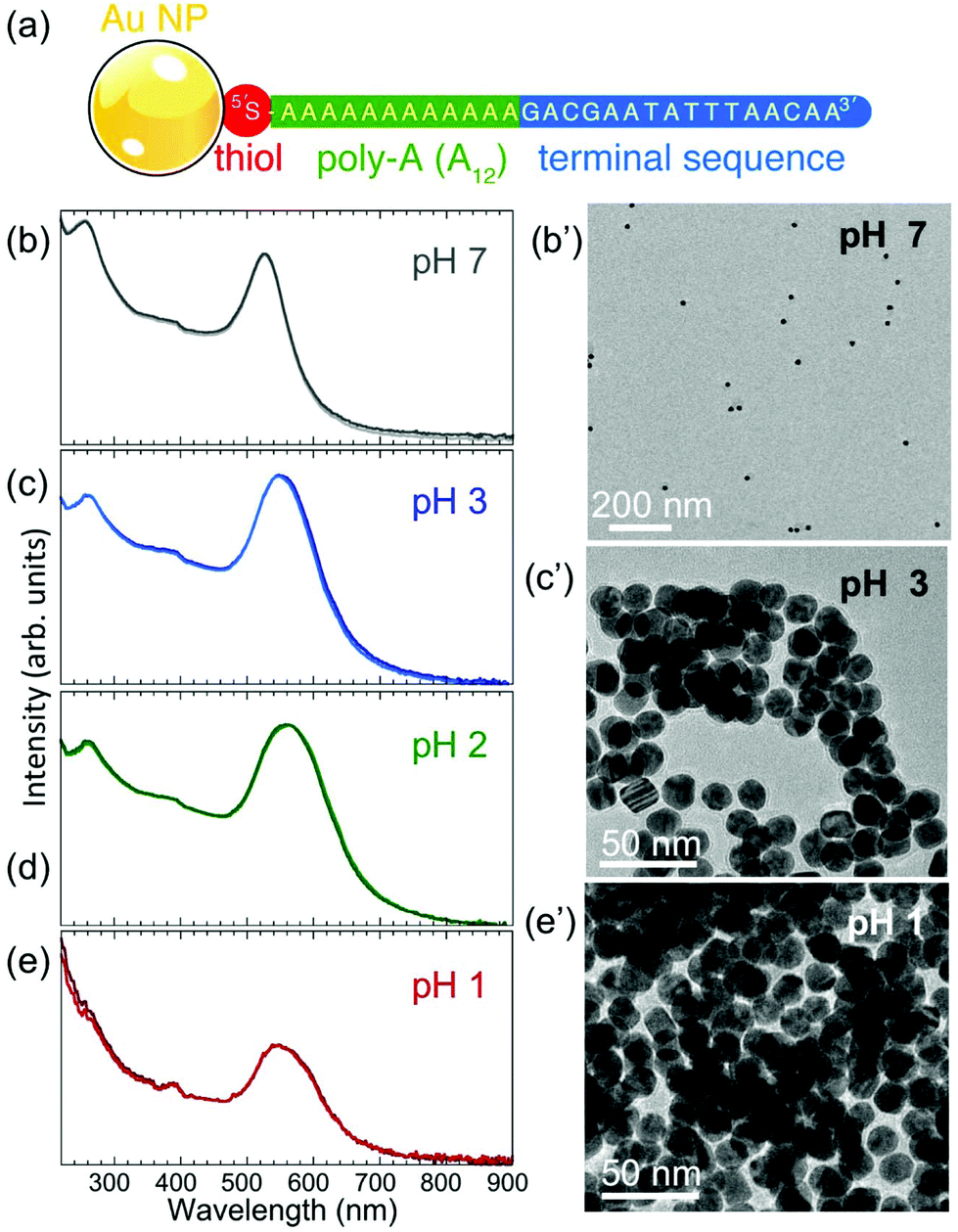 | ||
| Fig. 1 (a) Structure of the ssDNA–Au nanoparticle conjugates used in this study. (b)–(e) UV-VIS spectroscopy and ex situ TEM imaging of ssDNA-functionalized Au nanoparticles at different pH: (b) pH 7, (c) pH 3, (d) pH 2, and (e) pH 1. (b′) TEM image of a ssDNA–Au nanoparticle sample imaged dry after being extracted from a solution at pH 7. (c′) TEM image of a ssDNA–Au nanoparticle sample imaged dry after being extracted from a solution at pH 3. (e′) TEM image of a ssDNA–Au nanoparticle sample extracted from a solution at pH 1. | ||
A lowering of the pH of the solution by adding HCl causes significant changes to the nanoparticle suspension, as shown by UV-VIS spectra (Fig. 1(c)–(e)). The main absorption peak widens and is progressively red-shifted to 548 nm (pH 3) and 560 nm (pH 2). Since the main UV-VIS absorption peaks (and the color) of Au nanoparticle suspensions are mainly determined by the localized surface plasmon resonance of the nanostructures, which is affected by aggregation or size changes,32,33 the observed evolution at low pH suggests either coalescence or aggregation/self-assembly of the ssDNA–Au nanoparticle conjugates. TEM images of Au particle samples extracted from solution at different pH values and imaged dry after drop-casting onto TEM grids (Fig. 1(b′), (c′), (e′); Fig. S2(a), (b); Fig. S1†) show that the individual ssDNA functionalized Au nanoparticles remain intact (i.e., do not undergo coalescence) to very low pH (pH 1), and while at pH 7 the nanoparticles are dispersed, at lower pH they aggregate to form assemblies. This behavior is distinctly different from that of the original citrate-terminated Au nanoparticles prior to conjugation with thiolated ssDNA, which under the same conditions coalesce completely (Fig. S2(c) and (d)†). We conclude that thiolated ssDNA confers significant protection against coalescence of the Au nanoparticles, similar to non-thiolated DNA strands.11
The TEM images of Fig. 1(c′) and (e′) show that for pH < 4 the ssDNA-conjugated Au nanoparticles aggregate or self-assemble into clusters that differ in size depending on the pH of the solution. At pH 3, the assemblies contain a small number of ssDNA–Au nanoparticles (Fig. 1(c′)) in two- or three-dimensional arrangements, while at pH 1 the individual clusters are significantly larger. When the pH is adjusted to 1, the solutions turn clear and the aggregates settle at the bottom of the cuvettes.
Single-stranded DNA anchored to Au nanoparticles via a thiol group are not expected to wrap around the nanoparticles but to extend into the solution, in particular when the loading on the particle surface is high.34 In our case, the loading obtained by the conventional salt-ageing process27 was estimated to 80–130 strands per particle for Au particles with 17.5 nm mean diameter (see Methods). To explore the effects of an acidic environment on the conformation of the DNA shell, we carried out nanoparticle tracking analysis (NTA). In the measurement, which used somewhat larger (21 nm) Au nanoparticles capped by citrate or ssDNA, the rate of particle movement is related to a sphere equivalent hydrodynamic radius (or diameter) as calculated through the Stokes–Einstein equation (see Methods).35
The measurements of hydrodynamic diameters are summarized in Fig. 2. For citrate capped 21 nm Au particles, NTA yields a hydrodynamic diameter dH = 26.0 nm with a narrow full width at half maximum (FWHM) of 7.5 nm. This diameter is larger than the sum of the particle diameter plus twice the expected physical length of the citrate ligand (∼0.8–1.0 nm from the Au surface),36 consistent with solvent effects on the particle motion. The hydrodynamic diameter of the same Au nanoparticles conjugated with ssDNA (with base sequence as shown in Fig. 1(a)) is dH = 36.9 nm, and the diameter distribution is now substantially broader (FWHM: 14.8 nm). The ∼16 nm difference between this value and the Au nanoparticle diameter, which corresponds to the contribution of the DNA to the hydrodynamic diameter, is quite substantial and suggests that steric effects due to the high ssDNA loading on the Au nanoparticle surface forces the strands to extend into the solution. In a third measurement, the pH of the solution of ssDNA–Au nanoparticle conjugates was lowered to 4, where effects due to the acidic environment should be measurable but nanoparticle aggregation (which could complicate the analysis of the NTA data) is still minimal. At pH 4, we obtain a hydrodynamic diameter dH = 30.9 nm (FWHM: 13.1 nm), significantly smaller than for the same ssDNA–Au particle conjugates at neutral pH. This finding demonstrates that a reduction in pH causes measurable changes in the conformation of the ssDNA shell, in particular an overall contraction in length compared to the extended geometry at pH 7.
