
Palladium-catalysed ring-opening [3 + 2]-annulation of spirovinylcyclopropyl oxindole to diastereoselectively access spirooxindoles†
Jun-An
Xiao
 ab,
Xiu-Liang
Cheng
a,
Yu-Chun
Li
a,
Yi-Miao
He
ab,
Jin-Lian
Li
a,
Zhi-Ping
Liu
ab,
Peng-Ju
Xia
c,
Wei
Su
*ab and
Hua
Yang
*c
ab,
Xiu-Liang
Cheng
a,
Yu-Chun
Li
a,
Yi-Miao
He
ab,
Jin-Lian
Li
a,
Zhi-Ping
Liu
ab,
Peng-Ju
Xia
c,
Wei
Su
*ab and
Hua
Yang
*c
aCollege of Chemistry and Materials Science, Guangxi Teachers Education University, Nanning 530001, P. R. China
bGuangxi Key Laboratory of Natural Polymer Chemistry and Physics, Guangxi Teachers Education University, Nanning, 530001, P. R. China. E-mail: suwmail@163.com
cCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, P. R. China. E-mail: hyangchem@csu.edu.cn
First published on 29th November 2018
Abstract
A novel palladium-catalysed ring-opening [3 + 2]-annulation of spirovinylcyclopropyl oxindole with α,β-unsaturated nitroalkenes is reported. A series of spirooxindole derivatives were synthesized in high yields and good to excellent diastereoselectivities. This developed protocol offers a new and efficient pathway for the assembly of spirooxindoles.
Introduction
Donor–acceptor cyclopropane (DAC) has attracted significant attention recently, due to its versatile reactivities and high efficiency in the construction of cyclic compounds.1 In general, ring-opening [3 + n]-cycloadditions/annulations are the major work modes, which usually involve the generation of highly reactive dipoles promoted by a Lewis acid,2 protic acid,3 Lewis base4 and palladium(0) catalyst.5 Among all the DACs, vinylcyclopropanes (VCPs) have attracted special attention due to their unparalleled reactivities in the context of the ring-opening strategy (Scheme 1).6 In particular, VCPs can be used to access multicyclic or spirocyclic derivatives with the aid of a palladium(0) complex or Lewis acid.6a,b Several elegant protocols have been established for the formation of spirocyclic compounds via the ring-opening [3 + 2]-annulation of VCPs with dipolarophiles.6c,7 Despite these encouraging advances in the synthetic utilization of VCPs, the exploitation of a new VCP-based methodology is still extremely attractive to meet the demand in the diversity and complexity of ring systems and allow for the facile construction of a wide range of cyclic scaffolds.8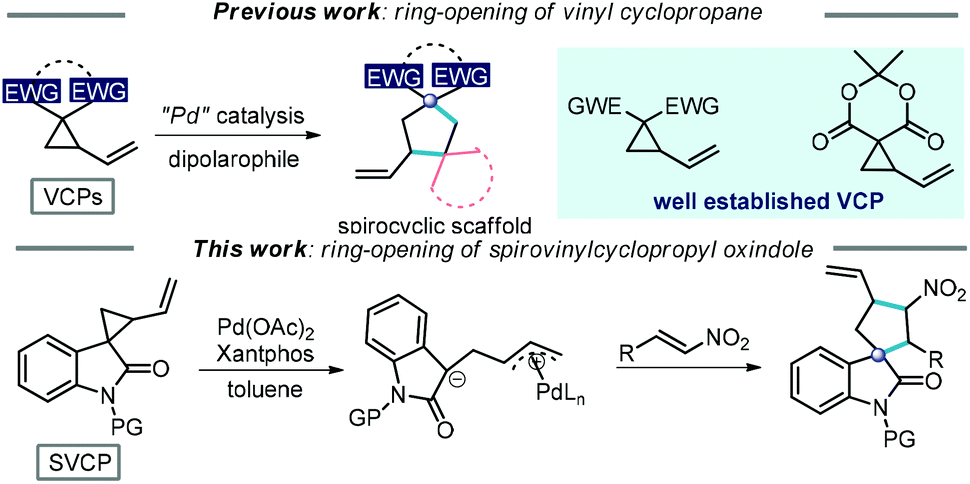 | ||
| Scheme 1 Construction of spirocyclic scaffolds via [3 + 2]-cycloaddition/annulation of vinyl cyclopropanes. | ||
Spirooxindoles are important core skeletons in biologically active alkaloids and relevant natural products (Scheme 2).9 Compounds bearing spirooxindole units have proven to possess interesting biological activities such as antitumor, antibiotic and antiparasitic activities.10 For example, citrinadine B, isolated from Penicillium citrinum N059 by Kobayashi, displays cytotoxicity against murine leukemia L1210 cells.11 Marcfortine A is a fungal metabolite of Penicillium roqueforti showing potent antiparasitic activity.12 Noticing the existence of a spiro(cyclopentyl-3-oxindole) scaffold in these bioactive compounds, we envisaged that ring-opening of spirovinylcyclopropane oxindole (SVCP) might offer an efficient approach to access these spirooxindole structures. Unfortunately, this strategy has been rarely applied to construct spirooxindoles.13 Herein we describe a practical protocol via a palladium-catalysed ring-opening [3 + 2] annulation of SVCP for the diastereoselective synthesis of spirooxindole derivatives under mild conditions. This novel protocol would offer alternative pathways stereoselectively accessing spirocyclic systems.
