
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimised approach to albumin–drug conjugates using monobromomaleimide-C-2 linkers†
Archie
Wall
a,
Karl
Nicholls
b,
Mikael B.
Caspersen
b,
Stig
Skrivergaard
c,
Kenneth A.
Howard
c,
Kersti
Karu
a,
Vijay
Chudasama
 *ad and
James R.
Baker
*a
*ad and
James R.
Baker
*a
aDepartment of Chemistry, UCL, 20 Gordon St, London, WC1H 0AJ, UK. E-mail: j.r.baker@ucl.ac.uk; v.chudasama@ucl.ac.uk
bAlbumedix Ltd, Castle Court, 59 Castle Boulevard, Nottingham NG7 1FD, UK
cInterdisciplinary Nanoscience Center (iNANO), Department of Molecular Biology and Genetics, Aarhus University, 8000 Aarhus C, Denmark
dResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal
First published on 14th August 2019
Abstract
Conjugation of therapeutics to human serum albumin (HSA) using bromomaleimides represents a promising platform for half-life extension. We show here that the Cys-34 crevice substantially reduces the rate of serum stabilising maleimide hydrolysis in these conjugates, necessitating reagent optimisation. This improved reagent design is applied to the construction of an HSA-paclitaxel conjugate, preventing drug loss during maleimide hydrolysis.
Human serum albumin (HSA) represents the most abundant protein in blood plasma, and plays numerous roles including as a transporter of fatty acids and various other endogenous and exogenous ligands around the body. It represents an ideal candidate for use in drug delivery systems as it is highly soluble and stable, has an extremely long circulatory half-life, and accumulates in tumours due to the enhanced permeability and retention effect.1,2 Several albumin based therapeutics have reached the clinic. For example Abraxane®, which is a nanoparticle formulation of paclitaxel with albumin, is used in the treatment of breast, pancreatic and non-small cell lung cancers.3 Other strategies exploited include the non-covalent association of drugs to albumin, fusion proteins, and covalent conjugation.4
HSA contains Cys-34 as a convenient handle for site-selective chemical conjugation, and is commonly targeted with maleimide reagents for the attachment of drugs.5–8 However, such maleimide conjugates are now known to be unstable over prolonged periods in vivo, undergoing retro-conjugate additions to release the cargo from the protein.9–12 Whilst hydrolysis of the maleimide can generate stable constructs, this process can lead to concomitant loss of an amount of the attached drug.9,13 To overcome this limitation we recently described monobromomaleimides (MBMs)14 as a promising platform for HSA conjugation (Scheme 1).13 By forming thiomaleimides 1, which have maintained the conjugated double bond and hence precluded any mechanistic possibility of spontaneous deconjugation (no acidic α-hydrogen), we showed that these conjugates undergo quantitative hydrolysis to afford serum stable maleamic acids 2.
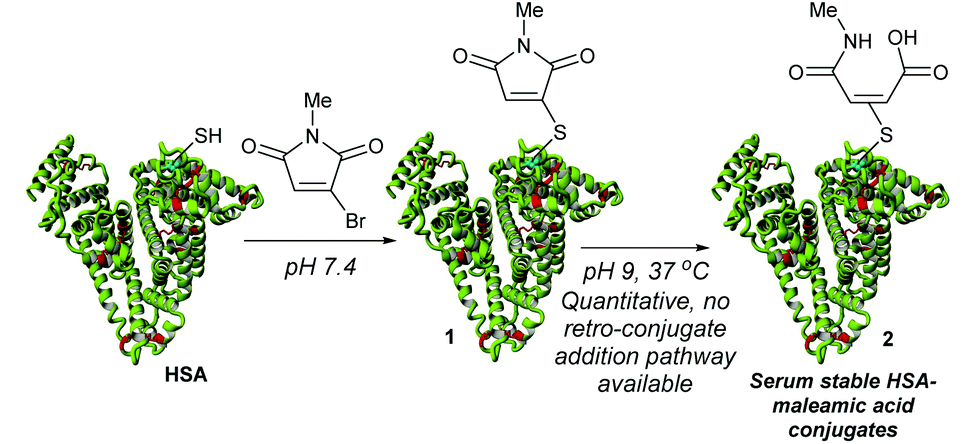 | ||
| Scheme 1 Previous work describing monobromomaleimides for human serum albumin (HSA) conjugation.13 | ||
To test the applicability of this approach to the construction of albumin–drug conjugates, we targeted the attachment of paclitaxel (PTX). PTX has poor solubility, and for clinical use requires a formulation using polyethoxylated castor oil (Cremophor EL) and ethanol, which are considered to contribute to side-effects such as hypersensitivity reactions. The conjugation of PTX to albumin would preclude the need for such formulations, whilst also offering an extended in vivo half-life and selective accumulation in tumours. Such albumin–PTX conjugates have previously been targeted using sub-optimal unselective lysine conjugation15,16 and cysteine conjugation with unstable maleimide reagents.17 Notably PTX–albumin conjugates are commonly prepared as esters of the C-2′ PTX hydroxyl,15,17 and hence are designed to release the free drug through hydrolysis. As such it is even more important that the albumin–drug attachment chemistry is robustly stable, to allow careful analysis and interpretation of the PK of drug release.
