Catalytic direct amidations in tert-butyl acetate using B(OCH2CF3)3†
Received
2nd May 2019
, Accepted 12th June 2019
First published on 14th June 2019
Abstract
Catalytic direct amidation reactions have been the focus of considerable recent research effort, due to the widespread use of amide formation processes in pharmaceutical synthesis. However, the vast majority of catalytic amidations are performed in non-polar solvents (aromatic hydrocarbons, ethers) which are typically undesirable from a sustainability perspective, and are often poor at solubilising polar carboxylic acid and amine substrates. As a consequence, most catalytic amidation protocols are unsuccessful when applied to polar and/or functionalised substrates of the kind commonly used in medicinal chemistry. In this paper we report a practical and useful catalytic direct amidation reaction using tert-butyl acetate as the reaction solvent. The use of an ester solvent offers improvements in terms of safety and sustainability, but also leads to an improved reaction scope with regard to polar substrates and less nucleophilic anilines, both of which are important components of amides used in medicinal chemistry. An amidation reaction was scaled up to 100 mmol and proceeded with excellent yield and efficiency, with a measured process mass intensity of 8.
Introduction
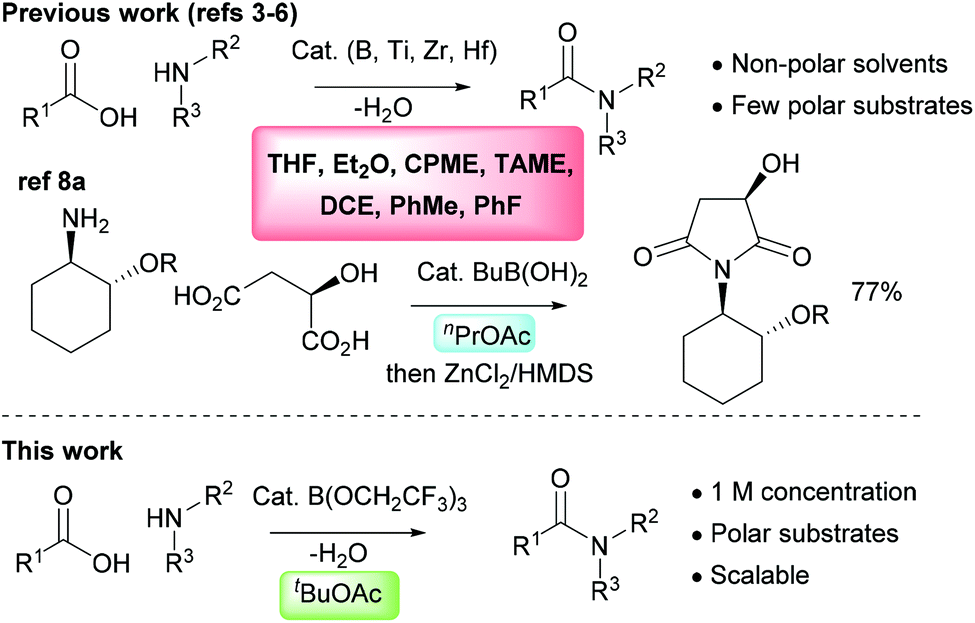
The direct amidation reaction between carboxylic acids and amines is one of the most common processes employed in the synthesis of organic molecules in both academic and industrial organisations. It remains a highly inefficient reaction, however, due to the widespread use of stoichiometric activating agents that leads to the generation of high molecular weight by-products as well as increased solvent use during both work-up and purification.1,2 There has consequently been considerable interest in recent years in the development of catalytic direct amidation reactions which enable amides to be produced from carboxylic acids and amines with a molecule of water as the only stoichiometric by-product. Common catalytic systems include those based around boronic acids,3 boric acid derivatives4 or boron heterocycles,5 as well as salts of the group(IV) metals titanium, zirconium or hafnium.6 However, to date these catalytic amidation methods have failed to become widely adopted.7 This is partly due to the fact that they cannot often be applied to common amide targets which incorporate polar functional groups such as heterocyclic rings. It is also a consequence of the poor efficiency of many catalytic amidation reactions due to the large quantities of solvents employed both in the reaction itself and in the work-up procedure.7 The use of molecular sieves to efficiently remove water from the reaction mixture exacerbates these problems as higher dilution reaction conditions are normally required, along with excess solvent during work-up for washing the molecular sieves to recover the amide product. For larger scale reactions, Dean–Stark water removal is employed as this is considerably more efficient in terms of solvent usage and scalability, and there are a few reports of direct amidation reactions catalysed by boric acid or simple boronic acids being employed on an industrial scale.8 The solvents used for catalytic amidation reactions are typically aromatic hydrocarbons (toluene,3a,4a fluorobenzene3b), chlorinated solvents (1,2-dichloroethane3i) or ethers (Et2O,6d THF,5a,b CPME,4e TAME4g,h), and these are often sub-optimal from a safety and/or sustainability perspective (Scheme 1).9 In addition, these relatively non-polar solvents do not effectively solubilise many polar functionalised carboxylic acids or amines. In this paper, we outline a highly effective and scalable method for performing catalytic direct amidation reactions in an ester solvent, and demonstrate its application to challenging substrates including polar heterocycles and poorly nucleophilic anilines.