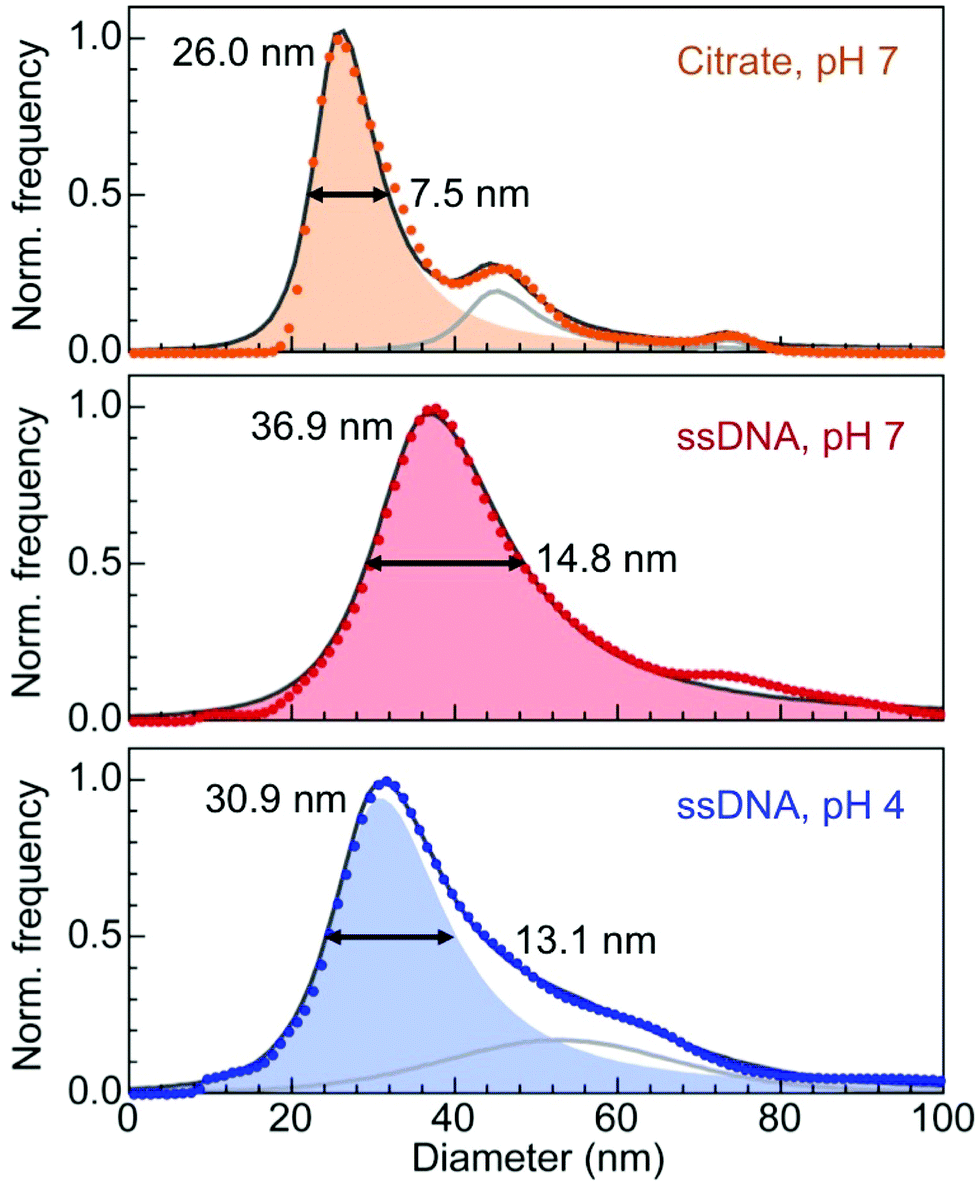 | ||
| Fig. 2 Analysis of the hydrodynamic diameter of citrate and ssDNA-capped Au nanoparticles. The mean particle diameter determined by TEM image analysis is 21 nm. Citrate capped particles were tracked in water. Single-stranded DNA capped particles were tracked in phosphate buffer solution at pH 7, and after adjusting the pH to 4 by addition of HCl. Numbers give the measured mean diameter and full width at half maximum (FWHM) for all three cases. Shaded regions are fits of the primary peaks using asymmetric Lorentzian functions. In some cases, secondary fit components were used, as indicated by gray lines. | ||
The response of Au nanoparticles conjugated with ssDNA to changes in pH can be understood as the result of interactions between protonated adenine bases (i.e., AH+–H+A) in acidic conditions, as shown schematically in Fig. 3.
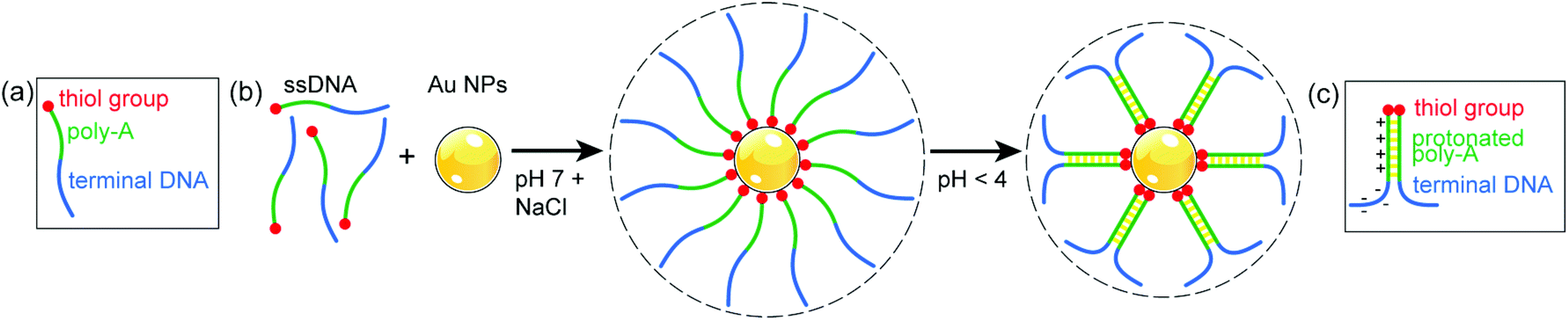 | ||
| Fig. 3 (a) Schematic structure of the thiolated ssDNA with A12 spacer used in this study. (b) Salt-assisted conjugation of Au nanoparticles (NPs) with ssDNA at pH 7, and changes due to interaction between protonated adenine bases in the A12 spacer upon lowering the pH to 4. (c) Structure and charge distribution in parallel duplexed ssDNA, as suggested in ref. 17. | ||
At neutral pH, ssDNA functionalized Au nanoparticles remain suspended in solution indefinitely due to the repulsion of the negatively charged sugar-phosphate backbone of DNA strands on different particles.28 The A12 spacer of the conjugated ssDNA shell (Fig. 3(a)), which at neutral pH extends into the solution, forms a protonated poly-A parallel duplex as the pH is lowered to 4 (Fig. 3(b)).17 This effect stiffens the duplex region and changes the charge distribution on the DNA. Due to the protonation, the net charge on the DNA strands is significantly reduced (from ∼30 e at pH 7 to ∼0 e at pH 1),37 resulting in a decrease in electrostatic repulsion. In addition, ssDNA with partial duplexation divides into a cationic poly-A duplex region and an anionic terminal sequence (Fig. 3(c)),17 which governs the further conformational changes of the terminal DNA. Assuming for simplicity a length per base of the poly-A duplex of 0.334 nm as in double helical B-DNA,38 the A12 sections alone would contribute ∼8 nm to yield a total (physical) diameter of ∼29 nm. The measured hydrodynamic diameter at pH 4 of ∼31 nm, again somewhat larger than this expected physical size, is consistent with this scenario if we assume that the terminal DNA folds back toward the particles and contributes negligibly to the Stokes diameter (Fig. 3(b) and (c)).