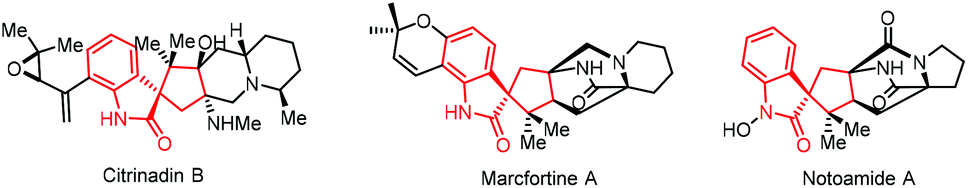 | ||
| Scheme 2 Representative natural products bearing spirooxindole motifs. | ||
Results and discussion
We initiated our studies by testing a model reaction of spirovinylcyclopropane oxindole 1a and β-nitrostyrene 2a catalysed by Pd(OAc)2 and S-BINAP. It is noteworthy that 1a was synthesized from N-benzyl-2-oxindole and 1,4-dibromobut-2-ene in a moderate yield with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Unfortunately, only 41% of diastereomer 1a′ was isolated and the desired annulation product 3a was unable to be accessed (Scheme 3).
:1 dr. Unfortunately, only 41% of diastereomer 1a′ was isolated and the desired annulation product 3a was unable to be accessed (Scheme 3).
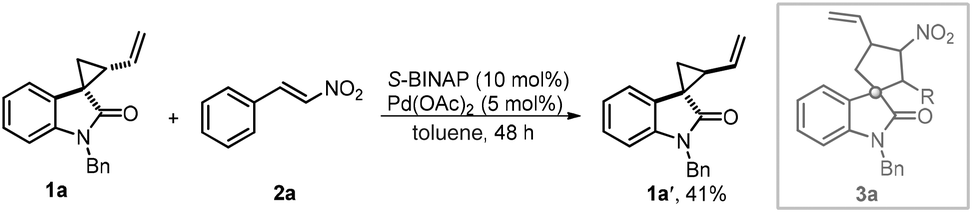 | ||
| Scheme 3 Preliminary study of the designed strategy. | ||
In order to establish a viable protocol for this strategy, a variety of Pd(II) salts and ligands were tested in this annulation process (as shown in Table 1). Interestingly, using 10 mol% of Pd(PPh3)4 as the catalyst in toluene afforded the corresponding product 3a in 44% yield with 74:26 dr (Table 1, entry 1). Next, various ligands were also evaluated by using Pd(OAc)2 as the catalyst. Lower yields of 3a were obtained in the presence of 1,3-bis(diphenylphosphino)propane (DPPP) or PPh3 (Table 1, entries 2 and 3). Pleasingly, the employment of xantphos gave the desired product 3a in 89% yield and 86:14 dr within 12 h (Table 1, entry 4). The replacement of Pd(OAc)2 with Pd2(dba)3 led to a slightly decreased yield (81%), but with unchanged dr (Table 1, entry 5). Subsequently, solvents were also extensively examined (entries 6–11) and toluene was proven to be the most suitable solvent for this transformation in terms of yield and diastereoselectivity. It is worth noting that the protonic solvent was found to have a dramatic impact on the efficiency of the annulation reaction (Table 1, entry 9). Presumably, methanol has an effect on the catalytic activity of the palladium catalyst and thus leads to the spirooxindole 3a in a poor yield and diastereoselectivity. Finally, the ratio of spirocyclopropane 1a and nitroalkenes 2a was also modulated and the yield of 3a was slightly eroded when the amount of nitroalkenes 2a was decreased from 2.0 equiv. to 1.0 equiv. (entries 12–14).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Ligand | Solvent | Time (h) | Yieldb (%) | drc (%) |
|
a Reaction conditions: 1a (0.2 mmol), 2a (0.24 mmol), Pd salts (5 mol%), ligand (10 mol%), solvent (2.0 mL), rt, 12–24 h.
