
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuper-resolution microscopy can identify specific protein distribution patterns in platelets incubated with cancer cells†
Jan
Bergstrand
a,
Lei
Xu
a,
Xinyan
Miao
a,
Nailin
Li
b,
Ozan
Öktem
c,
Bo
Franzén
d,
Gert
Auer
d,
Marta
Lomnytska‡
d and
Jerker
Widengren
 *a
*a
aRoyal Institute of Technology (KTH), Department of Applied Physics, Experimental Biomolecular Physics, Albanova Univ Center, SE-106 91 Stockholm, Sweden. E-mail: jwideng@kth.se
bKarolinska Institutet, Department of Medicine-Solna, Clinical Pharmacology, L7:03, Karolinska University Hospital-Solna, SE-171 76 Stockholm, Sweden
cRoyal Institute of Technology (KTH), Department of Mathematics, Lindstedsvägen 25, SE-100 44 Stockholm, Sweden
dKarolinska Institutet, Department of Oncology–Pathology, K7, Z1:00, Karolinska University Hospital, 171 76 Stockholm, Sweden
First published on 6th May 2019
Abstract
Protein contents in platelets are frequently changed upon tumor development and metastasis. However, how cancer cells can influence protein-selective redistribution and release within platelets, thereby promoting tumor development, remains largely elusive. With fluorescence-based super-resolution stimulated emission depletion (STED) imaging we reveal how specific proteins, implicated in tumor progression and metastasis, re-distribute within platelets, when subject to soluble activators (thrombin, adenosine diphosphate and thromboxane A2), and when incubated with cancer (MCF-7, MDA-MB-231, EFO21) or non-cancer cells (184A1, MCF10A). Upon cancer cell incubation, the cell-adhesion protein P-selectin was found to re-distribute into circular nano-structures, consistent with accumulation into the membrane of protein-storing alpha-granules within the platelets. These changes were to a significantly lesser extent, if at all, found in platelets incubated with normal cells, or in platelets subject to soluble platelet activators. From these patterns, we developed a classification procedure, whereby platelets exposed to cancer cells, to non-cancer cells, soluble activators, as well as non-activated platelets all could be identified in an automatic, objective manner. We demonstrate that STED imaging, in contrast to electron and confocal microscopy, has the necessary spatial resolution and labelling efficiency to identify protein distribution patterns in platelets and can resolve how they specifically change upon different activations. Combined with image analyses of specific protein distribution patterns within the platelets, STED imaging can thus have a role in future platelet-based cancer diagnostics and therapeutic monitoring. The presented approach can also bring further clarity into fundamental mechanisms for cancer cell–platelet interactions, and into non-contact cell-to-cell interactions in general.
Introduction
Increasing evidence suggests cross-talk between tumor cells and circulating platelets as a major driving force behind cancer progression and metastasis.1 This cross-talk is highly multi-faceted: on the one hand, platelets can adhere to circulating tumor cells (CTCs). Thereby, platelets help CTCs evade immune surveillance, survive shear stress of blood flow, and promote their tethering and arrest to capillary blood vessel walls. Moreover, by direct binding or via soluble mediator molecules, activated platelets can induce tumor growth, epithelial–mesenchymal transition, and promote angiogenesis and tumor cell establishment at distant sites.1–5Vice versa, tumor cells can also activate platelets in multiple ways, resulting in so-called tumor-educated platelets (TEPs).3,6 CTCs can bind directly to specific molecules on the platelet plasma membrane (PM), including P-selectin, integrins, glycoproteins, and mucin-binding receptors. Alternatively, CTCs can also activate platelets by extracellular release of bioactive compounds, such as thrombin, adenosine diphosphate (ADP) and thromboxane A2 (TXA2). Moreover, extracellular vesicles (EVs)2–4 from tumor cells can adhere to P-selectin on the platelet PM,7 then merge with the PM, whereby their content of tumor-derived proteins or RNA can be sequestered by the platelets.Altered contents of specific proteins in circulating platelets have been found both in mice bearing human malignant tumor xenografts,8,9 and in patients with different, newly diagnosed metastatic diseases.10 Such alterations have been ascribed to activation-specific protein storage, release and uptake in the platelets,11–14 but also to RNA uptake from EVs, followed by altered protein expression by the platelets themselves.2,3,6 They can be found by several methods, including 2D-electrophoresis, mass spectrometry, fluorescence-based flow cytometry and confocal laser scanning microscopy (CLSM).10,11,15 However, the mere content of specific proteins in the platelets is not sufficient to identify specific activation states of platelets. Additional information is also required to resolve the open question of how platelets specifically can regulate uptake and release of certain proteins.
In platelets, alpha-granules are the largest (200–500 nm in diameter) and most abundant secretory granules (50–80 per cell), containing a variety of proteins regulating angiogenesis, coagulation, cell proliferation, adhesion, and immune responses.3,12,13,16 Platelet releasate15 and CLSM co-localization studies11 suggest that selective release of proteins is possible because there are different sets of proteins in the individual alpha-granules, which then can be selectively released depending on stimulus. Higher resolution studies by electron microscopy (EM)17 and fluorescence super-resolution microscopy (SRM)18,19 however, rather indicate that proteins are stored in clusters within individual alpha-granules, down to 50 nm in diameter, and with highly segregated protein cargo. Other studies suggest kinetic differences in the release as a mechanism for selectivity,13,20 and intra-granular protein cargo segregation with alternative routes to and fusion with the open canalicular system (OCS) and the PM.12
Such mechanisms can be expected to be reflected in redistribution of P-selectin and other membrane proteins within the platelets upon activation. P-Selectin is a cell adhesion protein, found predominantly in endothelial cells and platelets. In resting platelets, P-selectin is mainly localized in the alpha-granule membranes. Upon platelet activation, it can get exposed on the platelet PM surface, thereby mediating platelet–tumor cell interactions and playing a major role in tumor cell thrombus formation, adhesion to blood vessel walls, extravasation and metastasis.16,21–23 P-Selectin is used as a biomarker for platelet activation, is often increased in cancer patients,4,24 while decreased numbers of thrombi and metastases have been observed in P-selectin-deficient colon carcinoma mice models.4,23,25,26 Taken together, many of the underlying mechanisms leading to TEPs are not fully understood, including the role of P-selectin. There is also limited evidence for alternative fusion routes of granules and how they may depend on type and character of platelet stimulation. In this study, we acquired and analyzed high-resolution SRM images on how P-selectin and other proteins redistribute in platelets upon different activations, as a means to resolve these questions.