To apply the MBM platform to such albumin–drug conjugates we identified that an improvement in the reagent design was required. The previous protocol (Scheme 1) required a stabilising hydrolysis be carried out at pH 9.0, 37 °C, overnight. Such prolonged alkaline conditions are known to lead to degradation of sensitive drugs such as PTX,18 and also to hydrolyse ester pro-drugs.19 A dramatic acceleration in this maleimide hydrolysis would be required to make this approach broadly applicable, and our investigations to this end are described.
It has been shown that antibody conjugates generated using bromomaleimides with electron withdrawing C-2 (–CH2C(O)–) linkers undergo rapid hydrolysis in 1–2 h post conjugation (pH 8.0–8.5, rt).20,21 To attempt to transfer this simple hydrophilic linker design to albumin conjugates we initially synthesised MBM-C-2-dansyl reagent 3. Dansyl was chosen as its fluorescent properties are quenched by attached maleimides, and then ‘turned-on’ by maleimide hydrolysis.22,23 This compound would thus allow a clear read-out of the rate of hydrolysis. Conjugation of this reagent to HSA afforded conjugate 4, with LCMS post-hydrolysis revealing that unmodified protein and a double addition adduct were also present as minor species (ESI Fig. S2 and S3†). Whilst clearly in need of optimisation, an initial hydrolysis study was carried out. MBM reagent 3, model cysteine adduct 5 and the conjugate 4 were compared, leading in each case to the expected increase in fluorescence at 559 nm (Fig. 1). Several interesting observations can be made. The bromomaleimide is ∼4-fold more susceptible to hydrolysis than the thiomaleimide conjugates, presumably due to the inductive electron-withdrawing effect of the bromine, which is consistent with the effect observed for dibromomaleimides (although to a reduced magnitude).20,24 The next is that the HSA causes a ∼20-fold decrease in the rate of hydrolysis of the attached thiomaleimide. This is likely due to the hydrophobic ∼10 Å crevice in which Cys-34 resides serving a substantive protective role.5 Finally we noted that the hydrolysis of monothiomaleimides demonstrates substantial regioselectivity, with a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of isomers (see ESI,† compares with 2.5:1 for the bromomaleimide). The carbonyl proximal to the thioether is hydrolysed preferentially, thought to be due to a combination of inductive activation and mesomeric deactivation of the two carbonyls respectively.
:1 ratio of isomers (see ESI,† compares with 2.5:1 for the bromomaleimide). The carbonyl proximal to the thioether is hydrolysed preferentially, thought to be due to a combination of inductive activation and mesomeric deactivation of the two carbonyls respectively.