From the NTA analysis of the hydrodynamic diameters, we thus conclude that the conformation shown schematically in Fig. 3(b) is the relevant geometry involved in the self-assembly of ssDNA–Au nanoparticle conjugates at low pH identified in Fig. 1 and S2, i.e., the initiation of nanoparticle clustering at pH < 4 and progressive increase in the cluster size with further lowering of the pH. While the ex situ TEM experiments on drop-cast, dry aggregates discussed so far showed no apparent ordering of the nanoparticles, in situ microscopy in the native aqueous solution, discussed below, suggests that they result from a controlled self-assembly process of ssDNA functionalized particles, in which distinct nucleation and growth pathways lead to well-defined, ordered nanoparticle coordination motifs.
2.b In situ liquid-cell electron microscopy of the self-assembly of ssDNA-functionalized Au nanoparticles
Liquid-cell electron microscopy, which has been developed for in situ imaging of a broad spectrum of solution-based processes including colloidal and plasmon-mediated nanocrystal synthesis,30,39–42 galvanic replacement,31,43 bio-mineralization,44 and nanocrystal self-assembly,45,46 can provide real-time observations of ssDNA-mediated self-assembly with single particle resolution. In recent work, we identified low-dose imaging conditions that largely avoid radiation damage to DNA,47 opening up the possibility of investigating DNA-mediated processes in their native solution environment by real space imaging rather than diffraction methods. The link between low pH and the initiation of self-assembly of ssDNA–Au nanoparticle conjugates raises questions about how to best design suitable in situ imaging experiments. The simplest approach, suspending the nanoparticles in a solution with low pH, loading them into the liquid cell, and finally imaging them, carries the risk that the assembly process may start or even run to completion before the first images are taken. A more elegant approach developed here involves utilizing local and transient modifications, specifically a lowering of the pH, of the aqueous ssDNA-nanoparticle solution due to radiolysis by the electron beam. The primary radiolysis process entails the formation of hydronium ions (H3O+) or protons (H+) in water, which renders the exposed regions more acidic than the surrounding solution. This ‘acid spike’ is observed immediately after the initial energy release and is found to be greatest for times <1 ns after the interaction with a relativistic electron. Previous work showed that after this initial decrease, the pH remains steady at ∼3.3, followed by a gradual recovery to pH 7 over ∼1 μs.48–53 Here we use both the nanoscale in situ imaging capability as well as the transient, local lowering of the solution pH to initiate and follow the nucleation and growth of self-assembled ssDNA–Au nanoparticle clusters.For the in situ imaging experiments, suspensions of ssDNA-functionalized 17.5 nm Au nanoparticles at neutral pH (identical to those used in the ex situ experiments) were loaded into the liquid cell and observed by high angle annular dark field scanning transmission electron microscopy (HAADF-STEM). Detailed imaging conditions are provided in the Methods. Fig. 4 shows representative real-time STEM observations (see Movie S1†) of the ssDNA–Au nanoparticles in solution and of the initial stages of their interaction.
 | ||
| Fig. 4 In situ microscopy of the self-assembly of ssDNA–Au nanoparticle conjugates into a compact 3D supracrystal. The time-lapse image sequence shows selected HAADF-STEM images of the formation of 3D assemblies of ssDNA–Au nanoparticle conjugates. Starting from single nanoparticles dispersed in the entire liquid volume, initial small (dimer) clusters progressively transform into 3D structures that aggregate near the top and bottom membranes of the liquid cell. Superstructures at one membrane are in focus (e.g., memA at t = 46 s), whereas those located close to the other membrane appear blurred (memB). Scale bars: 50 nm. | ||
Initially, monomers are the predominant species in the suspension, as seen by single particles that spontaneously move into the STEM focal plane (Fig. 4, t = 4 s). Self-assembly then sets in spontaneously, and at the early stage proceeds via monomer attachment and the formation of dimers (Fig. 4, 8–10 s). Dimers appear to be the stable nucleus since their dissociation into monomers is never observed. Typically, shortly after the formation of a dimer either a third Au monomer or a small cluster of particles attaches to generate a trimer or a larger assembly (Fig. 4, t ≥ 12 s). The aggregation of several particles leads first to the formation of a loosely packed (Fig. 4, 12 s) and later a close-packed supracrystal (Fig. 4, 17 s) via attachment and edge diffusion of particles. Whereas the initial small clusters tend to be planar, likely due to the templating effect of the SiNx liquid-cell membrane, additional particles are increasingly incorporated in a second layer where they tend to occupy higher-coordination hollow-sites (Fig. 4, 17 s). Second-layer particles are recognized by brighter STEM contrast due to an increased local thickness. In Fig. 4, the crystal at t = 17 s, for instance, consists of 10 nanoparticles, 8 of which are in the first layer and 2 in a second layer. The later-stage growth of this representative assembly involves incorporation of monomers (t = 32 s, 48 s, 50 s), trimers (87 s), and larger units (80 s). The larger units can either be planar or already contain particles in a second layer. Generally, the transition from 2D to 3D occurs early in the self-assembly process, suggesting that the interaction between the nanoparticles is stronger than their attraction to the SiNx liquid-cell membrane.
The initial stages of self-assembly and particle mobility are examined in more detail in Fig. 5. Early on, the solution contains mostly monomers (Fig. 5(a), t < 20 s). Self-assembly via monomer attachment leads to the formation of dimers (Fig. 5(a), t = 26 s). Our observations show that monomers and dimers are mobile in the solution, with both showing extensive Brownian motion. In addition to translational displacements, dimers undergo frequent rotations (e.g., the dimer marked with a white circle in Fig. 5(a)). An analysis of in situ microscopy image sequences shows substantially different Brownian mean-square displacements for monomers and dimers (Fig. 5(b)). As expected, the diffusion coefficient of monomers, Dmonomert ∼ 1.7 × 10−15 m2 s−1, is significantly larger than that of dimers, Ddimert ∼ 6.2 × 10−17 m2 s−1. The measured diffusion coefficient for monomers is several orders of magnitude larger than found previously for citrate-terminated Au particles of similar size in ultrathin liquid layers.54 But a comparison with our measurements by NTA (discussed above) shows that it is much smaller than the diffusion coefficient in an unrestricted PBS solution environment, 7.1 × 10−12 m2 s−1 at pH 4 (Fig. 2), likely due to interaction of the particles with the SiNx membrane of the liquid cell.