SRM techniques show promise to reveal many un-resolved details in the tumor cell–platelet interactions.13,27,28 In previous work,19,29 we introduced fluorescence-based, stimulated emission depletion (STED) SRM to study distribution patterns of specific proteins in platelets upon activation. For such analyses, STED imaging combines the major advantages of immunofluorescence CLSM and immuno-gold EM: high spatial resolution, high degree of labelling, extensive and perturbative sample preparations can be avoided, and studies of larger numbers of intact platelets are feasible. Among the proteins studied (pro-angiogenic VEGF, anti-angiogenic PF-4 and Fibrinogen (Fg)), we found clear differences in sizes, numbers and spatial distributions of their regional clusters. Also, no significant co-localization between the proteins was found, indicating that the proteins are stored differently in the platelets. Upon distinct activations by well-known platelet activators (thrombin and ADP), rearrangements occurred, which were specific for a certain protein and activation, indicating different release and uptake mechanisms. These platelet studies,19,29 along with recent SRM studies on cells in general,30–36 indicate the potential of images resolving high-resolution spatial distribution patterns of biomolecules in cells as a source of diagnostic information. This information is not within reach by CLSM, EM and other techniques.27,28,37
Here, we show that STED imaging can not only detect specific protein distribution patterns in platelets upon distinct activation by known agents, but also detect and identify the influence of different cancer cells on protein distribution patterns in platelets. Specifically, we studied the distribution patterns of the proteins P-selectin, VEGF, and Fg, implicated in tumor progression and metastasis, and Erp29, overexpressed in platelets from ovarian cancer patients.38 Freely diffusing platelets, incubated with cancer cells, showed distinctive changes in the spatial distribution patterns of P-selectin, but not for the other proteins. These changes were to a significantly lesser extent, if at all, found in platelets incubated with normal cells. Further, we developed an image analysis of the platelet STED images with their P-selectin distribution patterns, which allowed us to classify platelets exposed to cancer cells, non-activated platelets, and platelets exposed to non-cancer cells or to soluble activators, in an objective and automated manner. Finally, we discuss how our findings relate to previous observations of the interplay between tumor cells and platelets. We conclude that STED imaging, combined with image analyses of specific protein distribution patterns within the platelets, adds important information for identification of specific platelet activations, and can thus have a role in future platelet-based cancer diagnostics and therapeutic monitoring. The presented approach can also bring further clarity into fundamental mechanisms for cancer cell–platelet interactions, and offers extended possibilities to identify and analyse cells subject to non-contact cell-to-cell interactions in general.
Experimental
Platelet isolation, incubation of platelets with cell lines, and fixation
Fresh blood was drawn from healthy donors, having no medicine intake two weeks before sampling, collected in 3.8% (g ml−1) trisodium citrate, and centrifuged (150g, 20 min) at RT without break. The platelet rich plasma (PRP) fraction was then obtained from the supernatant, as previously described.29Five different cell-lines were used for co-culturing: the breast cancer cell-lines MCF7 and MDA-MB231, the ovarian high-grade serous cancer cell-line EFO21, and the immortalized non-cancer cell-lines 184A1 and MCF10A as control cells. 500 μl of the cell suspensions (2 × 105 cells per ml) was seeded on polylysine-coated coverslips in a 24-well plate and incubated at 37 °C for 6 hours. 5 × 107 platelets in enriched PRP were then added to each well, together with 600 μl serum-absent medium to avoid nonspecific activation of the platelets. Suspensions of the freely diffusing platelets were incubated with the cells for 2 hours at 37 °C, and then fixed with 2% paraformaldehyde. The fixed platelets were spun down (1000g, 10 min, 22 °C), and then diluted with an equal amount of BSA buffer (pH 7). As additional controls, platelet samples were also prepared without co-culturing, but otherwise as described above, in three such samples, ADP (10 mM for 5 min immediately before fixation), thrombin or TXA2 (0.1 U ml−1 and 1 μM, respectively for 10 min immediately before fixation) was separately added as a platelet activator, as previously described.19
Immunostaining
The immunostaining essentially followed previously established protocols.19 Sheep anti-mouse and goat anti-rabbit antibodies (Dianova, Hamburg, Germany) were conjugated with the fluorophores ATTO 594 and ATTO 647N, respectively (Atto-Tec GmbH, Siegen, Germany). Mouse monoclonal antibodies were used for targeting human P-selectin (Novus Biologicals, Europe), Erp29 (BD Science, Europe) and Fg (Santa Cruz Biotechnology, Heidelberg, Germany), rabbit polyclonal antibodies (Santa Cruz Biotechnology, Heidelberg, Germany) were used for human VEGF and Fg. Unless otherwise stated, DPBS was used throughout the study as buffer. Fixed platelets were permeabilized with Triton X-100 (0.5%, 10 min), blocked for nonspecific binding with bovine serum albumin (1% w/v, Sigma, Sweden) for 1 hour at RT, then washed 3 times with DPBS, and incubated overnight (4 °C, moist atmosphere) with primary antibodies against Erp29 (2 μg mL−1) and VEGF (2.5 μg mL−1), of Fg (2.5 μg mL−1) and P-selectin (2 μg mL−1). The samples were then washed 3 times with DPBS, incubated with the secondary antibody (3 μg mL−1) for 1 h in RT, and then washed three times before being mounted onto microslides with Mowiol-Dabco mounting medium (Sigma, Sweden).STED imaging
STED imaging was performed with an instrument (Abberior Instruments, Göttingen, Germany), built on a stand from Olympus (IX83), with a four-mirror beam scanner (Quad scanner, Abberior Instruments), and modified for two-color STED imaging: two fiber-coupled, pulsed (20 MHz) diode lasers emitting at 637 nm (LDH-D-C, PicoQuant AG, Berlin) and 594 nm (Abberior Instruments) are used for excitation (excitation by the two lasers and gating of the detectors alternated in each pixel during scanning to minimize cross-talk). The beam of a pulsed fiber laser (MPB, Canada, model PFL-P-30-775-B1R, 775 nm emission, 40 MHz repetition rate, 1.2 ns pulse width, 1.25 W maximum average power, 30 nJ pulse energy), reshaped by a phase plate (VPP-1c, RPC Photonics) into a donut profile, is used for stimulated emission. The three laser beams are overlapped and then focused by an oil immersion objective (Olympus, UPLSAPO 100XO, NA 1.4) into the sample. The fluorescence is collected through the same objective, passed through a dichroic mirror, a motorized confocal pinhole (MPH16, Thorlabs, set at 50 μm diameter) in the image plane, split by a second dichroic mirror, and then detected by two single-photon counting detectors (Excelitas Technologies, SPCM-AQRH-13), equipped with separate emission filters (FF01-615/20 and FF02-685/40-25, Semrock) and a common IR-filter (FF01-775/SP-25, Semrock) to suppress any scattered light from the STED laser. In this study, a spatial resolution (FWHM) of about 25 nm could be reached. Image acquisition, including laser timing/triggering and detector gating is controlled via a FPGA-card and by the Imspector software (Abberior Instruments). To reduce noise in the STED images a Gaussian smoothing filter (built-in smoothing function in ImageJ Fiji) was used. In the STED images, protein cluster sizes and numbers of clusters within the platelets were determined by an automatized algorithm written in MATLAB R2013b, as previously described.29Computer analyses
All code was custom written in MATLAB R2013b except for the dictionary learning. The dictionary learning was written in Python 3.5 implementing the Scikit package39 which is a free Python package for utilizing different kinds of machine learning tools. This package includes several options for tuning different parameters within the dictionary learning algorithm, the Orthogonal Matching Pursuit algorithm (OMP)40 and the structural similarity (SSIM) norm,41 which affects the outcome of the analysis. However, we found that the default setting of these parameters and with SSIM set to ‘dynamic range’ gave the best results without further need for tuning.Results
STED imaging of VEGF, Erp29, and Fibrinogen in resting platelets, when incubated with cancer and non-cancer cells, and upon activation by ADP
Distribution patterns of VEGF, Erp29 and Fibrinogen in platelets were separately imaged by confocal and high-resolution STED imaging, with a resolution down to 25 nm (see Methods section). Images were acquired from about 100 individual platelets (see caption Fig. 1 for exact numbers), for each of the proteins and for each of the following activation conditions (see Methods section); platelets exposed to ADP, non-exposed platelets (resting), and platelets incubated with three different tumor cell-lines: MCF7, MDA-MB231, EFO21. In the STED images, spatial distribution patterns of the studied proteins within the platelets could be resolved on a sub-granular level (Fig. 1A and B). VEGF and Fibrinogen were found to be spatially confined to smaller sub-granules/clusters less than 100 nm in diameter, presumably within the alpha-granules, as previously reported.19,29 Similar small clusters in the platelets were also found for Erp29. | ||
| Fig. 1 Imaging and cluster analysis for the proteins Erp29, VEGF and Fibrinogen. A. Representative raw STED images of platelets stained for Erp29 (green) and VEGF (red) for all activation conditions we investigated for these proteins. B. Representative raw STED images of platelets stained for fibrinogen for all activation conditions we investigated for these proteins. The platelets in A and B are not the same platelets since the STED setup we used is limited to at most two colors. Scale bars: 1 μm. C. Number of clusters per platelet of the proteins VEGF, Erp29, and Fibrinogen, and the average diameters of the clusters within a platelet, as determined by STED imaging (see main text). In the figures, the number and average diameter of the clusters, are plotted by their mean values and by their standard error of the mean (bars) for the different platelet categories: resting (non-exposed) platelets (blue), platelets activated by ADP (red), and platelets incubated with tumor cells (MCF7, MB231, EFO21). Number of platelets for each condition included in the analysis: resting: 117, ADP: 88, EFO21: 107, MDA-MB231: 108, MCF7: 100. | ||
The distribution patterns of VEGF, Erp29 and Fibrinogen in non-activated platelets and in platelets subject to the different types of activation were analyzed, and protein cluster sizes and numbers of clusters within the individual platelets were determined, as previously described (Fig. 1C).29 For platelets subject to ADP activation, distinct differences in protein cluster size and/or number of clusters could be observed for VEGF and Fibrinogen, in agreement with our previous studies,19 and also for Erp29. However, in platelets incubated with tumor cells, no significant differences compared to non-activated platelets were found in the cluster sizes or numbers for these three proteins (Fig. 1C), and also not in their overall spatial distribution patterns within the platelets.