b Isolated yield.
c Determined by 1H NMR, based on the isolated product.
d The ratio of 1a:2a = 1:1.5.
e The ratio of 1a:2a = 1:1.2.
f The ratio of 1a:2a = 1:1.
|
||||||
| 1 | Pd(PPh3)4 | — | Toluene | 24 | 44 | 74:26 |
| 2 | Pd(OAc)2 | DPPP | Toluene | 24 | 31 | 86:14 |
| 3 | Pd(OAc)2 | PPh3 | Toluene | 24 | 27 | 75:25 |
| 4 | Pd(OAc)2 | Xantphos | Toluene | 12 | 89 | 86:14 |
| 5 | Pd2(dba)3 | Xantphos | Toluene | 12 | 81 | 86:14 |
| 6 | Pd(OAc)2 | Xantphos | CH2Cl2 | 12 | 67 | 72:28 |
| 7 | Pd(OAc)2 | Xantphos | DCE | 12 | 90 | 71:29 |
| 8 | Pd(OAc)2 | Xantphos | THF | 12 | 80 | 58:42 |
| 9 | Pd(OAc)2 | Xantphos | MeOH | 12 | 16 | 52:48 |
| 10 | Pd(OAc)2 | Xantphos | DMSO | 12 | 83 | 75:25 |
| 11 | Pd(OAc)2 | Xantphos | MeCN | 12 | 77 | 61:39 |
| 12d | Pd(OAc)2 | Xantphos | Toluene | 12 | 88 | 85:15 |
| 13e | Pd(OAc)2 | Xantphos | Toluene | 12 | 85 | 82:18 |
| 14f | Pd(OAc)2 | Xantphos | Toluene | 24 | 77 | 82:18 |
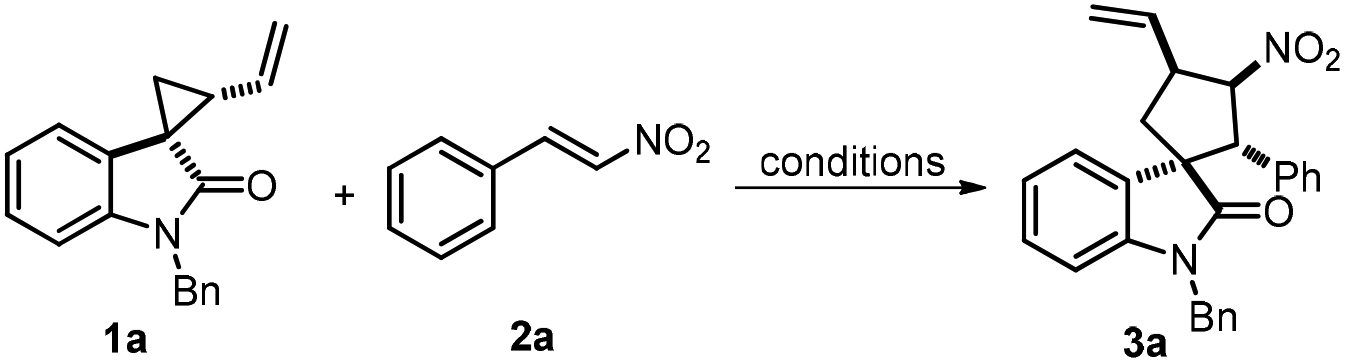
With the optimal reaction conditions established, we next investigated the substrate scope of the title reaction by employing a series of spirovinylcyclopropyl oxindoles and α,β-unsaturated nitroalkenes. Firstly, different N-substituted spirocyclopropanes 1 were evaluated. As expected from our optimization efforts, N-protected spirocyclopropanes 1a–1d gave moderate to excellent yields (56%–89%) and good dr (83:17–88:12) (PG = Me, Bn, Allyl and Boc). However, the reaction of unsubstituted spirocyclopropane 1e and nitroalkenes 2a afforded 67% yield but with a significantly decreased diastereomeric ratio (51:49). Subsequently, various substituted spirocyclopropanes 1g–1l were examined. In general, high to good yields (64%–84%) and moderate to excellent drs (57:43–95:5) were achieved with the substitution at the C5, C6 or C7 position on the oxindole