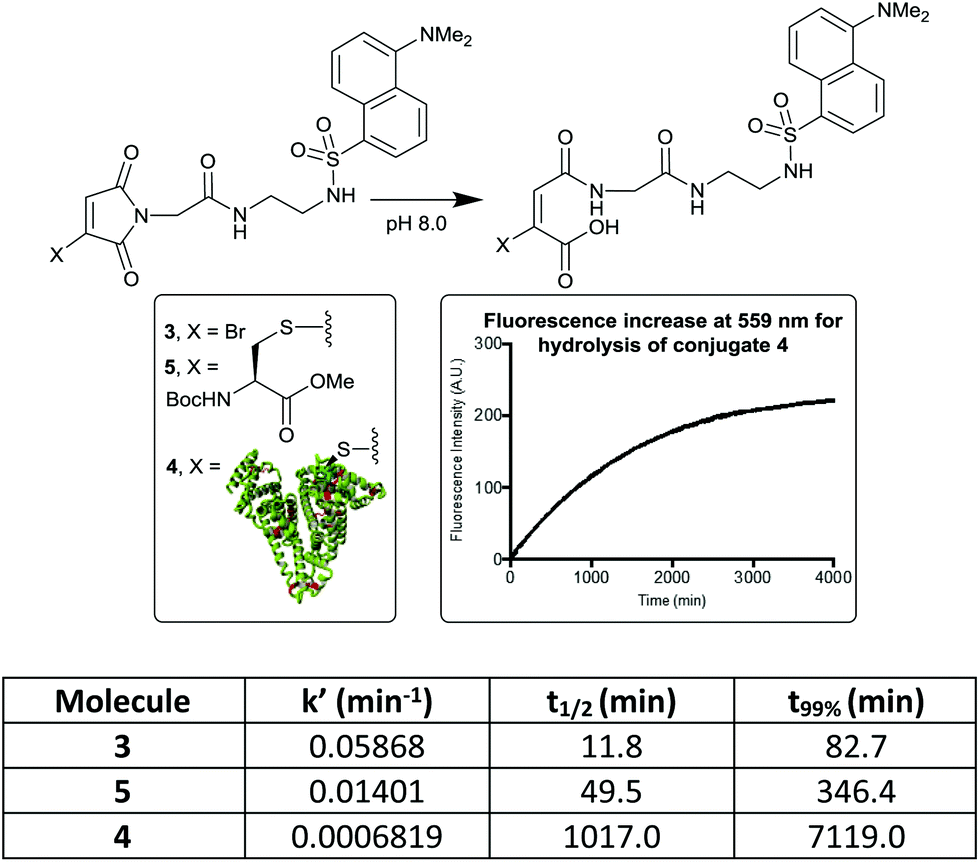 | ||
| Fig. 1 Hydrolysis rates of bromo- and thiomaleimides, measured by ‘turn-on’ of dansyl fluorescence. t99% is the time taken for the hydrolysis to be 99% complete. | ||
To improve upon this initial design, we chose to incorporate a PEG spacer in future molecules, postulating that it would improve the efficiency of the conjugation to the sterically hindered Cys-34. We also identified that the thiomaleimide absorbance at 350 nm could be monitored to afford the rate of conjugation, and its subsequent disappearance would reveal hydrolysis,20 negating the requirement to have a fluorophore attached. Pleasingly treatment of HSA with MBM 6 (2 equiv.) led to extremely clean conjugation, with no unmodified HSA or bis-modification (Fig. 2). Furthermore, monitoring the increasing absorbance at 350 nm allowed us to identify that the reaction was >99% complete after 10 min. This confirmed the rapid nature of bromomaleimide HSA conjugation, despite the relatively hindered location of Cys-34.