P-Selectin frequently forms into circular nanostructures in platelets incubated with cancer cells
In contrast to VEGF, Erp29 and Fibrinogen, P-selectin was not found to be confined to smaller clusters. Moreover, differences in the spatial distribution patterns were found not only upon activation by ADP, but also when incubated with the three tumor cell types. The STED images from this latter category of platelets reveal circular patterns in a significant fraction of the platelets (Fig. 2A). These circular patterns have a diameter of about 200–400 nm (Fig. S1†), consistent with the size of platelet alpha-granules.3,12,13,16 These patterns were found to a much lesser extent, if at all, in platelets not incubated with tumor cells, with or without ADP added. This indicates that tumor cells influence the platelets to re-distribute their content of P-selectin and possibly also their alpha-granules. It can further be noted that the P-selectin re-distribution, readily observable with high-resolution STED imaging, cannot be detected by confocal microscopy with diffraction-limited resolution (Fig. 2B). The same protein cluster analysis as for VEGF, Erp29 and Fibrinogen (Fig. 1C) was also applied on the distribution patterns of P-selectin (Fig. 2C). However, only a minor effect upon tumor cell incubation could be found via this analysis. In the following, we instead investigated to what extent circular nanoscale patterns of P-selectin appeared in platelets incubated with non-cancer cells, or under other forms of platelet activation. We further studied if these circular patterns, together with the overall peripheral distribution of P-selectin within the platelets, could be used as a strategy to identify platelets subject to different, specific activations.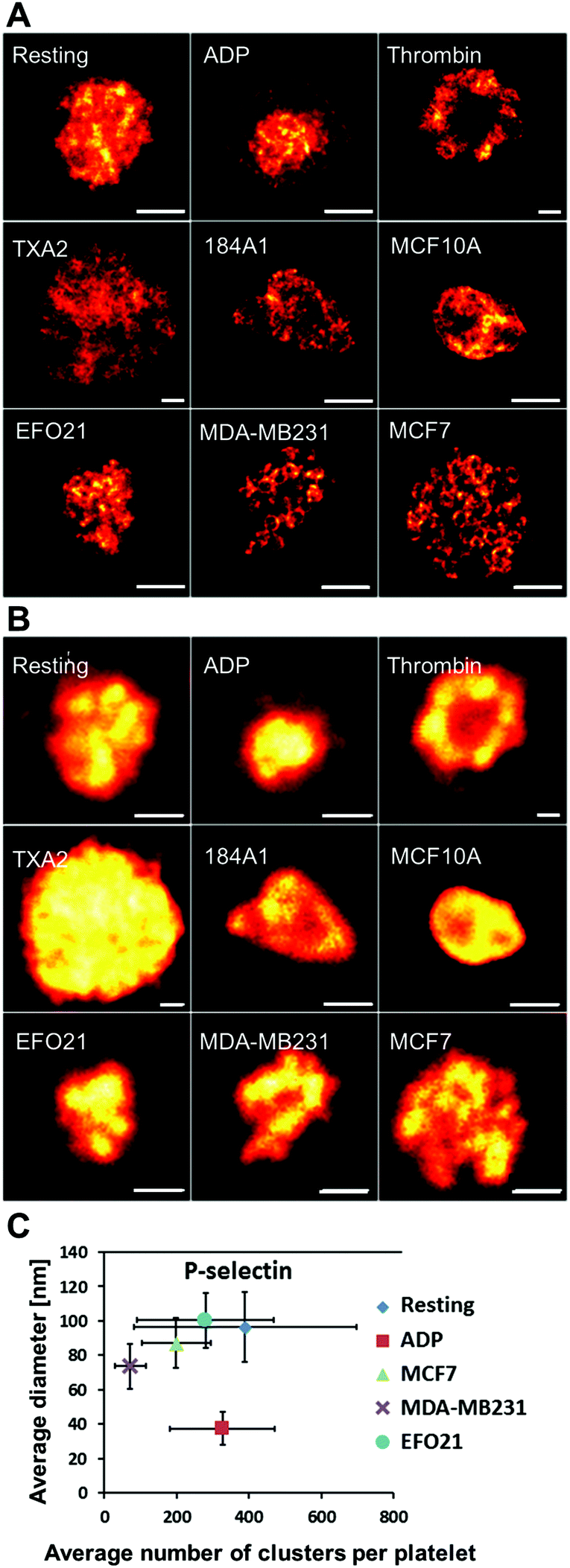 | ||
| Fig. 2 Representative images of P-selectin labeled platelets for all the different activation conditions. A. High resolution STED images with a resolution down to 25 nm. B. Corresponding confocal images, imaged from the same samples as shown in A. With the resolution achieved by confocal microscopy (∼250 nm) it is difficult to see any differences in the detailed distribution patterns of P-selectin in the platelets. However, with the resolution offered by STED imaging (∼25 nm) clear circular patterns is revealed for some of the platelets. In the images shown in A, a circular P-selectin pattern is clearly seen in the images of platelets incubated with MB231 and MCF7 cells. Scale bars 1 μm. C. Analysis of P-selectin cluster size and number of clusters per platelet as in Fig. 1C (see caption in Fig. 1 for number of platelets in each category used for this analysis). The plot shows the average cluster diameter and the mean number of clusters per platelet. Bars indicate the standard error of the mean. | ||
P-Selectin in platelets incubated with non-cancer cells do not show the frequent circular patterns found in platelets incubated with cancer cells
To investigate if formation of the circular patterns of P-selectin are particularly triggered by the presence of the tumor cells, or if presence of corresponding benign cells also yields similar P-selectin patterns, we imaged P-selectin in platelets, prepared in the same way, but now instead incubated with immortalized non-cancer cells (184A1, MCF10A). Similar to ADP-activated platelets, the STED images of these platelets give at hand that P-selectin organizes into circular patterns to a far lesser extent than in platelets exposed to the tumor cells (Fig. 2A).P-Selectin in platelets activated by ADP, thrombin and TXA2 do not show the frequent circular patterns found in platelets incubated with cancer cells
ADP has been found to be released by several different tumor types,3,42 and the same differential release of VEGF have been reported for platelets activated by either ADP or by MCF7 breast cancer cells,15 suggesting similar activation mechanisms. In our study however, the protein distribution patterns in platelets exposed to ADP were clearly distinguished from those in platelets incubated with tumor cells. Differences were evident both from sizes and numbers of VEGF, Fibrinogen and Erp29 clusters in the platelets (Fig. 1C), and in the spatial distribution of P-selectin (Fig. 2A). This suggests different re-distribution mechanisms and that the re-distribution of P-selectin in platelets incubated with tumor cells is not primarily mediated by ADP. To further explore possible mediators of platelet activation, we investigated how the spatial distribution of P-selectin in the platelets is altered upon activation by the major platelet activation agents, TXA2 and thrombin, reported to be directly secreted by different cancer cells.3,4,42 Upon thrombin activation, a clear accumulation of P-selectin in the periphery of the platelets was observed (Fig. 2A), well in agreement with the common view of thrombin, mobilizing P-selectin to the platelet surface.3,4 With TXA2 activation, we could also see a clear difference in the P-selectin distribution, with P-selectin then distributed over a significantly larger area in the platelets (Fig. 2A). However, neither thrombin- nor TXA2-activation generated the frequent circular patterns of P-selectin found in platelets incubated with cancer cells.Manual, blind classification of platelets, based on their P-selectin distribution patterns