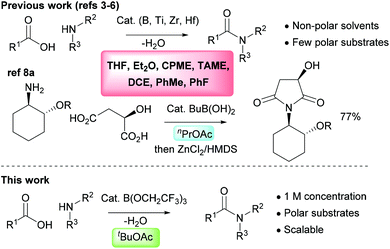 |
| | Scheme 1 Solvents previously employed in catalytic amidation reactions and our new approach using tert-butyl acetate. CPME = cyclopentyl methyl ether; TAME = tert-amyl methyl ether; DCE = 1,2-dichloroethane. | |
Results & discussion
Background
We have recently reported that B(OCH2CF3)3 can be employed as a very effective catalyst for direct amidation reactions which is applicable to a wide range of substrates.4g Remarkably, it can also be used for the catalytic direct amidation of many unprotected amino acids.4h The reactions were largely performed in tert-amyl methyl ether (TAME) as solvent, although selected reactions involving less nucleophilic but relatively apolar amines were more efficient in toluene. Important advantages of this catalytic system include an extremely wide substrate scope, together with a relatively high reaction concentration (0.5 M) which leads to efficient and scalable reactions. B(OCH2CF3)3 is available commercially, or can be synthesised on a large scale from B2O3 and CF3CH2OH.4d,g
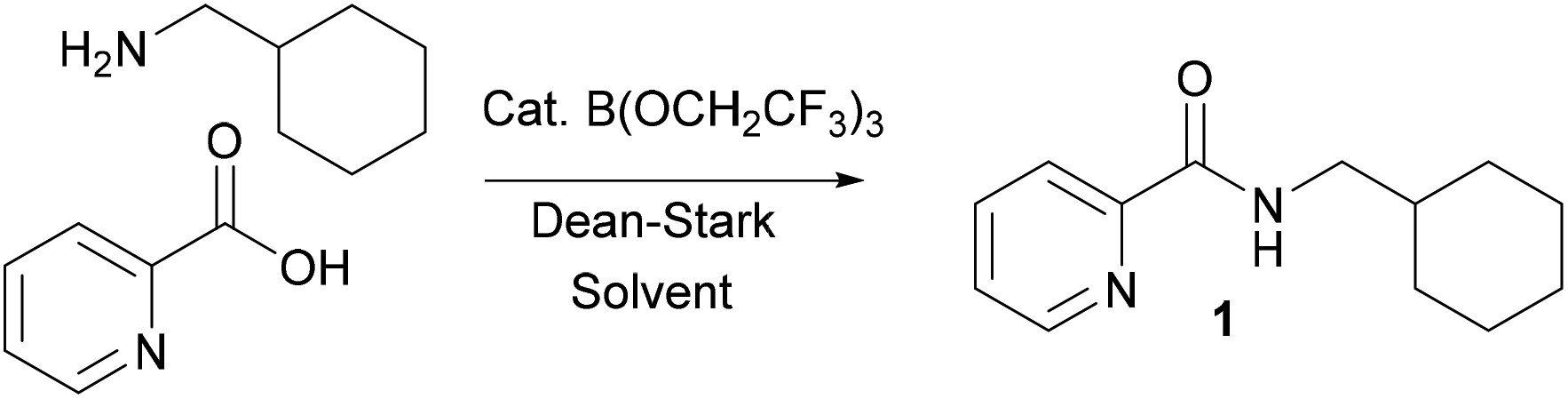
During the course of an ongoing research project on Pd-catalysed direct C–H arylation reactions,10 we needed to prepare a series of amides derived from picolinic acid such as 1 (Scheme 2). Initial evaluation of our catalytic amidation method using TAME as a solvent led to a moderate 71% yield of the desired amide 1 with 20 mol% catalyst (Table 1, entry 1).