moiety. It is worth noting that the diastereoselectivity obviously dropped upon increasing the steric hindrance of the substituting group (1g–1i). The introduction of an electron-donating group onto the oxindole moiety would slightly decrease the yield and increase the diastereoselectivity (1j). In contrast to C5 and C6 substituted spirocyclopropanes (1h and 1k), the substrate bearing a chloro group at the C7 position afforded a good yield (67%) and an excellent diastereoselectivity (91:9 dr). Next, various α,β-unsaturated nitroalkenes 2a–2l were also investigated. Pleasingly, para-substituted nitroalkenes 2a–2d gave good yields (80%–87%) and drs (81:11–94:6) (Table 2, 3m–3p). It was found that the introduction of a strong electron-donating group onto the phenyl group in nitroalkene 2, no matter on the para-position or the meta-position, would decrease the yield and diastereoselectivity (Table 2, 3q–3r). Interestingly, the ortho-substituted nitroalkenes gave the corresponding product 3s in good dr but the yield was remarkably decreased. Presumably, the difference in reactivity might be majorly due to the steric effect induced by the ortho-substituent. Besides, other aromatic substituted α,β-unsaturated nitroalkenes were also well tolerated (3t–3v), except for (1E)-2-phenylethenyl nitroalkene (3w). Additionally, other dipolarophiles, such as cinnamonitrile, ethyl cinnamate, 2-cyclohexen-1-one, N-tosyl-3-nitroindole, 2-nitrobenzofuran and alkyne derivatives, were tested under the standard conditions, but essentially no reaction was observed. Consequently, it can be concluded that the above-mentioned dipolarophiles demonstrate relatively lower reactivity toward [3 + 2]-annulation of SVCP than nitroalkene 2. Finally, the chemical structure and relative configuration of spirooxindole 3l were unambiguously confirmed by X-ray crystal structure analysis (CCDC 1857258†).