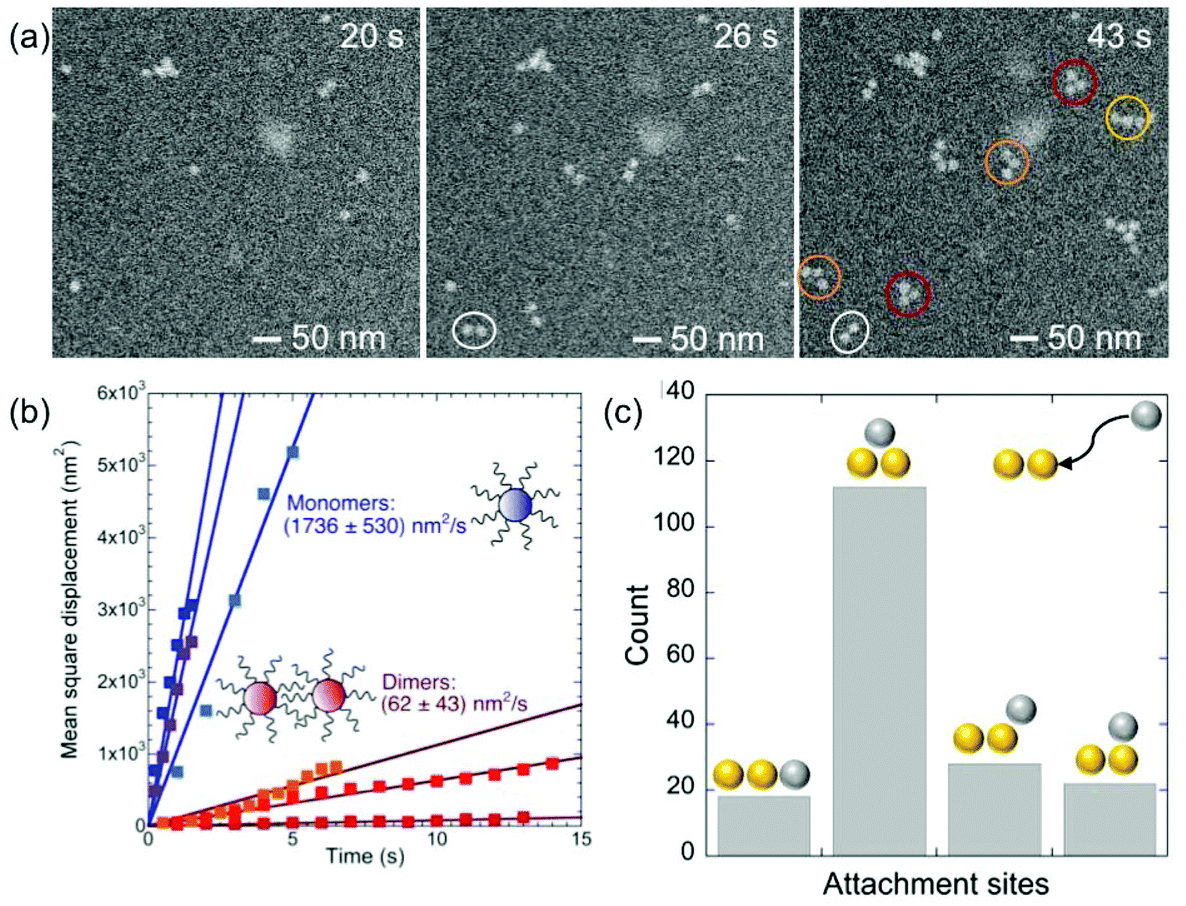 | ||
| Fig. 5 Initial stages of single-stranded DNA–Au nanoparticle interaction and self-assembly. (a) Selected HAADF-STEM images illustrating the initial stages of self-assembly of DNA–Au nanoparticle conjugates: 20 s: predominant monomers in the suspension. 26 s: monomer attachment and formation of dimers, the stable nucleus. t = 43 s: formation of trimers and multimers. (b) Measured Brownian mean-square displacements of monomers and dimers of single-stranded DNA–Au nanoparticle conjugates in solution. (c) Preferred attachment sites for a third particle joining a dimer. | ||
Shortly after the formation of a dimer, a third Au monomer typically attaches to generate a trimer (examples are marked with red circles in Fig. 5(a), t = 43 s). The third particle can attach in different sites, thus forming trimers with different spatial motifs as seen in Fig. 5(a). From an analysis of a large number of events, we conclude that the predominant trimer configuration is triangular, even though a substantial number of linear and armchair attachments are also observed (Fig. 5(c)). This preference indicates the existence of a preferred nearest-neighbor arrangement of self-assembled ssDNA–Au nanoparticle superlattices.
The specific ordered two-dimensional (2D) structures observed in the solution at the early stages of self-assembly are summarized in Fig. 6. They include triangular lattices (Fig. 6(a)), square lattices (Fig. 6(b)), and linear chains (Fig. 6(c)). An analysis of a large number of 2D clusters observed in situ provides evidence for a predominant triangular (i.e., close-packed, Fig. 6(a)) arrangement with (60 ± 5)° nearest-neighbor angles (Fig. 6(d)). A smaller maximum in the distribution corresponds to an angle of (90 ± 5)°, i.e., a square lattice as shown in Fig. 6(b). While the more open square packing appears to be stable for planar clusters up to ∼9 particles in size, it is observed much less frequently than the denser hexagonal motif. Fig. S5† suggests that this lower abundance is due to the ultimate rearrangement of small planar square lattices due to further added monomers occupying positions associated with denser in-plane packing or in the second layer.
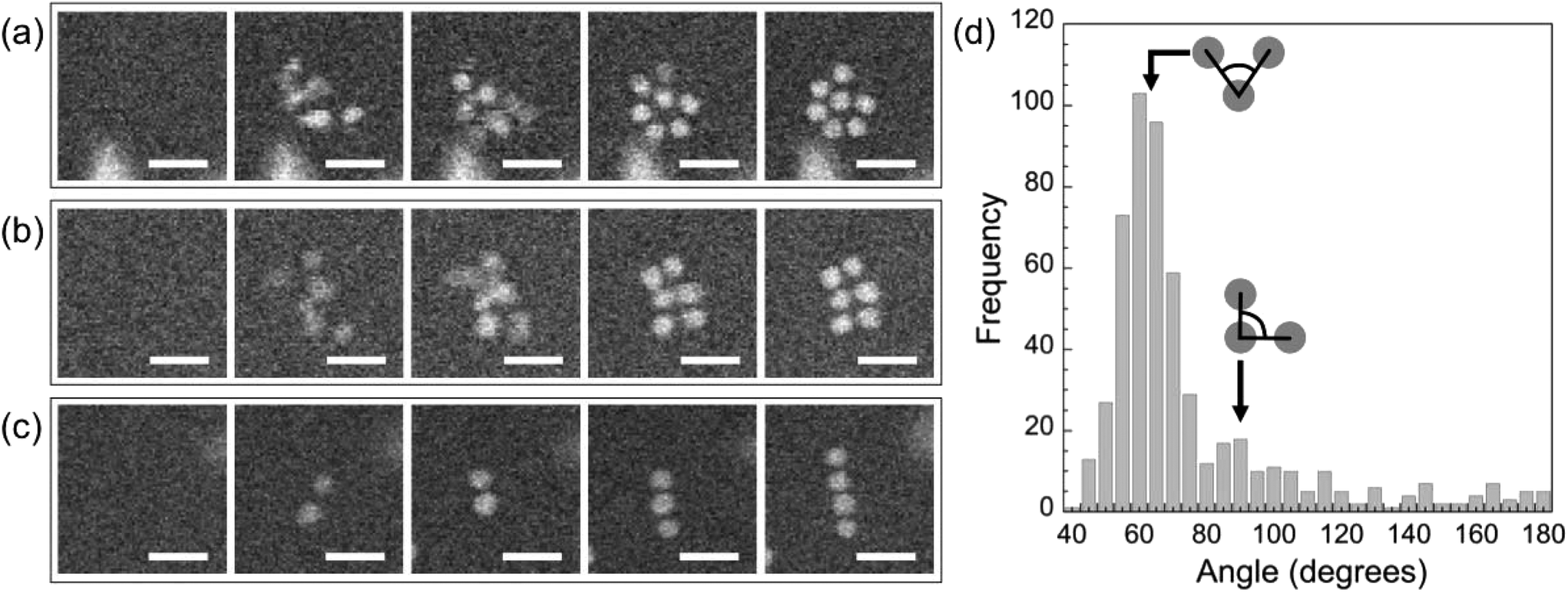 | ||
| Fig. 6 Predominant 2D structural motifs in ssDNA–Au nanoparticle self-assembly. (a) Triangular lattice. (b) Square lattice. (c) Linear chain. (d) Analysis of nearest neighbor angles for a large number of self-assembled clusters observed in situ. Scale bar: 50 nm. | ||
In many instances, one-dimensional chains of ssDNA–Au nanoparticle conjugates form instead of compact clusters. The initial linear chain growth persists up to a length of 5–6 particles, typically followed by a further expansion away from the chain axis either in the plane or in the second layer. The trajectories of the incorporation of nanoparticles into the first and second layer for such a 1D chain are shown in detail in Fig. S3,† where a linear tetramer seeds the assembly of a longer chain. As particles move close to the chain, they frequently are loosely attached away from the chain axis but then find stable configurations within the chain (Fig. S3;†t = 18 s, trajectory 1). In other cases, particles first diffuse into contact within the plane of the membrane but then occupy final top- (t = 22 s, trajectory 2) or hollow-sites (t = 45 s, trajectory 3) in the second layer. Often, however, the interactions of the ssDNA nanoparticles lead to transition from a short linear chain (e.g., a trimer) to a more close-packed configuration. One such example is shown in Fig. S4,† where the addition of a somewhat larger particle to a trimer initiates the transition from a linear chain into an armchair configuration, and further to a more densely packed 3D assembly.