We next investigated if the re-distribution of P-selectin in platelets incubated with tumor cells can be used to identify such platelets among other platelets. First, STED images of P-selectin in platelets were subject to a blind classification procedure: by visual inspection of all P-selectin STED images, we defined three categories of platelets from the P-selectin distribution patterns in the platelets, based on the visibility of circular patterns in the platelets: not visible, discernible, and clearly visible (Fig. 3A). All platelet images were then classified into one of these three categories, in a blind manner, with each image displayed at random, and with no information about what activation condition the imaged platelet was subject to. The outcome of this classification (Fig. 3B), confirms that platelets exposed to tumor cells display a higher incidence of circular patterns of P-selectin than platelets exposed to benign cells, or to no cells at all, and that a manual classification is feasible.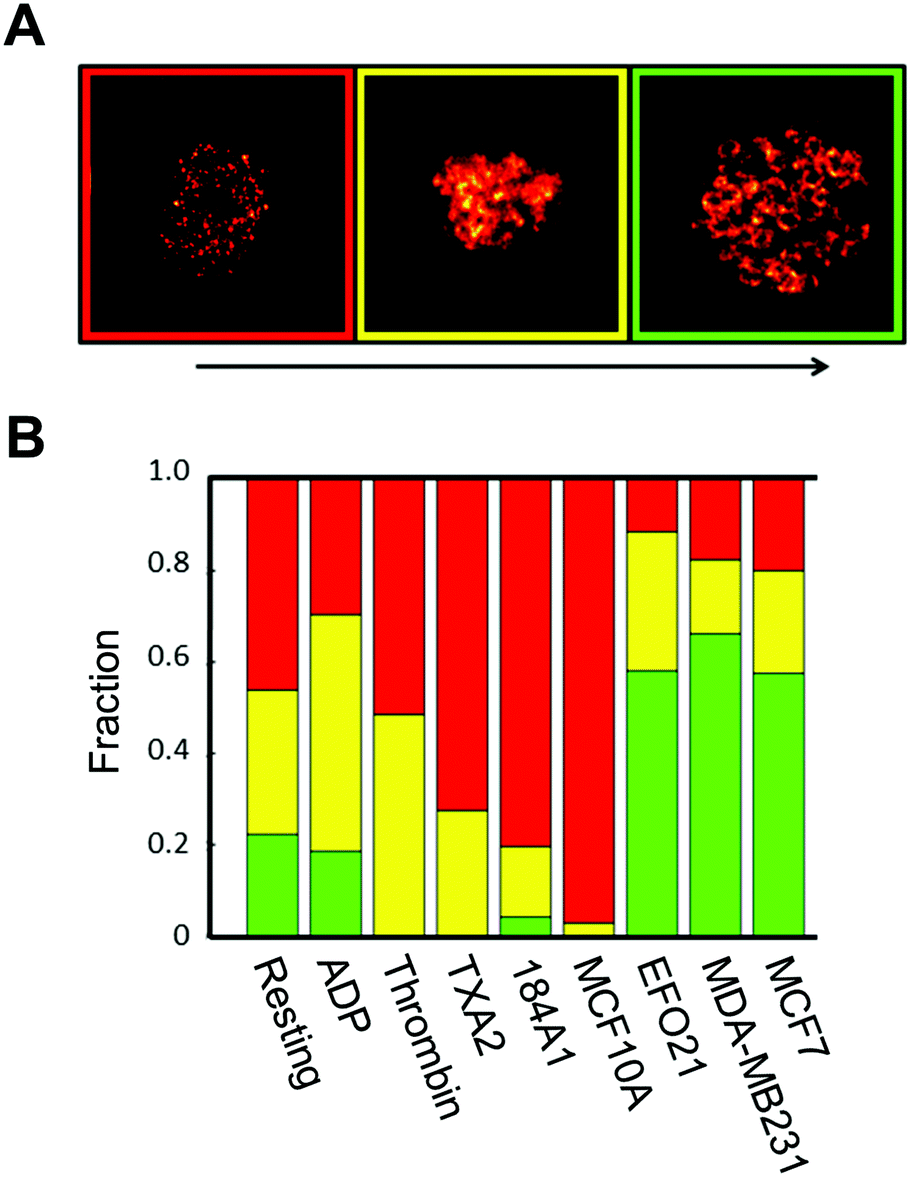 | ||
| Fig. 3 Manual classification of P-selectin images. A. Upon visual inspection of the P-selectin images three different categories were identified based on the circular P-selectin pattern. Red category: no visible circular pattern and mostly cluster like structures. Yellow category: some visible circular pattern but sometimes fuzzy and not so easily identified circles. Green category: clearly visible and easily identifiable circular patterns of P-selectin. B. Outcome of the blind manual classification, based on the three categories, as defined in A. Number of platelets for each condition that was included in the classification: resting: 117, ADP: 88, thrombin: 100, TXA2: 103, 184A1: 99, MCF10A: 98 EFO21: 107, MDA-MB231: 108, MCF7: 100. | ||
Development of an objective, automatic classification of platelet activation states, based on dictionary learning algorithms, image reconstruction and structure similarity estimates
Next, we investigated if the manual classification procedure above could be replaced by an automatized, objective classification of the platelet images. For this purpose, we implemented an algorithm based on dictionary learning (Python, Scikit package).39 Here, a dictionary of image elements is built (trained) from a larger number (≥104) of example images (training data), in our case ideally from STED images of platelets incubated with tumor cells. However, not having this large number of experimental example images at hand, we instead used simulated images as basis for training the dictionary used in the classification (schematically described in Fig. 4A). These computer-simulated platelet images were generated to have cluster-like structures randomly distributed in the platelets (further details in S2†). While training the dictionary, we enforced sparsity, i.e. that images in the training data are expressible as a linear combination of the elements in the dictionary using the least possible number of elements. The recorded STED images could then be classified, based on how well they could be reconstructed from the best possible linear combination of dictionary elements, i.e. to what extent their P-selectin distribution patterns deviate from the semi-random distributions in the images used to create the dictionary. The sparse representation of a platelet image in terms of elements in a trained dictionary was computed using the Orthogonal Matching Pursuit (OMP) algorithm,40 which iteratively selects the best dictionary elements based on a certain similarity measurement to approximately obtain the sparsest representation of the image. | ||
| Fig. 4 Objective classification of P-selectin images using dictionary learning. A. Schematic outline of the dictionary training process used in this study, and the reconstruction of experimental STED images based on the trained dictionary. The obtained dictionary consists of 9 × 9 image patches of 30 × 30 pixels. The dictionary is constructed to allow least possible terms (sparsity) in the linear combinations necessary to represent an image in the training set. See S3† for further details. With a trained dictionary at hand, each experimental STED image of a platelet and its distribution of P-selectin is reconstructed using the Orthogonal Matching Pursuit (OMP) algorithm. Finally, the reconstructed image is compared with the original image using the SSIM norm, yielding a value between 0 and 1, depending on how well the reconstructed image reproduces the original image (see the ESI† for further details). B. Fraction of platelets (color-coded) reaching a certain SSIM norm value with the dictionary used in this study, and for the different platelet activation conditions. | ||
The platelet image reconstructed from the trained dictionary is then compared with the original image, based on a Structural Similarity (SSIM) norm,41 which yields a number between 0 and 1, depending on the degree of similarity. An SSIM norm value is thereby assigned to every experimental P-selectin platelet image. Plotting the cumulative fraction of images reaching a certain SSIM norm value for the different types of platelet activation allows one to clearly distinguish platelets exposed to tumor cells from platelets exposed to benign cells, or to no cells at all, and also from ADP-activated platelets. Interestingly, the resulting classification pattern for these categories of platelets (Fig. 4B) is quite similar to that obtained from the manual classification (Fig. 3B), but is based on an automatized, objective classification. However, for thrombin- and TXA2-activated platelets there is a difference between the two categorizations. While the manual categorization suggests no clearly visible circular P-selectin patterns, the automatized categorization indicates that the distribution of P-selectin in these platelets significantly deviates from a random one.