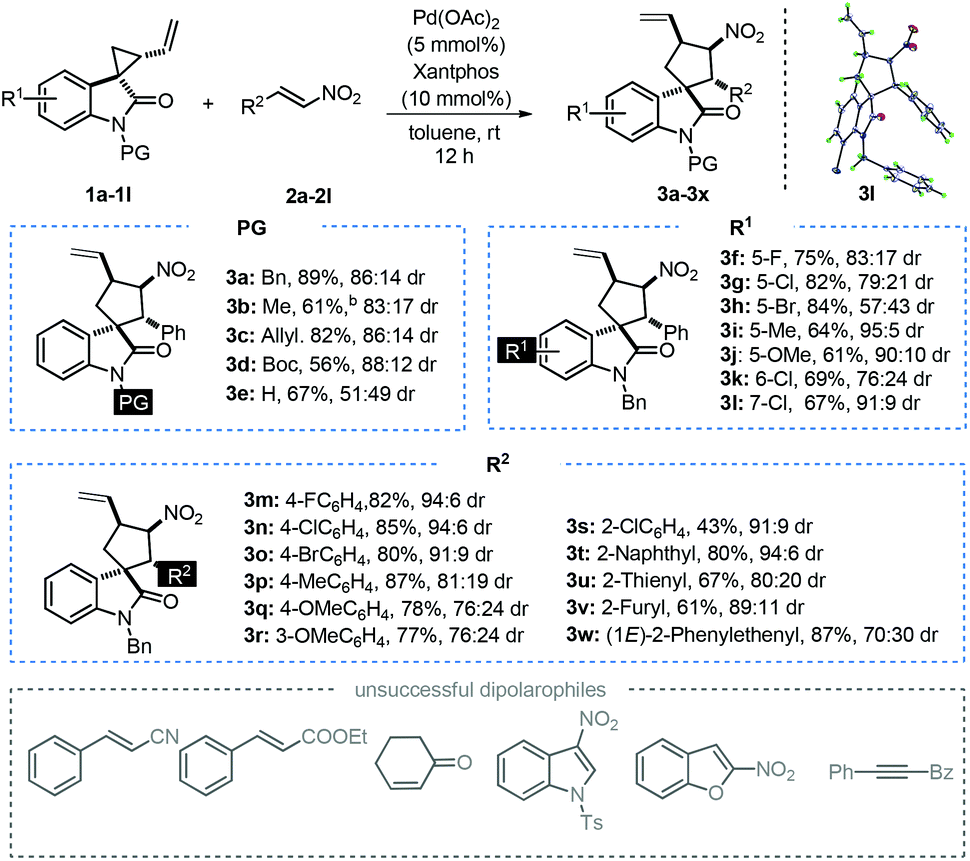
At this stage, we were curious about the effect of the conversion from 1a and 1a′ on the [3 + 2]-annulation process. As a result, a series of monitoring experiments were carried out in the presence of Pd(PPh3)4. Surprisingly, over half of spirocyclopropane 1a was epimerized to its diastereomer 1a′ within 30 minutes, and an equilibrium was achieved after 6 hours with a 39:61 diastereomeric ratio (Scheme 4, eqn (1)). Interestingly, cyclopropane 1a′ was successfully employed in the title reaction and the desired spirooxindole 3a was obtained in 78% yield with 85:15 dr, which are paralleled with the results by utilizing 1a as the substrate (Scheme 4, eqn (2)). These unexpected results suggest that spirocyclopropane could be epimerized to the thermodynamically stable diastereomer upon the promotion by a palladium complex via intramolecular cyclization, which would hardly impact the annulation process.
 | ||
| Scheme 4 Monitoring experiments and comparison experiments of SVCP. | ||
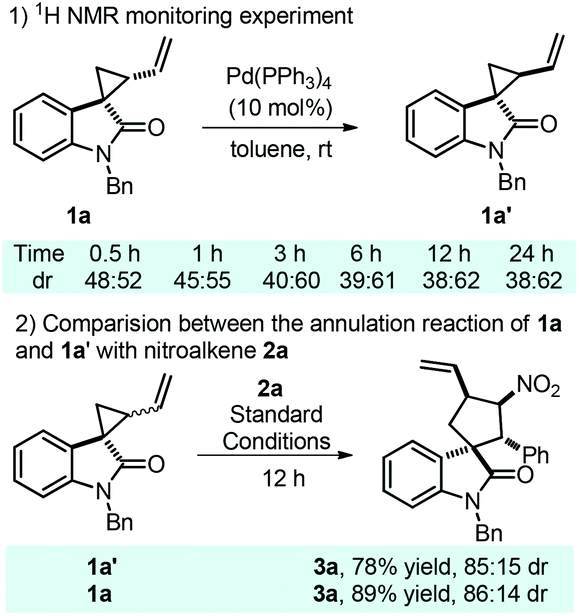
Next, in order to better help us understand the mechanistic pathway, three independent control experiments were performed. Firstly, spirocyclopropane 4 without a terminal alkene moiety attached was tested. Unfortunately, the desired spirooxindole was unable to be obtained (Scheme 5, eqn (a)). In contrast, spirocyclopropane 5 bearing an internal alkene moiety gave spirooxindole 6 in 92% yield with good diastereoselectivity (84:16 dr, Scheme 5, eqn (b)). Finally, nonaromatic nitroalkene 7 was also examined in this annulation reaction, but a complex reaction was observed (Scheme 5, eqn (c)). Presumably, the nonaromatic nitroalkene might be unstable under these conditions, leading to complex results.