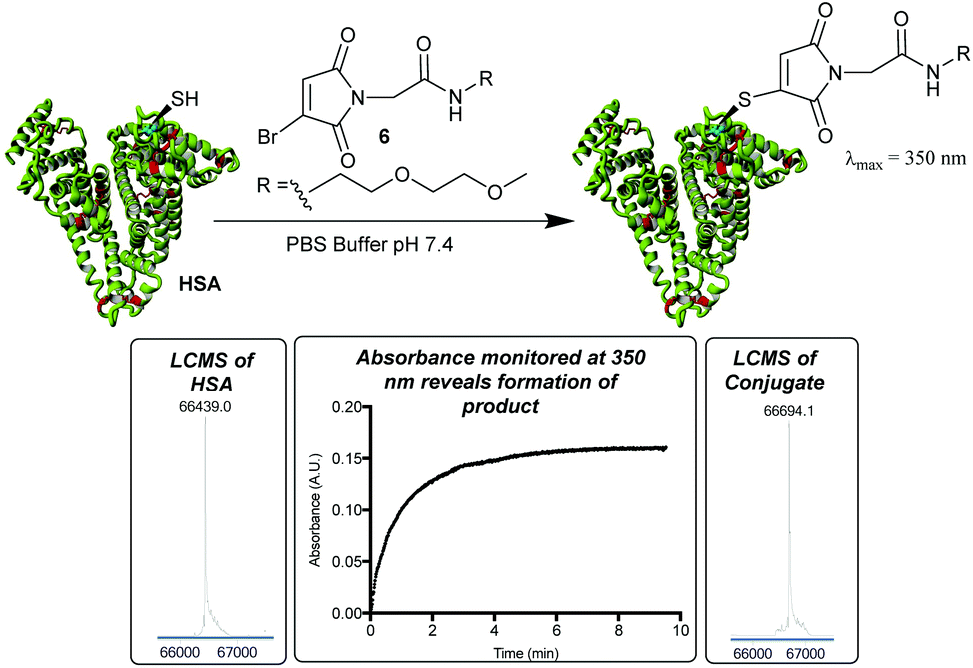 | ||
| Fig. 2 Incorporation of a PEG moiety into the reagent improves efficiency of conjugation as observed by LCMS. Native HSA: 66439 Da (expected 66439 Da); cysteine modification: 66694 Da (expected: 66694 Da). UV absorbance measurements at 350 nm used to reveal reaction progress. | ||
By monitoring the disappearance of the absorbance at 350 nm it was then possible to reveal the rates of hydrolysis of the thiomaleimide conjugates at pH 8.0 (Fig. 3). It was interesting to note that a simple ethylene glycol conjugate 7 showed an initial ∼1.5 fold acceleration (entry 1) relative to the hydrophobic dansyl conjugates. But the much more substantial increase was observed upon increasing the temperature, with an ∼8 fold increase upon shifting from 20 °C to 37 °C (entry 2). Further increases to the temperature could lead to extremely rapid hydrolyses (entries 3 and 4); however, we selected the optimised conditions as being 37 °C for 8.5 h. This represented a reasonable timescale for the reaction, whilst being conditions known to be well tolerated by protein conjugates.
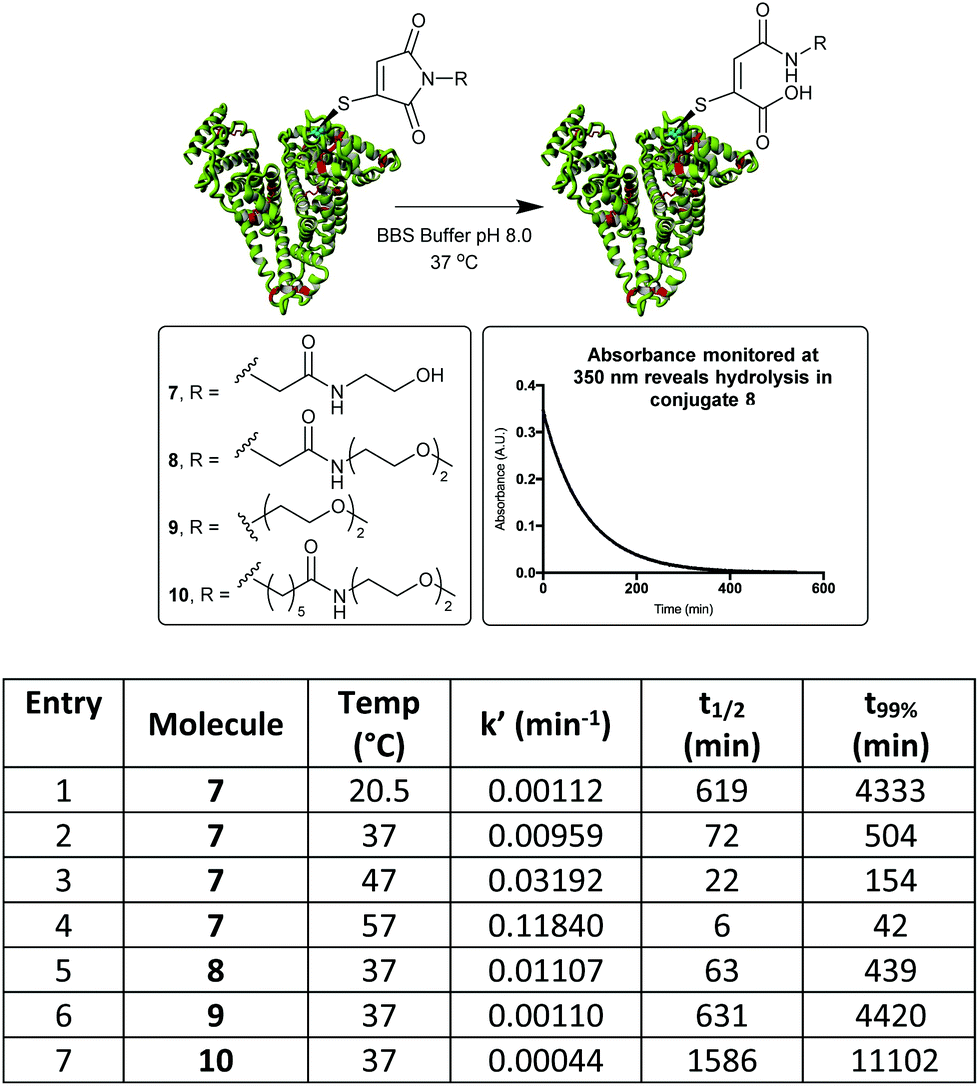 | ||
| Fig. 3 Hydrolysis study on HSA-MBM conjugates, by monitoring loss of absorbance at 350 nm. t99% is the time taken for the hydrolysis to be 99% complete. | ||
To further explore the effect of linker design on hydrolysis, a conjugate derived from the C-2-PEG reagent (conjugate 8) was compared with a direct PEG (9) and a C-6-PEG (10) analogue. The advantage of the electron-withdrawing C-2 moiety was clear, with the hydrolysis occurring in just under 8 h compared to 74 h for the direct PEG and 185 h for the C-6 analogue (Fig. 3).