Radial distribution of P-selectin used as an additional classification feature
To further improve the classification, and allow this classification to comprise all platelet categories in this study, we investigated if the radial distributions of P-selectin in the platelets could be used as an additional classification feature, obtained as intensity traces along lines drawn in the platelets, from their centers of mass to their peripheries, in 72 different directions within each platelet (Fig. 5A and S3†).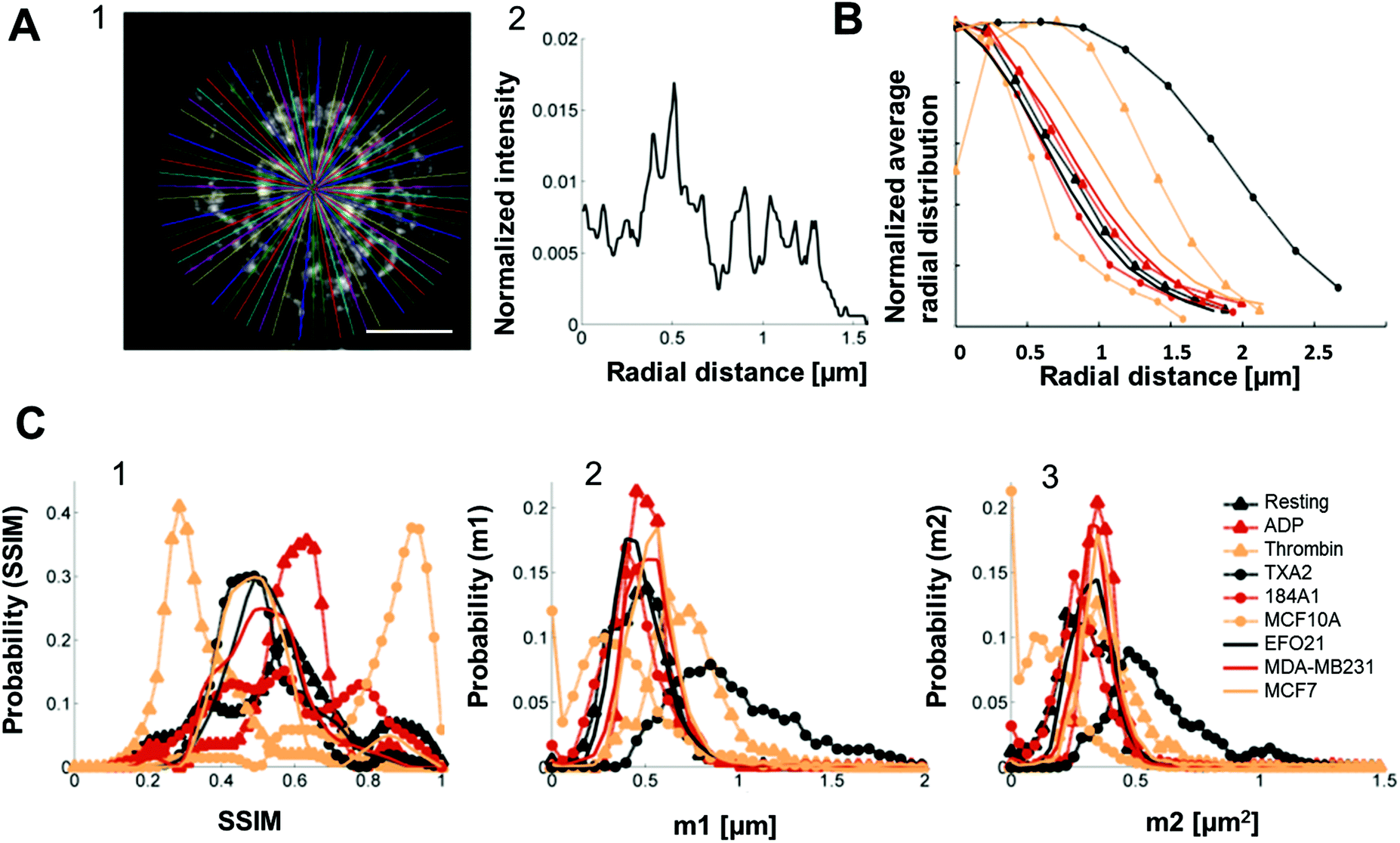 | ||
| Fig. 5 Radial distribution analysis of P-selectin labeled platelets and histograms constructed from both the dictionary learning outcome and the radial distribution. A1. 72 lines, with an angle of 5° between them, drawn from the center of mass of a representative platelet stained for P-selectin (here the platelet was activated by incubation with tumor cell-line EFO21). Scale bar: 1 μm. A2. An example of an intensity trace taken along one of the lines in A1 (this particular intensity trace corresponds to the horizontal line going from the center out to the right). B. Average radial distribution of platelets for all the different activation conditions normalized such that maximum value = 1 for easier comparison. C1. Histograms constructed of all of the SSIM values for all the different activation conditions separately. C2. Histograms of the m1 parameters (as calculated by eqn (S2)†) constructed for each activation condition separately. C3. Histograms of the m2 parameters (as calculated by eqn (S3)†) constructed for each activation condition separately. All histograms were normalized such that the area underneath them equals unity. | ||
The average radial distributions of P-selectin, calculated for the different platelet categories (Fig. 5B) are all very similar, except for the distributions in thrombin- and TXA2-activated platelets. In these platelets, P-selectin was distributed over a significantly larger area (average diameter of 4–5 μm and 3 μm for TXA2- and thrombin-activated platelets, respectively, about twice as large as observed for the other activation conditions). Therefore, if both the radial distribution and dictionary learning (SSIM) analyses are taken into account, a more accurate categorization should be possible, comprising also the thrombin- and TXA2-activated platelets. For each platelet, we calculated the first and second order moments (m1,m2) of their radial distributions. The m1 and m2 values were calculated from the pixel intensities along a line, from the center of mass of individual platelets (determined by eqn (S1)†) to their periphery, and in 72 different radial directions in each platelet image. The first order moment m1 along a certain radial direction (calculated from eqn (S2)†) can be considered as the center of mass of the pixel intensities along that radial direction and could therefore be an indicator of the extension of the platelet in a particular radial direction. The second order moment m2 (given by eqn (S3)†) is the variance of the pixel intensity distribution around m1 for a certain radial direction. For example, a platelet with most of the P-selectin located at the periphery (e.g. thrombin activated platelets, see Fig. 2A) will have a smaller variation in the pixel intensity distribution in the radial direction, compared to a platelet where the P-selectin is more evenly distributed (e.g. in resting platelets, see Fig. 2A). Thus the former platelet would yield a smaller m2-value than the latter. We then constructed histograms of the obtained SSIM, m1 and m2 values for all platelet categories (Fig. 5C). Using these histograms as probability distributions the probability for a platelet with a given set of SSIM, m1 and m2 values to belong to a certain category can then be calculated and used for categorization (S4†). To test this categorization, we imaged 10 test platelets for each activation condition (their parameter values not included in the histograms) and calculated the corresponding SSIM, m1 and m2 values for each of these platelets, and the probabilities that they belonged to the different categories. The outcome of this analysis (Fig. 6) indicates that all platelets can be categorized in an automatized and accurate manner, including thrombin- and TXA2-activated platelets. Even for resting platelets, with the worst classification (5 of 10 categorized correctly as resting platelets), the probability that ≥5 platelets would be assigned to another specific category than the correct one is small (<4%).