 | ||
| Scheme 5 Control experiments. | ||
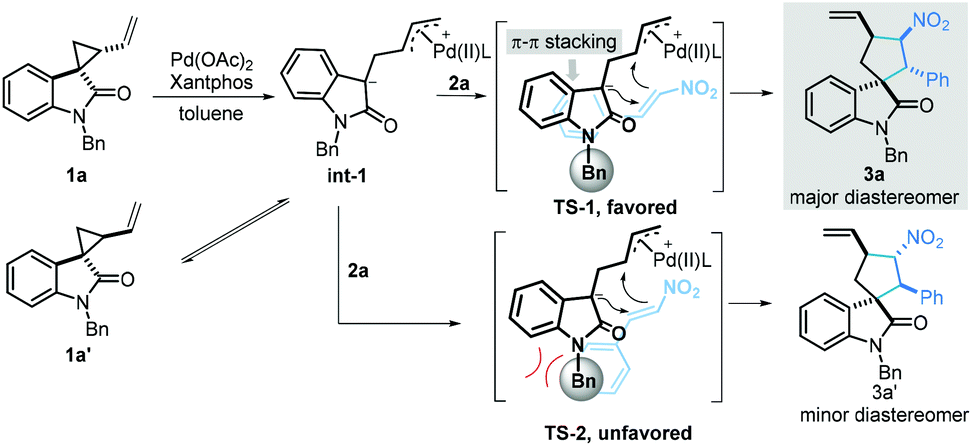
On the basis of the above experimental results, a plausible mechanism is proposed in Scheme 6. First, a Pd-stabilized zwitterionic 1,3-dipole intermediate int-1 was efficiently generated via a palladium-catalysed ring-opening process. Notably, intermediate int-1 could be converted into diastereomer 1a′. Subsequently, the intermediate int-1 underwent a [3 + 2] annulation to give the desired product via the transition state TS-1 or TS-2 to give the annulation product. As can be seen, the steric repulsion between the phenyl moiety in 2a and the protecting group in 1a rendered TS-2 unfavorable, leading to diastereomer 3a′ as the minor product. In contrast, the π–π stacking between two phenyl moieties of 1a and 2a facilitated the formation of TS-1, resulting in the generation of 3a as the major diastereomer.
 | ||
| Scheme 6 Plausible mechanism for the ring-opening [3 + 2]-annulation. | ||
Conclusions
In summary, we have developed a diastereoselective palladium-catalysed ring-opening [3 + 2] annulation reaction for the construction of spirooxindole scaffolds in high yields and good diastereomeric ratios. The developed protocol features broad substrate scope, mild reaction conditions and high efficiency. This work broadens the application of spirovinylcyclopropane derivatives as versatile useful building blocks in the assembly of valuable spirocyclic systems. Further investigation on asymmetric catalytic annulation of spirovinylcyclopropanes is currently being pursued in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21761006 and 21861008), the Guangxi Natural Science Foundation (2018GXNSFBA138037), the Basic Ability Improvement Project for Young Teachers in Guangxi Colleges and Universities (2018KY0364), the“BAGUI Scholar” Program of Guangxi Province of China and Central South University.Notes and references
- (a) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS; (b) H. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed.
- (a) Y. Matsumoto, D. Nakatake, R. Yazaki and T. Ohshima, Chem. – Eur. J., 2018, 24, 6062–6066 CrossRef CAS PubMed; (b) S. Tsunoi, Y. Maruoka, I. Suzuki and I. Shibata, Org. Lett., 2015, 17, 4010–4013 CrossRef CAS PubMed; (c) C. A. Carson and M. A. Kerr, J. Org. Chem., 2005, 70, 8242–8244 CrossRef CAS; (d) R. Dey and P. Banerjee, Org. Lett., 2017, 19, 304–307 CrossRef CAS PubMed; (e) D. Perrotta, M. Wang and J. Waser, Angew. Chem., Int. Ed., 2018, 57, 5120–5123 CrossRef CAS PubMed; (f) X.-f. Ma, J.-C. Zhang, Q. Tang, J. Ke, W. Zou and H.-W. Shao, Chem. Commun., 2014, 50, 3505–3508 RSC.
- B. Cui, J. Ren and Z.-W. Wang, J. Org. Chem., 2013, 79, 790–796 CrossRef.
- (a) S.-X. Lin, L. Li, F.-S. Liang and Q. Liu, Chem. Commun., 2014, 50, 10491–10494 RSC; (b) J. Wu, Y.-H. Tang, W. Wei, Y. Wu, Y. Li, J.-J. Zhang, Y.-S. Zheng and S.-L. Xu, Angew. Chem., Int. Ed., 2018, 57, 6284–6288 CrossRef CAS; (c) J.-M. Liu, W.-J. Ye, X.-S. Qing and C.-D. Wang, J. Org. Chem., 2016, 81, 7970–7976 CrossRef CAS.