With an optimised linker design established, we proceeded to construct the albumin–paclitaxel conjugate. The synthesis involved initial coupling of the PEG spacer to the MBM-C-2 acid 11, followed by a deprotection of the t-butyl ester and coupling to paclitaxel (Fig. 4). MBM-Paclitaxel reagent 12 was then conjugated to HSA, with post-conjugation hydrolysis affording the target conjugate 13. No detectable loss of payload was observed during the hydrolysis step. As expected, if no C-2 moiety was incorporated then prolonged incubation at higher pH (6.5 h, pH 8.5, 37 °C) was required and >30% of drug release was observed due to ester hydrolysis (ESI Fig. S8†). It is interesting to note that HSA again serves a protective role in these conjugates, as closely related paclitaxel-PEG esters have half-lives of 3–8 h at pH 7.4, 37 °C.25
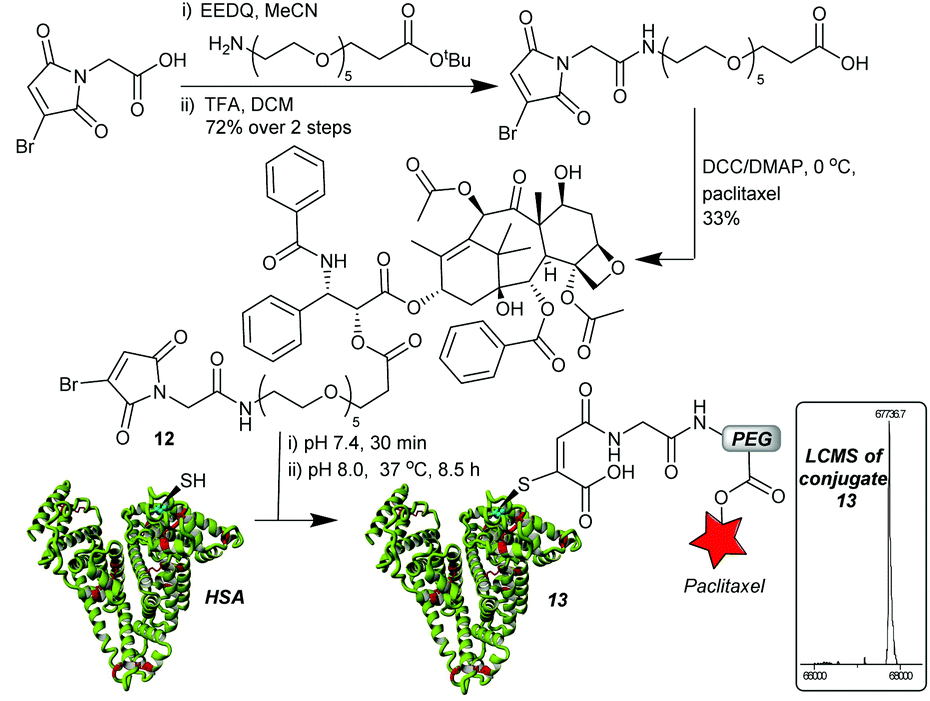 | ||
| Fig. 4 MBM-C-2-Paclitaxel synthesis and construction of albumin–drug conjugate. Conjugate 13: 67737 Da (expected 67738 Da). | ||
To evaluate the albumin–paclitaxel conjugate (HSA-PTX) cell killing potential, a MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) viability assay was performed to measure the metabolic activity of a human breast cancer cell line (MDA-MB-231-luc-D3H2LN)26 after treatment with either free paclitaxel (PTX) or the HSA-PTX conjugate (Fig. 5). The HSA-PTX conjugate performed similarly to that of free PTX, with the minor reduction of in vitro cytotoxicity consistent with comparable PTX–albumin pro-drug conjugates reported.17 Notably the ester linkage in such paclitaxel conjugates is designed to release in the tumor environment, not necessarily requiring internalisation. Indeed we confirmed that this mechanism was likely to be contributing to this in vitro cytotoxicity assay, as incubating the HSA-PTX conjugate in the cellular media led to >50% ester hydrolysis in 48 h at 37 °C (ESI Fig S9†).
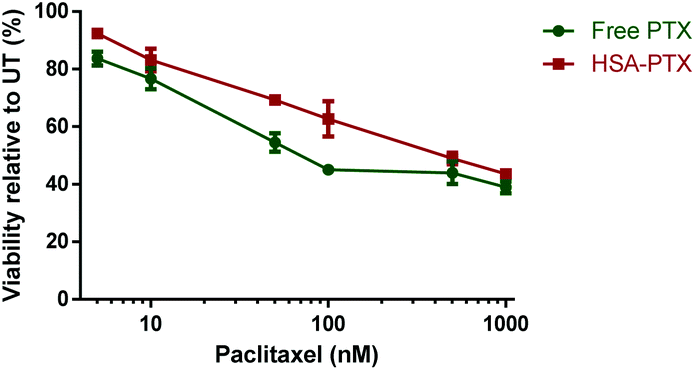 | ||