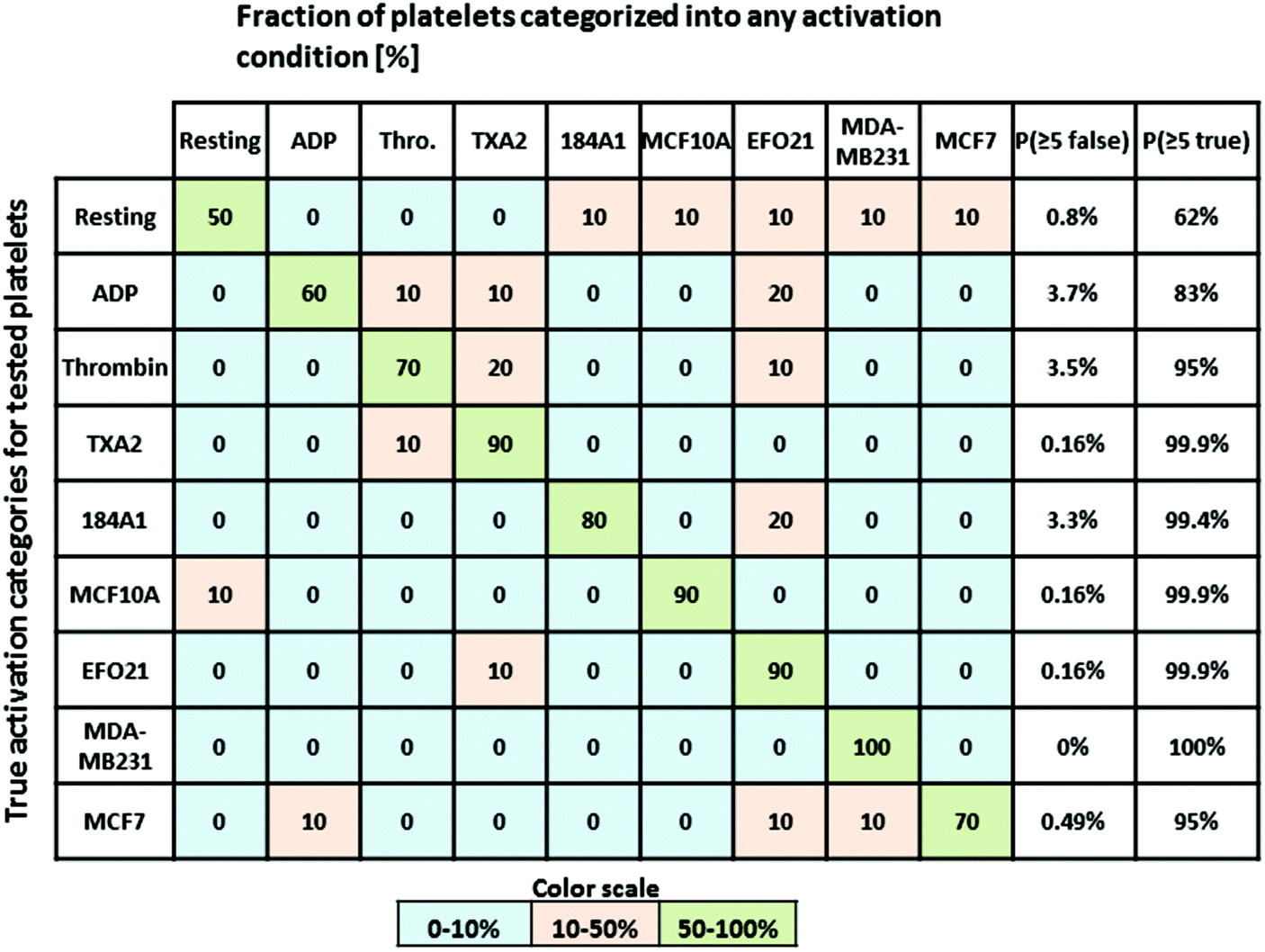 | ||
| Fig. 6 Categorization of different activation conditions based on SSIM values and first and second order moments of radial distributions, calculated from 10 individual platelets in each activation category. Rows show the activation category of the tested platelets. Columns show the activation category the tested platelet was categorized into. Values in the table are the fraction in % of platelets categorized into respective column. The two last columns show the probability, based on the outcome of this categorization, for categorizing 5 or more platelets into one of all the false categories, and to categorize 5 or more platelets into the correct category (i.e. 5 or more platelets are truly categorized). | ||
Discussion
In this study, we introduce STED SRM to investigate how tumor cells, incubated with freely diffusing platelets, influence how the platelets store proteins involved in tumor development and metastasis. While no significant effects could be observed on the storage patterns of VEGF, Erp29 or Fibrinogen, we found that tumor cells influence the P-selectin distribution within the platelets, in a way not found for benign cells. Moreover, the circular distribution patterns of P-selectin in tumor cell-exposed platelets were rarely found in platelets subject to ADP, TXA2 or thrombin activation, indicating that release of these compounds from the tumor cells is not a sole or major mechanism behind the altered P-selectin patterns in the platelets.The high incidence of circular patterns of P-selectin in platelets incubated with tumor cells may seem to contradict the view of P-selectin as a general surface biomarker of platelet activation.4,43 However, these patterns could be consistent with both accumulation of P-selectin in the membrane of the alpha-granules, and with transfer of P-selectin to the PM of the platelets. EM studies have shown that upon platelet activation the alpha-granules tend to migrate to the center, become closely apposed, and prior to exocytosis and cargo release, to fuse with one another, with the platelet PM, or with the tunneling membrane invaginations of the OCS.3,12–14,16,44 Similar microstructural changes have been observed in platelets from patients with non-small cell lung cancer.45 In platelets incubated with tumor cells, we see circles of P-selectin in the center, consistent with the reported migration of alpha-granules to this region. The fact that we do not see a peripheral pattern of P-selectin in these platelets, as we see in platelets activated by thrombin, could mean that the fusion of alpha-granules with the plasma membrane is halted at this stage. Alternatively, it has been reported that while the alpha-granule contents are released upon membrane fusion, the alpha-granule membrane “ghost” can remain as a pore or opening in the PM or OCS.13 EM studies do not exclude clustering of P-selectin in discrete regions in the PM following activation,43 and have also identified that platelets can display different routes of alpha-granule fusion upon activation.46 Platelet activation can trigger the exposure of the OCS.3,16 Thereby, P-selectin and other granule membrane proteins also get incorporated into the PM, but may remain within a circular shape of an alpha-granule membrane “ghost”, or within a circular “nozzle” of the OCS.