To complete the characterization of the ordering of self-assembled superstructures of ssDNA–Au nanoparticle conjugates we analyzed the interparticle spacing in solution, which can provide information about possible interaction mechanisms underlying the assembly process. It is worth noting that the interparticle spacing was measured only for planar 2D assemblies since, even though we can follow incorporation of nanoparticles in a second layer (see Fig. S6†), it is not possible to unambiguously determine the distance between the nanoparticles in different layers from 2D HAADF-STEM images. The interpretation of these data requires some caution due to the possibility of radiation damage to the DNA. While we have previously identified low-dose in situ imaging conditions that allow essentially damage-free electron microscopy of DNA-mediated nanoparticle superlattices,47 the need for a significant ‘acid spike’ to transiently lower the solution pH and trigger self-assembly forces the use of higher electron doses (see Methods for details). Thus, radiation damage cannot be ruled out completely and should manifest itself in a progressive reduction of the interparticle spacing, similar to the behavior identified for nanoparticles interacting via linker DNA.47Fig. 7 indeed shows this characteristic tightening of the particle spacing during extended STEM imaging for all ordered motifs, including 2D triangular and square lattices (Fig. 7(a) and (b)), as well as linear chains (Fig. 7(c) and (d)). Note that our previous work showed that the superlattice structure is insensitive to the early stages of DNA damage, i.e., even when clear signs of damage are observable via a tightening of the interparticle spacing, the nearest-neighbor configuration (e.g., ‘bond’ angles) remains unaffected.47 This implies that our conclusions regarding the assembly process and the predominant motifs (Fig. 6, based on short-term electron exposures) are robust even in the presence of limited radiation damage. Finally, coalescence of nanoparticles due to the de-protection of their surfaces, which would represent the final stage of ssDNA damage47 has never observed here even during much longer imaging and exposure to the electron beam, confirming limited radiation damage to the DNA strands during the much shorter imaging periods used in this study.
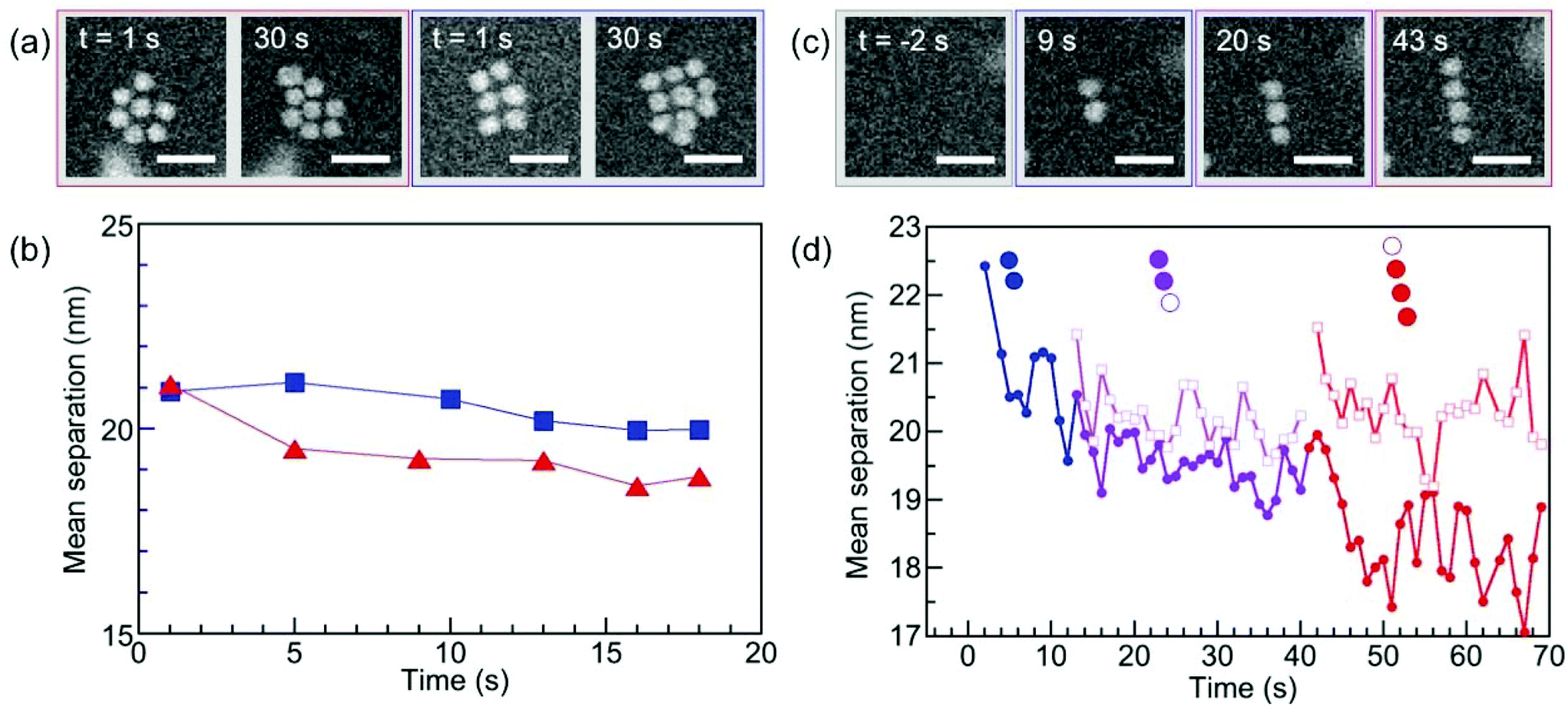 | ||
| Fig. 7 Evolution of the lattice parameter under electron irradiation during in situ STEM imaging. (a) Excerpts from time-lapse image series showing growing triangular and square 2D superstructures in solution. (b) Time-evolution of the mean interparticle spacing in triangular (red symbols) and square (blue symbols) superstructures. (c) Excerpt from a time-lapse image series showing a growing linear chain in solution. (d) Analysis of the mean interparticle spacing (solid symbols) and of the separation of the last-added monomer from its nearest neighbor in the linear chain (open symbols). A clear trend toward smaller interparticle spacing is found during extended STEM observation/electron irradiation. Scale bar: 50 nm. | ||
To obtain insight into possible interaction mechanisms, we thus analyzed the spacing of newly added particles in small existing assemblies as early as their incorporation into a high-symmetry site occurs, i.e., before the onset of significant radiation damage. This analysis of planar clusters shows that ssDNA-terminated (17.5 ± 1.0) nm Au particles self-assemble into 2D (and 3D) supracrystals with mean center-to-center particle spacing of (23.0 ± 1.4) nm (Fig. 8(a)), corresponding to a surface-to-surface spacing of (5.5 ± 0.9) nm (Fig. 8(b)). This separation, obtained for particles conjugated with ssDNA in the native (hydrated) state, is smaller than twice the length of extended poly-A duplexes formed through bonding of protonated adenine bases (i.e., AH+–H+A) in acidic solution, but is consistent with an interdigitation of the duplexed A12 strands on neighboring particles (Fig. 8(c)). In such a scenario, hydrogen bonding between complementary bases of the terminal DNA sequence (see Fig. 1(a)) or interaction via protonated bases (e.g., C–H+C26 or additional AH+–H+A in the terminal sequence) at low pH can stabilize the assemblies beyond the non-specific interaction of the particles and their interdigitated DNA shells. Future work, e.g., using theoretical simulations, could shed additional light on the competition between different interaction mechanisms involved in the self-assembly of ssDNA conjugated nanoparticles at low pH.