| Fig. 5 MTT viability assay performed with MDA-MB-231-luc-D3H2LN cells26 using either free paclitaxel (PTX) or HSA-paclitaxel (HSA-PTX) conjugates at concentrations ranging from 5 nM–1000 nM after 48 h of incubation. Data points represent the mean ± S.D (n = 3). | ||
Conclusions
In conclusion, we have shown that monobromomaleimide-C-2-PEG linkers offer an ideal approach to generate serum stable HSA conjugates. The UV absorbance of the thiomaleimides formed upon conjugation can be used to reveal the rapid nature of the reaction with Cys-34, and the subsequent rate of stabilising hydrolysis. The protective nature of the Cys-34 cleft is found to substantially reduce the speed of hydrolysis; but with the inclusion of the electron withdrawing C-2 linker along with conducting the reaction at 37 °C, pH 8.0, it is complete in 8 h. This optimisation allows the quantitative construction of an HSA-paclitaxel conjugate without any hydrolysis of the labile ester bond. Furthermore, the cytotoxicity of the HSA-paclitaxel conjugate was retained, which combined with the drug delivery properties of HSA indicates the potential of this conjugate as a prospective anti-cancer therapeutic.Conflicts of interest
J. B. and V. C. are directors of UCL spin-out company Thiologics.Acknowledgements
The authors gratefully acknowledge EPSRC and Albumedix for funding. Also CEMBID (Center for Multifunctional Biomolecular Drug Design, Grant Number: NNF17OC0028070) for funding to KAH and SS. We also acknowledge Recipharm where the cytotoxic paclitaxel reagent coupling and conjugation were carried out, the EPSRC UK National Mass Spectrometry Facility (NMSF), Swansea and Abil Aliev in UCL department of chemistry for NMR support.Notes and references
- D. Sleep, J. Cameron and L. R. Evans, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5526–5534 CrossRef CAS PubMed.