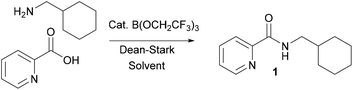 |
| | Scheme 2 Preparation of amide 1via B(OCH2CF3)3-catalysed amidation. | |
Table 1 Optimisation of reaction solvent and conditions for the synthesis of amide 1. Reaction conditions: 5 mmol scale, 0.5 M concentration in indicated solvent, 10 mol% B(OCH2CF3)3, reflux, Dean–Stark. 24 h
| Entry |
Catalyst |
Solvent |
Bp |
Yielda |
|
Isolated yields.
20 mol% B(OCH2CF3)3.
1 M concentration.
0.75 M concentration.
Ar = 3,5-bis-(trifluoromethyl)phenyl.
|
| 1b |
B(OCH2CF3)3 |
TAME |
86 °C |
71% |
| 2 |
B(OCH2CF3)3 |
EtOAc |
77 °C |
9% |
| 3c |
B(OCH2CF3)3 |
n
PrOAc |
102 °C |
27% |
| 4 |
B(OCH2CF3)3 |
n
BuOAc |
126 °C |
20% |
| 5 |
B(OCH2CF3)3 |
iPrOAc |
89 °C |
52% |
| 6 |
B(OCH2CF3)3 |
t
BuOAc |
97 °C |
75% |
|
7c
|
B(OCH
2
CF
3
)
3
|
t
BuOAc
|
97 °C
|
91%
|
| 8c |
B(OCH2CF3)3 |
EtCN |
97 °C |
76% |
| 9c |
B(OCH2CF3)3 |
n
PrCN |
117 °C |
64% |
| 10d |
B(OMe)3 |
t
BuOAc |
97 °C |
8% |
| 11 |
B(OEt)3 |
t
BuOAc |
97 °C |
10% |
| 12c |
Ti(OiPr)4 |
t
BuOAc |
97 °C |
14% |
| 13e |
ArB(OH)2 |
t
BuOAc |
97 °C |
73% |
We hypothesised that this was due to the relatively poor solubility of picolinic acid (and the corresponding ammonium salt) in TAME. We therefore decided to explore other reaction solvents which might circumvent these problems. Esters were identified as potentially suitable, as they are typically greener and more sustainable solvents than ethers or aromatic hydrocarbons, and offer a better safety profile (i.e. they are typically not prone to peroxide formation).9 They are also considerably more polar so should be much more effective at solubilising polar substrates and/or ammonium salts derived from them. Importantly, we had previously observed that stoichiometric amidation mediated by B(OCH2CF3)3 could be carried out in ethyl acetate as a solvent without any significant amidation of the ester being observed.4e To date, there is only a single report of a catalytic amidation reaction performed in an ester solvent, using BuB(OH)2 as a catalyst in n-propyl acetate.8a Aside from this, esters have not been explored as solvents for catalytic amidation reactions.
Reaction optimisation
We selected the synthesis of amide 1 as a test reaction for evaluating a range of ester solvents. Using 10 mol% B(OCH2CF3)3 catalyst, the reaction could be performed in a range of ester solvents under Dean–Stark reaction conditions though the yields were somewhat variable (Table 1, entries 2–7). A very low yield was obtained in ethyl acetate (entry 2), and n-propyl acetate (entry 3) or n-butyl acetate (entry 4) offered relatively little improvement. Pleasingly, isopropyl acetate (entry 5) and tert-butyl acetate (entry 6) gave significantly improved yields with the latter being particularly effective, even though these solvents have lower boiling points than their unbranched isomers. The yield of amide 1 could be further improved in tert-butyl acetate by increasing the concentration of the reaction mixture to 1 M, leading to a reduction in the solvent requirements of the process. Finally, the nitrile solvents propionitrile (entry 8) and butyronitrile (entry 9) were also effective for the amidation reaction, potentially offering useful more-polar alternative solvents.11 It should be noted that the most effective solvents have relatively low boiling points and form azeotropes with a high proportion of water present, with tert-butyl acetate (bp 97 °C; 22 wt% H2O in azeotrope) and propionitrile (bp 97 °C; 24 wt% H2O in azeotrope) being most effective.12 This may reflect a balance between the need for efficient water removal from the reaction and the greater levels of catalyst decomposition which may take place at higher reaction temperatures.‡ A selection of other simple amidation catalysts (entries 10–13) were also evaluated in tBuOAc, with only an arylboronic acid being effective.