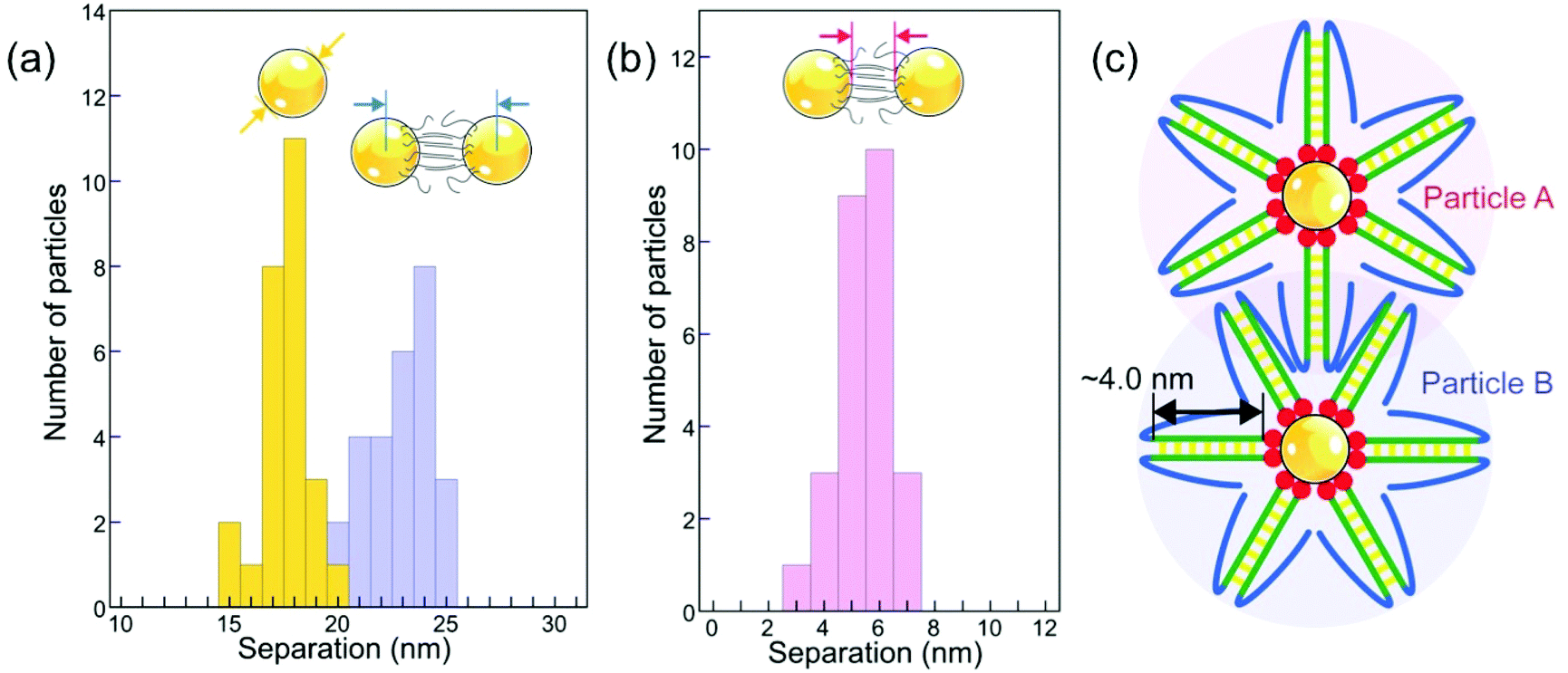 | ||
| Fig. 8 Interparticle spacing of ssDNA–Au nanoparticle conjugates in solution. (a) Nanoparticle diameter (yellow, (17.5 ± 1.0) nm) and center-to-center separation histograms (blue, (22.95 ± 1.37) nm) analyzed from in situ microscopy data obtained in solution. (b) Histogram of surface-to-surface interparticle spacing in solution (5.5 ± 0.9) nm, determined from the data shown in (a). (c) Interdigitation of poly-A duplexes (green) as a potential mechanism for stabilizing ssDNA–Au nanoparticle assemblies. Note the possibility of fortuitous pairing of complementary bases between back-folded terminal DNA strands (blue). | ||
3. Conclusions
A combination of ex situ and in situ imaging by conventional and liquid-cell transmission electron microscopy was used to investigate the behavior of Au nanoparticles conjugated with thiolated ssDNA at neutral and low pH. The ssDNA shell effectively protects the Au particles against coalescence at low pH. An analysis of the hydrodynamic radius of the ssDNA–Au nanoparticle conjugates demonstrates that a reduction in pH causes measurable changes in the conformation of the ssDNA shell, in particular an overall contraction in length compared to an extended geometry at pH 7. This contraction, which can be linked to the formation of a duplex of protonated adenine bases adjacent to the thiol group, allows the ssDNA–Au nanoparticle conjugates to approach and interact at low pH, thus enabling self-assembly processes that give rise to ordered superstructures. Liquid cell electron microscopy was used to follow this self-assembly of ssDNA conjugated nanoparticles in real time in their native solution environment. A low pH environment in the solution was achieved locally in the field of view using transient, local lowering of the solution pH due to the radiolysis of water by the high-energy electrons. Real-time observations, which showed that monomers and dimers of ssDNA–Au nanoparticle conjugates are mobile in solution, were used to establish the nucleation and growth mechanism of ordered assemblies. The stable nucleus is a dimer, and the further assembly proceeds via incorporation of monomers, dimers, and larger multimers and results in the formation of ordered crystallites with predominant triangular and square structures, as well as linear chains. The observed interparticle distances in solution are consistent with an interaction based on the interdigitation of poly-adenine duplexes on neighboring particles. Our results show the self-assembly of Au nanoparticles conjugated with thiolated single-stranded DNA in acidic solutions as an alternative to approaches for building DNA-directed nanoparticle superlattices using conventional base-pairing interactions.4. Materials and methods
Citrate-capped Au nanoparticles (Pelco NanoXact and BioPure) with 20 nm nominal diameter and concentration of 1.3 × 1011 particles per mL were used as starting material. Oligonucleotides with the structure 5′thiol modifier-A10-AAGACGAATATTTAACAA3′ (see Fig. 1(a)) were purchased from Integrated DNA Technologies. The functionalization of the Au nanoparticles was performed following the protocol described in ref. 16 and 27. UV-Vis spectroscopy was measured using a PerkinElmer Lambda 265 spectrophotometer. The ssDNA loading on the nanoparticles was estimated from the UV-VIS measurements on the supernatant collected after the salt-ageing process.Real-time (S)TEM experiments were carried out in an in situ specimen holder (Hummingbird Scientific) using liquid cells comprising two 30 nm thick, electron transparent SiNx membrane windows with 50 × 50 μm2 window area. The spacing between the windows was controlled using 100 nm or 200 nm SiO2 spacers. Despite the use of defined spacers, we always obtain a continuous range of liquid thicknesses within the same cell due to bowing of the thin membranes.41 STEM imaging was performed in a FEI Talos F200X microscope operated at 200 kV, with ∼2 Å probe size and beam currents in the range between 1–20 pA, measured in vacuum before introduction of the liquid cell. Typical conditions for the acquisition of time lapse image series were electron dose rates of 0.1–30 e− (Å2 s)−1, 512 × 512 pixels, and pixel dwell times of 2 μs (0.52 s per frame) or 4 μs (1.05 s per frame). In addition to the in situ LCEM experiments with ssDNA conjugated Au nanoparticles (Movie S1†) we performed control experiments with citrate-terminated Au nanoparticles under identical conditions. In contrast to the ssDNA nanoparticles, which assembled in solution, the citrate-terminated nanoparticles remained dispersed in solution during the in situ experiments (Movie S2†). The analysis of the assemblies was performed in the software package ImageJ,55 using built-in threshold and particle analysis functions to determine the centroid coordinates of the particles. Hydrodynamic radii were measured by nanoparticle tracking analysis (NTA) performed on a Malvern NanoSight NS300 instrument with a high sensitivity CMOS camera, temperature control, and a choice of laser block. NTA is a method of visualizing and analyzing the Brownian motion of particles with diameters between 10 nm–2000 nm in solution.35 Illuminated by laser light, citrate-capped Au nanoparticles and DNA–Au nanoparticle conjugates were tracked via their scattered light, with frame-to-frame particle positions captured by the CMOS camera. The Brownian motion was related to a sphere-equivalent hydrodynamic diameter, dH, through the Stokes–Einstein equation, 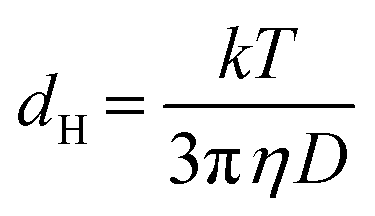 , where D is the translational diffusion coefficient, k is Boltzmann's constant, T the absolute temperature, and η denotes the viscosity of the solvent, either water with η(H2O) = 9 × 10−4 Pa s for citrate-capped Au nanoparticles, or (0.1 M phosphate buffer, 0.5 M NaCl) aqueous solution with η(PBS) = 1 × 10−3 Pa s for ssDNA–Au nanoparticle conjugates. The NTA measurements were carried out immediately (within 5 minutes) after adjustment of the pH by adding acid to the solution.