- (a) A. T. Parsons, M. J. Campbell and J. S. Johnson, Org. Lett., 2008, 10, 2541–2544 CrossRef CAS PubMed; (b) W.-K. Li, Z.-S. Liu, L. He, T.-R. Kang and Q.-Z. Liu, Asian J. Org. Chem., 2015, 4, 28–32 CrossRef CAS; (c) A. F. G. Goldberg and B. M. Stoltz, Org. Lett., 2011, 13, 4474–4476 CrossRef CAS PubMed.
- (a) J.-A. Xiao, J. Li, P.-J. Xia, Z.-F. Zhou, Z.-X. Deng, H.-Y. Xiang, X.-Q. Chen and H. Yang, J. Org. Chem., 2016, 81, 11185–11194 CrossRef CAS PubMed; (b) M.-S. Xie, Y. Wang, J.-P. Li, C. Du, Y.-Y. Zhang, E.-J. Hao, Y.-M. Zhang, G.-R. Qu and H.-M. Guo, Chem. Commun., 2015, 51, 12451–12454 RSC; (c) L.-Y. Mei, Y. Wei, Q. Xu and M. Shi, Organometallics, 2012, 31, 7591–7599 CrossRef CAS.
- (a) F. Wei, C.-L. Ren, D. Wang and L. Liu, Chem. – Eur. J., 2015, 21, 2335–2338 CrossRef CAS PubMed; (b) M. Laugeois, S. Ponra, V. Ratovelomanana-Vidal, V. R. Michelet and M. R. Vitale, Chem. Commun., 2016, 52, 5332–5335 RSC.
- (a) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed; (b) D.-J. Cheng, Y. Ishihara, B. Tan and C. F. Barbas, ACS Catal., 2014, 4, 743–762 CrossRef CAS; (c) J. Bariwal, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3831–3848 RSC; (d) R. Rios, Chem. Soc. Rev., 2012, 41, 1060–1074 RSC; (e) K. A. Miller and R. M. Williams, Chem. Soc. Rev., 2009, 38, 3160–3174 RSC.
- (a) L. Wang, S. Li, M. Blümel, R. Puttreddy, A. Peuronen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2017, 56, 8516–8521 CrossRef CAS; (b) B. M. Trost, D. A. Bringley, T. Zhang and N. Cramer, J. Am. Chem. Soc., 2013, 135, 16720–16735 CrossRef CAS PubMed; (c) C. J. A. Ribeiro, J. D. Amaral, C. M. P. Rodrigues, R. Moreira and M. M. M. Santos, Med. Chem. Commun., 2016, 7, 420–425 RSC; (d) X. Z. Wearing and J. M. Cook, Org. Lett., 2002, 4, 4237–4240 CrossRef CAS PubMed; (e) N. Kogure, N. Ishii, M. Kitajima, S. Wongseripipatana and H. Takayama, Org. Lett., 2006, 8, 3085–3088 CrossRef CAS; (f) M. Pérez-Gómez, S. Hernández-Ponte, D. Bautista and J. García-López, Chem. Commun., 2017, 53, 2842–2845 RSC; (g) R. Samineni, J. Madapa, S. Pabbaraja and G. Mehta, Org. Lett., 2017, 19, 6152–6155 CrossRef CAS PubMed.
- K. Wang, X.-Y. Zhou, Y.-Y. Wang, M.-M. Li, Y.-S. Li, L.-Y. Peng, X. Cheng, Y. Li, Y.-P. Wang and Q.-S. Zhao, J. Nat. Prod., 2011, 74, 12–15 CrossRef CAS PubMed.
- K. Kong, J. A. Enquist, M. E. McCallum, G. M. Smith, T. Matsumaru, E. Menhaji-Klotz and J. L. Wood, J. Am. Chem. Soc., 2013, 135, 10890–10893 CrossRef CAS PubMed.
- S. Afewerki, G. Ma, I. Ibrahem, L. Liu, J. Sun and A. Córdova, ACS Catal., 2015, 5, 1266–1272 CrossRef CAS.
- C. Marti and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 11505–11515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1857257 and 1857258. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob02859a |
| This journal is © The Royal Society of Chemistry 2019 |