Reaction scope
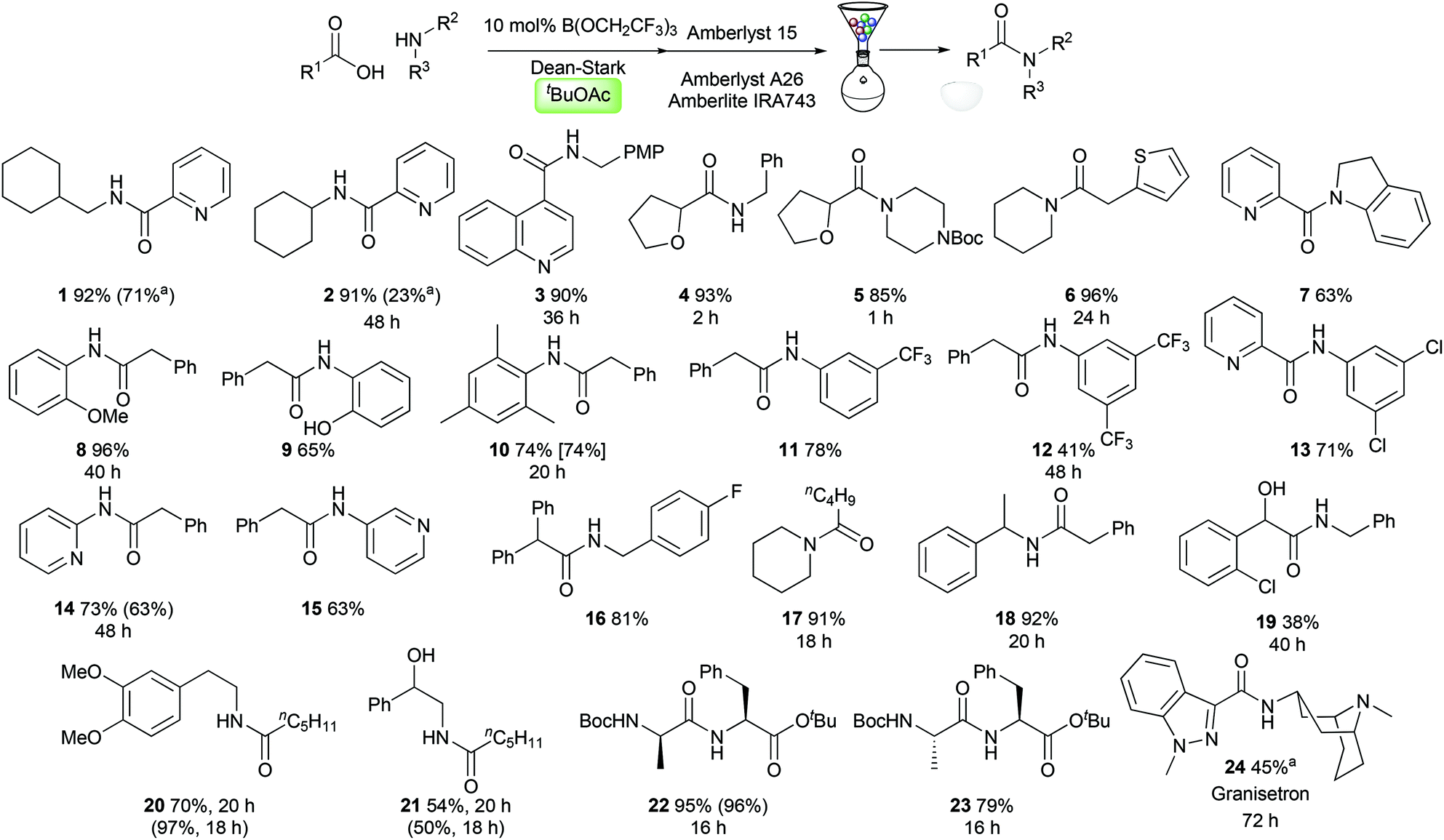
With optimised conditions in hand, we evaluated them in the synthesis of a selection of challenging amides 1–24. We were particularly interested in examining the reactivity of medicinally relevant substrates such as polar heterocycles and poorly nucleophilic amines. Pleasingly, amides could be prepared effectively from heterocyclic carboxylic acids, containing a pyridine (1–2, 7), a quinoline (3), a tetrahydrofuran (4–5), and a thiophene (6). A range of anilines and related derivatives could also be employed including indoline (7), electron-rich anilines (8–10), electron-deficient anilines (11–13) and aminopyridines (14–15). Aliphatic amines including simple alkylamines (1–4), electron-deficient benzylamines (16), secondary amines (5–6, 17), and branched systems (18, 22–24) were also good substrates. The amide 19 derived from 2-chloromandelic acid was obtained in only 38% yield, demonstrating the challenging nature of this particular substrate. Amides derived from hexanoic acid and dimethyldopamine (20) or 1-phenylethanolamine (21) were synthesised effectively, with the latter reaction demonstrating that the amidation reaction is chemoselective for acylation of the amine over the alcohol. Dipeptide synthesis from Boc-protected D- or L-alanine and L-phenylalanine tert-butyl ester proceeded efficiently, giving diastereomeric amides 22/23 with no observable epimerisation. Finally, the serotonin 5-HT3 receptor antagonist Granisetron 24 (used commonly as an antiemetic)13 could be prepared in 45% yield using only 20 mol% catalyst. Notably, in several cases amides were prepared from reactants where both the carboxylic acid and the amine can be considered as challenging substrates for catalytic amidation (5, 7, 13, 22–24).