, where D is the translational diffusion coefficient, k is Boltzmann's constant, T the absolute temperature, and η denotes the viscosity of the solvent, either water with η(H2O) = 9 × 10−4 Pa s for citrate-capped Au nanoparticles, or (0.1 M phosphate buffer, 0.5 M NaCl) aqueous solution with η(PBS) = 1 × 10−3 Pa s for ssDNA–Au nanoparticle conjugates. The NTA measurements were carried out immediately (within 5 minutes) after adjustment of the pH by adding acid to the solution.
Author contributions
E.S. and P.S. designed and planned the experiments, analyzed the results and wrote the paper. E.S. performed the ex situ and in situ electron microscopy experiments. B.Z. performed conjugation of some of the samples, carried out NTA measurements and their analysis. S.S. performed conjugation of some of the samples, measured the UV-VIS spectra and participated in the analysis of the in situ microscopy data. All authors discussed the results, and read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Army Research Laboratory and the U.S. Army Research Office under grant number W911NF-17-1-0141. The authors acknowledge seed funding from the Nebraska Public Power District through the Nebraska Center for Energy Sciences Research, and technical support by Q. Wu.References
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, A DNA-based method for rationally assembling nanoparticles into macroscopic materials, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, DNA-programmable nanoparticle crystallization, Nature, 2008, 451, 553 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, DNA-guided crystallization of colloidal nanoparticles, Nature, 2008, 451, 549 CrossRef CAS PubMed.
- Y. Zhang, S. Pal, B. Srinivasan, T. Vo, S. Kumar and O. Gang, Selective transformations between nanoparticle superlattices via the reprogramming of DNA-mediated interactions, Nat. Mater., 2015, 14, 840 CrossRef CAS PubMed.
- M. M. Maye, M. T. Kumara, D. Nykypanchuk, W. B. Sherman and O. Gang, Switching binary states of nanoparticle superlattices and dimer clusters by DNA strands, Nat. Nanotechnol., 2009, 5, 116 CrossRef PubMed.
- Y. Kim, R. J. Macfarlane, M. R. Jones and C. A. Mirkin, Transmutable nanoparticles with reconfigurable surface ligands, Science, 2016, 351, 579–582 CrossRef CAS PubMed.
- J. A. Mason, C. R. Laramy, C.-T. Lai, M. N. O'Brien, Q.-Y. Lin, V. P. Dravid, G. C. Schatz and C. A. Mirkin, Contraction and Expansion of Stimuli-Responsive DNA Bonds in Flexible Colloidal Crystals, J. Am. Chem. Soc., 2016, 138, 8722–8725 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- P. Polak, Z. Zalevsky and O. Shefi, Gold nanoparticles-based biosensing of single nucleotide DNA mutations, Int. J. Biol. Macromol., 2013, 59, 134–137 CrossRef CAS PubMed.
- H. Li and L. Rothberg, Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef CAS PubMed.
- L. Sun, Z. Zhang, S. Wang, J. Zhang, H. Li, L. Ren, J. Weng and Q. Zhang, Effect of pH on the Interaction of Gold Nanoparticles with DNA and Application in the Detection of Human p53 Gene Mutation, Nanoscale Res. Lett., 2009, 4, 216–220 CrossRef CAS PubMed.
- L. Li, L. Wang, Q. Xu, L. Xu, W. Liang, Y. Li, M. Ding, A. Aldalbahi, Z. Ge, L. Wang, J. Yan, N. Lu, J. Li, Y. Wen and G. Liu, Bacterial Analysis Using an Electrochemical DNA Biosensor with Poly-Adenine-Mediated DNA Self-Assembly, ACS Appl. Mater. Interfaces, 2018, 10, 6895–6903 CrossRef CAS PubMed.
- R. Lao, S. Song, H. Wu, L. Wang, Z. Zhang, L. He and C. Fan, Electrochemical Interrogation of DNA Monolayers on Gold Surfaces, Anal. Chem., 2005, 77, 6475–6480 CrossRef CAS PubMed.
- E. M. Nelson and L. J. Rothberg, Kinetics and Mechanism of Single-Stranded DNA Adsorption onto Citrate-Stabilized Gold Nanoparticles in Colloidal Solution, Langmuir, 2011, 27, 1770–1777 CrossRef CAS PubMed.
- K. M. Koo, A. A. I. Sina, L. G. Carrascosa, M. J. A. Shiddiky and M. Trau, DNA-bare gold affinity interactions: mechanism and applications in biosensing, Anal. Methods, 2015, 7, 7042–7054 RSC.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Maximizing DNA Loading on a Range of Gold Nanoparticle Sizes, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- Z. Huang, B. Liu and J. Liu, Parallel Polyadenine Duplex Formation at Low pH Facilitates DNA Conjugation onto Gold Nanoparticles, Langmuir, 2016, 32, 11986–11992 CrossRef CAS PubMed.
- A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez Jr. and P. G. Schultz, Organization of 'nanocrystal molecules' using DNA, Nature, 1996, 382, 609 CrossRef CAS PubMed.
- K. A. Brown, S. Park and K. Hamad-Schifferli, Nucleotide−Surface Interactions in DNA-Modified Au−Nanoparticle Conjugates: Sequence Effects on Reactivity and Hybridization, J. Phys. Chem. C, 2008, 112, 7517–7521 CrossRef CAS.
- J. M. Carnerero, J.-R. Aila, P. M. Castillo and P.-G. Rafael, Covalent and Non-Covalent DNA–Gold-Nanoparticle Interactions: New Avenues of Research, ChemPhysChem, 2017, 18, 17–33 CrossRef CAS.
- D. H. M. Dam, H. Lee, R. C. Lee, K. H. Kim, N. L. Kelleher and T. W. Odom, Tunable Loading of Oligonucleotides with Secondary Structure on Gold Nanoparticles through a pH-Driven Method, Bioconjugate Chem., 2015, 26, 279–285 CrossRef CAS PubMed.