- D. Sleep, Expert Opin. Drug Delivery, 2015, 12, 793–812 CrossRef CAS PubMed.
- W. J. Gradishar, Expert Opin. Pharmacother., 2006, 7, 1041–1053 CrossRef CAS PubMed.
- B. Elsadek and F. Kratz, J. Controlled Release, 2012, 157, 4–28 CrossRef CAS PubMed.
- F. Kratz, A. Warnecke, K. Scheuermann, C. Stockmar, J. Schwab, P. Lazar, P. Druckes, N. Esser, J. Drevs, D. Rognan, C. Bissantz, C. Hinderling, G. Folkers, I. Fichtner and C. Unger, J. Med. Chem., 2002, 45, 5523–5533 CrossRef CAS.
- K. Thibaudeau, R. Leger, X. C. Huang, M. Robitaille, O. Quraishi, C. Soucy, N. Bousquet-Gagnon, P. van Wyk, V. Paradis, J. P. Castaigne and D. Bridon, Bioconjugate Chem., 2005, 16, 1000–1008 CrossRef CAS PubMed.
- J. G. Mehtala, C. Kulczar, M. Lavan, G. Knipp and A. Wei, Bioconjugate Chem., 2015, 26, 941–949 CrossRef CAS PubMed.
- E. S. Akinboye, O. C. Rogers and J. T. Isaacs, Prostate, 2018, 78, 655–663 CrossRef CAS PubMed.
- L. N. Tumey, M. Charati, T. He, E. Sousa, D. S. Ma, X. G. Han, T. Clark, J. Casavant, F. Loganzo, F. Barletta, J. Lucas and E. I. Graziani, Bioconjugate Chem., 2014, 25, 1871–1880 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saadi, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, N. Trung, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawlil, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- M. E. B. Smith, M. B. Caspersen, E. Robinson, M. Morais, A. Maruani, J. P. M. Nunes, K. Nicholls, M. J. Saxton, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2015, 13, 7946–7949 RSC.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- F. Dosio, P. Brusa, P. Crosasso, S. Arpicco and L. Cattel, J. Controlled Release, 1997, 47, 293–304 CrossRef CAS.
- F. Dosio, S. Arpicco, P. Brusa, B. Stella and L. Cattel, J. Controlled Release, 2001, 76, 107–117 CrossRef CAS PubMed.
- J. Yang, Q. Lv, W. Wei, Z. Yang, J. Dong, R. Zhang, Q. Kan, Z. He and Y. Xu, Drug Delivery, 2018, 25, 807–814 CrossRef CAS PubMed.
- J. Tian and V. J. Stella, J. Pharm. Sci., 2008, 97, 3100–3108 CrossRef CAS PubMed.
- M. Skwarczynski, Y. Hayashi and Y. Kiso, J. Med. Chem., 2006, 49, 7253–7269 CrossRef CAS PubMed.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- J. P. M. Nunes, V. Vassileva, E. Robinson, M. Morais, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 24828–24832 RSC.
- J. Guy, K. Caron, S. Dufresne, S. W. Michnick, W. G. Skene and J. W. Keillor, J. Am. Chem. Soc., 2007, 129, 11969–11977 CrossRef CAS PubMed.
- J. Youziel, A. R. Akhbar, Q. Aziz, M. E. B. Smith, S. Caddick, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2014, 12, 557–560 RSC.
- N. Forte, M. Livanos, E. Miranda, M. Morais, X. Yang, V. S. Rajkumar, K. A. Chester, V. Chudasama and J. R. Baker, Bioconjugate Chem., 2018, 29, 486–492 CrossRef CAS PubMed.
- R. B. Greenwald, A. Pendri, D. Bolikal and C. W. Gilbert, Bioorg. Med. Chem. Lett., 1994, 4, 2465–2470 CrossRef CAS.
- L. M. Zasadil, K. A. Andersen, D. Yeum, G. B. Rocque, L. G. Wilke, A. J. Tevaarwerk, R. T. Raines, M. E. Burkard and B. A. Weaver, Sci. Transl. Med., 2014, 6, 229ra43 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob00721k |
| This journal is © The Royal Society of Chemistry 2019 |