§ As can be seen in Scheme 3, with the exception of amide 20, tert-butyl acetate offers comparable or improved amide yields over amide syntheses previously performed in TAME or toluene (1, 2, 10, 14, 21). In most cases, the amides could be purified using a solid-phase work-up procedure employing scavenger resins to remove unreacted amine (Amberlyst 15), carboxylic acid (Amberlyst A-26) and boron compounds (Amberlite IRA743), with no requirement for aqueous work-up or chromatography.4d
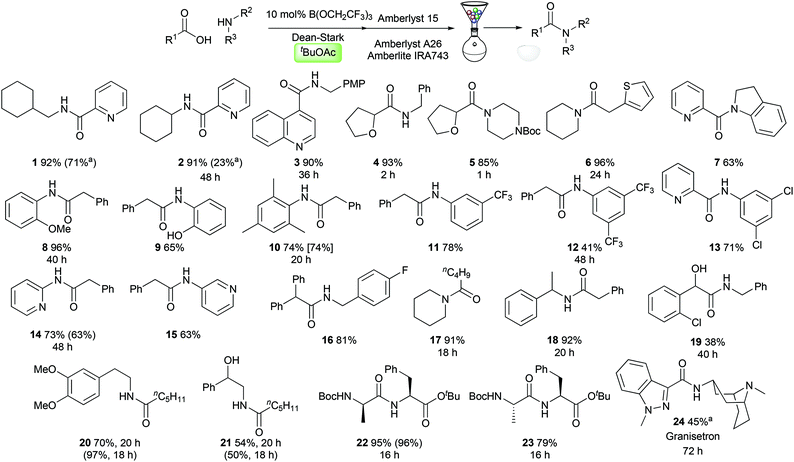 |
| | Scheme 3 Scope of the catalytic amidation reaction in tert-butyl acetate. All reactions were run for 24 h unless otherwise indicated. Yields given in parentheses/brackets are for the synthesis of the same amide in TAME/PhMe respectively. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 mol% B(OCH2CF3)3 used. 20 mol% B(OCH2CF3)3 used. | |
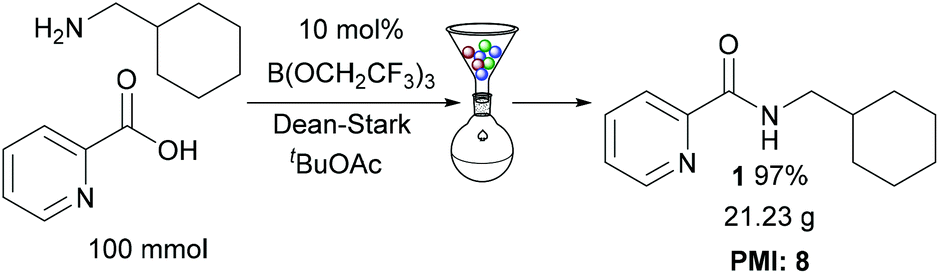
Multigram scale reaction
To demonstrate the efficiency of our protocol, amide 1 was prepared on a 100 mmol scale (Scheme 4), using the solid-phase work-up procedure to purify the amide. This gave 21.23 g of amide 1 in 97% overall yield. The process mass intensity14 for the reaction was calculated to be 8, showing that this catalytic amidation method is extremely competitive for use in large scale preparations (e.g. a typical PMI for stoichiometric amidation reactions used in the synthesis of pharmaceutical intermediates is ∼43 (ref. 7a)).