- S. Chakraborty, S. Sharma, P. K. Maiti and Y. Krishnan, The poly dA helix: a new structural motif for high performance DNA-based molecular switches, Nucleic Acids Res., 2009, 37, 2810–2817 CrossRef CAS PubMed.
- A. G. Petrovic and P. L. Polavarapu, Structural transitions in polyriboadenylic acid induced by the changes in pH and temperature: Vibrational circular dichroism study in solution and film states, J. Phys. Chem. B, 2005, 109, 23698–23705 CrossRef CAS PubMed.
- K.-K. Yu, W.-B. Tseng, M.-J. Wu, A. S. K. K. Alagarsamy, W.-L. Tseng and P.-C. Lin, Polyadenosine-based fluorescent probe for reversible pH sensing based on protonated adenine-adenine base pairs: Applications to sensing of enzyme-substrate system and enzymatic logic gates, Sens. Actuators, B, 2018, 273, 681–688 CrossRef CAS.
- C. Colominas, F. J. Luque and M. Orozco, Tautomerism and Protonation of Guanine and Cytosine. Implications in the Formation of Hydrogen-Bonded Complexes, J. Am. Chem. Soc., 1996, 118, 6811–6821 CrossRef CAS.
- A. I. S. Holm, L. M. Nielsen, B. Kohler, S. V. Hoffmann and S. B. Nielsen, Electronic coupling between cytosine bases in DNA single strands and i-motifs revealed from synchrotron radiation circular dichroism experiments, Phys. Chem. Chem. Phys., 2010, 12, 3426–3430 RSC.
- H. D. Hill and C. A. Mirkin, The bio-barcode assay for the detection of protein and nucleic acid targets using DTT-induced ligand exchange, Nat. Protoc., 2006, 1, 324 CrossRef CAS PubMed.
- J. I. Cutler, K. Zhang, D. Zheng, E. Auyeung, A. E. Prigodich and C. A. Mirkin, Polyvalent Nucleic Acid Nanostructures, J. Am. Chem. Soc., 2011, 133, 9254–9257 CrossRef CAS PubMed.
- E. Sutter, K. Jungjohann, S. Bliznakov, A. Courty, E. Maisonhaute, S. Tenney and P. Sutter, In situ Liquid Cell Electron Microscopy of Ag-Pd Galvanic Replacement Reactions on Ag Nanoparticles, Nat. Commun., 2014, 5, 4946–4954 CrossRef CAS.
- E. A. Sutter and P. W. Sutter, Determination of Redox Reaction Rates and Orders by In Situ Liquid Cell Electron Microscopy of Pd and Au Solution Growth, J. Am. Chem. Soc., 2014, 136, 16865–16870 CrossRef CAS.
- E. A. Sutter and P. W. Sutter, In situ liquid cell electron microscopy of Ag-Au galvanic replacement reactions, Nanoscale, 2017, 9, 1271–1278 RSC.
- M. Quinten and U. Kreibig, Optical properties of aggregates of small metal particles, Surf. Sci., 1986, 172, 557–577 CrossRef CAS.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, One-pot colorimetric differentiation of polynucleotides with single base imperfections using gold nanoparticle probes, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, Spherical Nucleic Acids, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
- J.-W. Park and J. S. Shumaker-Parry, Structural Study of Citrate Layers on Gold Nanoparticles: Role of Intermolecular Interactions in Stabilizing Nanoparticles, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS PubMed.
- P. Thaplyal and P. C. Bevilacqua, Experimental Approaches for Measuring pK(a)'s in RNA and DNA, Methods Enzymol., 2014, 549, 189–219 CAS.
- M. Levitt, How many base-pairs per turn does DNA have in solution and in chromatin? Some theoretical calculations, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 640–644 CrossRef CAS.
- H. Zheng, R. K. Smith, Y.-w. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Observation of Single Colloidal Platinum Nanocrystal Growth Trajectories, Science, 2009, 324, 1309–1312 CrossRef CAS.
- T. J. Woehl, J. E. Evans, I. Arslan, W. D. Ristenpart and N. D. Browning, Direct in Situ Determination of the Mechanisms Controlling Nanoparticle Nucleation and Growth, ACS Nano, 2012, 6, 8599–8610 CrossRef CAS PubMed.
- K. L. Jungjohann, S. Bliznakov, P. W. Sutter, E. A. Stach and E. A. Sutter, In Situ Liquid Cell Electron Microscopy of the Solution Growth of Au–Pd Core–Shell Nanostructures, Nano Lett., 2013, 13, 2964–2970 CrossRef CAS PubMed.
- P. Sutter, Y. Li, C. Argyropoulos and E. Sutter, In Situ Electron Microscopy of Plasmon-Mediated Nanocrystal Synthesis, J. Am. Chem. Soc., 2017, 139, 6771–6776 CrossRef CAS PubMed.
- E. Sutter, K. Jungjohann, S. Bliznakov, A. Courty, E. Maisonhaute, S. Tenney and P. Sutter, In situ liquid-cell electron microscopy of silver–palladium galvanic replacement reactions on silver nanoparticles, Nat. Commun., 2014, 5, 4946 CrossRef CAS PubMed.
- P. J. M. Smeets, K. R. Cho, R. G. E. Kempen, N. A. J. M. Sommerdijk and J. J. De Yoreo, Calcium carbonate nucleation driven by ion binding in a biomimetic matrix revealed by in situ electron microscopy, Nat. Mater., 2015, 14, 394 CrossRef CAS PubMed.
- J. Park, H. Zheng, W. C. Lee, P. L. Geissler, E. Rabani and A. P. Alivisatos, Direct Observation of Nanoparticle Superlattice Formation by Using Liquid Cell Transmission Electron Microscopy, ACS Nano, 2012, 6, 2078–2085 CrossRef CAS PubMed.
- E. Sutter, P. Sutter, A. V. Tkachenko, R. Krahne, J. de Graaf, M. Arciniegas and L. Manna, In situ microscopy of the self-assembly of branched nanocrystals in solution, Nat. Commun., 2016, 7, 11213 CrossRef CAS PubMed.
- P. Sutter, B. Zhang and E. Sutter, Radiation Damage During In situ Electron Microscopy of DNA-Mediated Nanoparticle Assemblies in Solution, Nanoscale, 2018, 10, 12674–12682 RSC.
- V. Kanike, J. Meesungnoen and J.-P. Jay-Gerin, Acid spike effect in spurs/tracks of the low/high linear energy transfer radiolysis of water: potential implications for radiobiology, RSC Adv., 2015, 5, 43361–43370 RSC.
- K. H. Schmidt and S. M. Ander, Formation and recombination of the hydronium ion (H3O + ) and hydroxide in irradiated water, J. Phys. Chem., 1969, 73, 2846–2852 CrossRef CAS.
- G. C. Barker, P. Fowles, D. C. Sammon and B. Stringer, Pulse radiolytic induced transient electrical conductance in liquid solutions. Part 1.-Technique and the radiolysis of water, Trans. Faraday Soc., 1970, 66, 1498–1508 RSC.
- R. F. Anderson, B. Vojnovic and B. D. Michael, The radiation-chemical yields of H3O+ and OH- as determined by nanosecond conductimetric measurements, Radiat. Phys. Chem., 1985, 26, 301–303 CrossRef CAS.
- B. Cercek and M. Kongshaug, Hydrogen ion yields in the radiolysis of neutral aqueous solutions, J. Phys. Chem., 1969, 73, 2056–2058 CrossRef CAS PubMed.
- A. K. Pikaev, S. A. Kabakchi and A. A. Zansokhova, Yields and reactions of hydrogen ions on radiolysis of water and aqueous solutions, Faraday Discuss. Chem. Soc., 1977, 63, 112–123 RSC.
- J. Y. Lu, Z. Aabdin, N. D. Loh, D. Bhattacharya and U. Mirsaidov, Nanoparticle Dynamics in a Nanodroplet, Nano Lett., 2014, 14, 2111–2115 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08421a |
| This journal is © The Royal Society of Chemistry 2019 |