This work shows the benefits of fluorescence-based STED imaging in studies of protein storage, uptake and release in platelets. For these studies, conventional CLSM, with a resolution of 250–300 nm, is clearly insufficient (Fig. 2B). EM has the necessary resolution, but requires extensive and perturbing sample preparations, and protein labelling with metallic nano-beads with orders of magnitude lower labelling efficiencies than in immunofluorescence labelling.47 STED imaging combines spatial resolution down to sub-granular level, high degrees of labelling, and no requirements for extensive and perturbing sample preparations. This suggests a major role for STED imaging in platelet studies, of their protein uptake, storage and release mechanisms, how they are influenced by tumor cells, and of how this can contribute to tumor growth and metastasis.19,28,29
This study also demonstrates that STED images of P-selectin in platelets can be used to identify platelets exposed to tumor cells, from platelets exposed to non-cancer cells, from platelets activated by ADP, TXA2 and thrombin, and from non-activated platelets. Apart from manual, blind classification, we show that classification can also be done in an automatic, objective manner, using dictionary learning. While classification in a clinical context would be much more complex, further improvements in the classification are also possible, by analysing more platelets, additional distributions of proteins in the platelets, and by improved analyses. In our analysis, we used 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 simulated images as training set to create a dictionary for image reconstruction. With a corresponding number of experimental STED images available, representing each of the different platelet activation conditions to be classified, considerable refinements of this classification will be possible.
000 simulated images as training set to create a dictionary for image reconstruction. With a corresponding number of experimental STED images available, representing each of the different platelet activation conditions to be classified, considerable refinements of this classification will be possible.
Platelets and their characteristics have in the last few years emerged as a potentially very valuable source of diagnostic information. In cancer patients, several platelet features are affected and can be analysed,48 including content of specific proteins and platelet mRNA,49 activation state (e.g. monitored via surface expression of P-selectin), and platelet counts. SRM STED imaging, and a platelet image classification procedure as outlined in this work, can be added to these features. STED imaging is currently quickly developing into a standard imaging technique, available also outside of the specialist labs. STED imaging of platelets, together with automated image analyses of specific protein distribution patterns within the platelets, can therefore become part of a platelet-based diagnostic battery for minimally invasive diagnostics and therapeutic monitoring of cancer.
Conclusions
It is well established that cross-talk between tumor cells and platelets plays a central role in tumor development and metastasis. Yet, many of the features and underlying mechanisms remain to be revealed and understood. In this study, we introduced STED SRM combined with dictionary learning and image reconstruction algorithms to explore if additional important clues can be obtained, by studying how specific proteins may re-distribute within the platelets upon various activations, including incubation of both cancer and non-cancer cells. We also set out to explore if cancer cell-induced activations of the platelets could be specifically identified from the protein distribution patterns in the STED images, as a possible basis for diagnostics.In platelets incubated with cancer cells, we found that the cell-adhesion protein P-selectin re-distributed into circular nano-structures, consistent with accumulation into the membrane of protein-storing alpha-granules within the platelets. These changes were to a significantly lesser extent, if at all, found in platelets incubated with normal cells, or in platelets subject to soluble platelet activators. Based on the imaged distribution patterns of P-selectin in the platelets, we developed a classification procedure, whereby platelets exposed to cancer cells, to non-cancer cells, soluble activators as well as non-activated platelets all could be identified in an automatic, objective manner. We thus conclude that STED imaging, combined with image analyses of specific protein distribution patterns within the platelets, can add important information for identification of specific platelet activations, and can have a role in future platelet-based cancer diagnostics and therapeutic monitoring. This study further demonstrates the potential of analyzing nanoscale spatial distribution patterns of biomolecules in cells, obtained by SRM, as a diagnostic strategy. This information is not within reach by CLSM, EM and other techniques, and can add diagnostic information to that obtained from mere up- or down-regulation of specific cellular disease biomarker molecules. The presented approach can also bring further clarity into fundamental mechanisms for cancer cell–platelet interactions. This study also suggests the use of SRM together with analyses of spatial distribution patterns of proteins in cells to detect, analyse and better understand non-contact cell-to-cell interactions in general.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The MCF10A cell line was kindly provided by Prof Aristidis Moustakas, Uppsala University. This project was funded by a HMT grant from Stockholm County Council and KTH (to ML and JW), and by a grant from Cancerfonden (CAN 2017/471, to JW).References
- B. Tesfamariam, Pharmacol. Ther., 2016, 157, 112–119 CrossRef CAS PubMed.
- C. K. S. Meikle, C. A. Kelly, P. Garg, L. M. Wuescher, R. A. Ali and R. G. Worth, Front. Cell Dev. Biol., 2017, 4, UNSP 147 Search PubMed.
- Q. Zhang, H. D. Liu, Q. Q. Zhu, P. Zhan, S. H. Zhu, J. Y. Zhang, T. F. Lv and Y. Song, Int. J. Biochem. Cell Biol., 2017, 90, 68–80 CrossRef CAS PubMed.
- S. Mezouar, C. Frère, R. Darbousset, D. Mege, L. Crescence, F. Dignat-George, L. Panicot-Dubois and C. Dubois, Thromb. Res., 2016, 139, 65–76 CrossRef CAS PubMed.
- M. J. Yan and P. Jurasz, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 392–400 CrossRef CAS PubMed.
- S. M. Feller and M. Lewitzky, Cell Commun. Signaling, 2016, 14, 24 CrossRef PubMed.
- G. M. Thomas, L. Panicot-Dubois, R. Lacroix, F. Dignat-George, D. Lombardo and C. Dubois, J. Exp. Med., 2009, 206, 1913–1927 CrossRef CAS.
- D. Cervi, T.-T. Yip, N. Bhattacharya, V. N. Podust, J. Peterson, A. Abou-Slaybi, G. N. Naumov, E. Bender, N. Almog, J. E. Italiano Jr., J. Folkman and G. L. Klement, Blood, 2008, 111, 1201–1207 CrossRef CAS PubMed.
- G. L. Klement, T.-T. Yip, F. Cassiola, L. Kikuchi, D. Cervi, V. Podust, J. E. Italiano, E. Wheatley, A. Abou-Slaybi, E. Bender, N. Almog, M. W. Kieran and J. Folkman, Blood, 2009, 113, 2835–2842 CrossRef CAS PubMed.
- T. Wiesner, S. Bugl, F. Mayer, J. T. Hartmann and H.-G. Kopp, Clin. Exp. Metastasis, 2010, 27, 141–149 CrossRef CAS PubMed.
- J. E. Italiano Jr., J. L. Richardson, S. Patel-Hett, E. Battinelli, A. Zaslavsky, S. Short, S. Ryeom, J. Folkman and G. L. Klement, Blood, 2008, 111, 1227–1233 CrossRef PubMed.
- H. Heijnen and P. van der Sluijs, J. Thromb. Haemostasis, 2015, 13, 2141–2151 CrossRef CAS PubMed.
- S. Yadav and B. Storrie, Platelets, 2017, 28, 108–118 CrossRef CAS PubMed.
- S. Joshi and S. W. Whiteheart, Platelets, 2017, 28, 129–137 CrossRef CAS PubMed.
- E. M. Battinelli, B. A. Markens and J. E. Italiano, Blood, 2011, 118, 1359–1369 CrossRef CAS PubMed.
- P. Blair and R. Flaumenhaft, Blood Rev., 2009, 23, 177–189 CrossRef CAS.
- H. v. N. T. Pannerden, F. de Haas, W. Geerts, G. Posthuma, S. van Dijk and H. F. G. Heijnen, Blood, 2010, 116, 1147–1156 CrossRef PubMed.
- J. Kamykowski, P. Carlton, S. Sehgal and B. Storrie, Blood, 2011, 118, 1370–1373 CrossRef CAS PubMed.
- D. Rönnlund, Y. Yang, H. Blom, G. Auer and J. Widengren, Adv. Healthcare Mater., 2012, 1, 707–713 CrossRef.
- D. Jonnalagadda, L. T. Izu and S. W. Whiteheart, Blood, 2012, 120, 5209–5216 CrossRef CAS PubMed.
- H. Laubli and L. Borsig, Semin. Cancer Biol., 2010, 20, 169–177 CrossRef PubMed.
- L. A. Coupland, B. H. Chong and C. R. Parish, Cancer Res., 2012, 72, 4662–4671 CrossRef CAS PubMed.
- A. Korniluk, J. Kaminska, P. Kiszlo, H. Kemona and V. Dymicka-Piekarska, Biomarkers, 2017, 22, 629–634 Search PubMed.
- M. Mantur, H. Kemona, R. Kozlowski and I. Kemona-Chetnik, Neoplasma, 2003, 50, 262–265 Search PubMed.
- D. Sharma, K. E. Brummel-Ziedins, B. A. Bouchard and C. E. Holmes, J. Cell. Physiol., 2014, 229, 1005–1015 CrossRef CAS PubMed.
- S. Kohler, S. Ullrich, U. Richter and U. Schumacher, Br. J. Cancer, 2010, 102, 602–609 CrossRef CAS PubMed.
- H. Blom and J. Widengren, Curr. Opin. Chem. Biol., 2014, 20, 127–133 CrossRef CAS PubMed.
- H. Blom and J. Widengren, Chem. Rev., 2017, 117, 7377–7427 CrossRef CAS PubMed.
- D. Rönnlund, L. Xu, A. Perols, A. K. B. Gad, A. E. Karlstrom, G. Auer and J. Widengren, ACS Nano, 2014, 8, 4358–4365 CrossRef PubMed.
- D. Ronnlund, A. K. B. Gad, H. Blom, P. Aspenstrom and J. Widengren, Cytometry, Part A, 2013, 83, 855–865 CrossRef PubMed.
- H. Blom, D. Rönnlund, L. Scott, Z. Spicarova, V. Rantanen, J. Widengren, A. Aperia and H. Brismar, Microsc. Res. Tech., 2012, 75, 220–228 CrossRef CAS PubMed.
- J. L. Chen, J. Gao, J. Z. Wu, M. Zhang, M. J. Cai, H. J. Xu, J. G. Jiang, Z. Y. Tian and H. D. Wang, Nanoscale, 2015, 7, 3373–3380 RSC.
- J. Gao, Y. Wang, M. J. Cai, Y. G. Pan, H. J. Xu, J. G. Jiang, H. B. Ji and H. D. Wang, Nanoscale, 2015, 7, 2511–2519 RSC.
- J. L. Chen, T. Z. Liu, J. Gao, L. Gao, L. L. Zhou, M. J. Cai, Y. Shi, W. Y. Xiong, J. G. Jiang, T. Tong and H. D. Wang, Adv. Sci., 2016, 3, 1600270 CrossRef PubMed.
- J. Gao, J. L. Chen, M. J. Cai, H. J. Xu, J. G. Jiang, T. Tong and H. D. Wang, Methods Appl. Fluoresc., 2017, 5, 024004 CrossRef PubMed.
- B. Angelov and A. Angelova, Nanoscale, 2017, 9, 9797–9804 RSC.
- J. M. Pullman, Front. Med., 2019, 6, 37 CrossRef PubMed.
- M. Lomnytska, R. Pinto, S. Becker, U. Engström, S. Gustafsson, C. Björklund, M. Templin, J. Bergstrand, L. Xu, J. Widengren, E. Epstein, B. Franzén and G. Auer, Biomark. Res., 2018, 6, 2 CrossRef PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- R. Rubinstein, T. Peleg and M. Elad, IEEE Trans. Signal Process., 2013, 61, 661–667 Search PubMed.
- Z. Wang, A. C. Bovik, H. R. Sheikh and E. P. Simoncelli, IEEE Trans. Image Process., 2004, 13, 600–612 CrossRef.
- N. L. Li, Int. J. Cancer, 2016, 138, 2078–2087 CrossRef CAS.
- P. E. Stenberg, R. P. McEver, M. A. Shuman, Y. V. Jacques and D. F. Bainton, J. Cell Biol., 1985, 101, 880–886 CrossRef CAS PubMed.
- P. E. Stenberg, M. A. Shuman, S. P. Levine and D. F. Bainton, J. Cell Biol., 1984, 98, 748–760 CrossRef CAS.
- Y. Zhuge, J. Y. Zhou, G. D. Yang, D. L. Zu, X. L. Xu, M. Q. Tian and G. H. Lu, Chin. Med. J., 2009, 122, 1026–1031 CAS.
- A. Eckly, J. Y. Rinckel, F. Proamer, N. Ulas, S. Joshi, S. W. Whiteheart and C. Gachet, Blood, 2016, 128, 2538–2549 CrossRef CAS PubMed.
- D. A. Meyer, J. A. Oliver and R. M. Albrecht, Microsc. Microanal., 2010, 16, 33–42 CrossRef CAS.
- S. Sabrkhany, M. J. E. Kuijpers, S. M. J. van Kuijk, L. Sanders, S. Pineda, S. Damink, A. M. C. Dingemans, A. W. Griffioen and M. Egbrink, Eur. J. Cancer, 2017, 80, 5–13 CrossRef CAS PubMed.
- M. G. Best, N. Sol, S. t Veld, A. Vancura, M. Muller, A. L. N. Niemeijer, A. V. Fejes, L. Fat, A. t Veld, C. Leurs, T. Y. Le Large, L. L. Meijer, I. E. Kooi, F. Rustenburg, P. Schellen, H. Verschueren, E. Post, L. E. Wedekind, J. Bracht, M. Esenkbrink, L. Wils, F. Favaro, J. D. Schoonhoven, J. Tannous, H. Meijers-Heijboer, G. Kazemier, E. Giovannetti, J. C. Reijneveld, S. Idema, J. Killestein, M. Heger, S. C. de Jager, R. T. Urbanus, I. E. Hoefer, G. Pasterkamp, C. Mannhalter, J. Gomez-Arroyo, H. J. Bogaard, D. P. Noske, W. P. Vandertop, D. van den Broek, B. Ylstra, R. J. A. Nilsson, P. Wesseling, N. Karachaliou, R. Rosell, E. Lee-Lewandrowski, K. B. Lewandrowski, B. A. Tannous, A. J. de Langen, E. F. Smit, M. M. van den Heuvel and T. Wurdinger, Cancer Cell, 2017, 32, 238–252 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr01967g |
| ‡ Present address: Department of Obstetrics and Gynaecology, Academical University Hospital, Uppsala, SE-75185, Sweden, Institute for Women and Child Health, Uppsala University, SE-75185, Sweden |
| This journal is © The Royal Society of Chemistry 2019 |