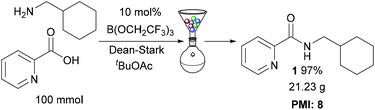 |
| | Scheme 4 Large scale synthesis of amide 1. | |
Conclusions
We have demonstrated that tert-butyl acetate is a very effective solvent for B(OCH2CF3)3-catalysed direct amidation reactions. These conditions were found to be particularly effective for challenging pharmaceutically relevant substrates including carboxylic acids containing polar heterocycles, as well as electron-deficient anilines and aminopyridines. The reactions can be run at 1 M concentration, and the amides can typically be purified using a simple resin-based filtration process which has low solvent requirements. A 100 mmol synthesis of an amide gave a 97% yield of product with process mass intensity of 8. tert-Butyl acetate is a readily available low-cost solvent, produced industrially from acetic acid and isobutylene, which has a good safety profile9 making it potentially suitable for widespread use in catalytic direct amidation reactions.
Experimental section
General amidation procedure
A suspension of carboxylic acid (5 mmol, 1 equiv.), amine (5 mmol, 1 equiv.) and B(OCH2CF3)3 (108 μL, 0.5 mmol, 10 mol%) in tBuOAc (5 mL, 1 M) with a Dean–Stark trap (side arm filled with tBuOAc) was heated to reflux. An air condenser was fitted and the reaction mixture heated for 1–48 h (see Scheme 3). The reaction was cooled to room temperature and water (0.5 mL), dimethyl carbonate (5 mL), Amberlite IRA-743 (0.25 g), Amberlyst A15 (0.5 g) and A-26(OH) (0.5 g) resins were added and the resulting suspension was stirred for 30 min. Anhydrous magnesium sulfate (∼0.5 g) was added, the mixture filtered, and the resins/solids washed with ethyl acetate (2 × 5 mL). The combined filtrates were concentrated in vacuo to yield the pure amide.
Further experimental details
Characterisation data for all amides 1–24, together with 1H and 13C spectra can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank AstraZeneca and the UCL MAPS faculty for providing an EPSRC CASE award to support a Ph.D. studentship for CEC, the Leverhulme Trust for providing a grant (RPG-2017-221) to support VL, and the Wellcome Trust (105342/Z/14/Z) for providing a PhD studentship to support LTM.
Notes and references
-
(a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC;
(b) L. Amarnath, I. Andrews, R. Bandichhor, A. Bhattacharya, P. Dunn, J. Hayler, W. Hinkley, N. Holub, D. Hughes, L. Humphreys, B. Kaptein, H. Krishnen, K. Lorenz, S. Mathew, G. Nagaraju, T. Rammeloo, P. Richardson, L. Wang, A. Wells and T. White, Org. Process Res. Dev., 2012, 16, 535 CrossRef;
(c) M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082 RSC;
(d) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- For recently reported stoichiometric amidation methods, see:
(a) T. Krause, S. Baader, B. Erb and L. J. Gooβen, Nat. Commun., 2016, 7, 11732 CrossRef PubMed;
(b) L. Hu, S. Xu, Z. Zhao, Y. Yang, Z. Peng, M. Yang, C. Wang and J. Zhao, J. Am. Chem. Soc., 2016, 138, 13135 CrossRef CAS PubMed;
(c) X. Xu, H. Feng, L. Huang and X. Liu, J. Org. Chem., 2018, 83, 7962 CrossRef CAS PubMed;
(d) D. C. Braddock, P. D. Lickiss, B. C. Rowley, D. Pugh, T. Purnomo, G. Santhakumar and S. J. Fussel, Org. Lett., 2018, 20, 950 CrossRef CAS PubMed;
(e) S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087 RSC;
(f) M. Cortes-Clerget, N. R. Lee and B. H. Lipshutz, Nat. Protoc., 2019, 14, 1108 CrossRef CAS PubMed.
-
(a) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196 CrossRef CAS;
(b) K. Arnold, B. Davies, R. L. Giles, C. Grosjean, G. E. Smith and A. Whiting, Adv. Synth. Catal., 2006, 348, 813 CrossRef CAS;
(c) R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876 CrossRef CAS PubMed;
(d) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124 RSC;
(e) K. Arnold, B. Davies, D. Hérault and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 2673 CrossRef CAS PubMed;
(f) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386 CrossRef CAS PubMed;
(g) S. Fatemi, N. Gernigon and D. G. Hall, Green Chem., 2015, 17, 4016 RSC;
(h) S. Arkhipenko, M. T. Sabatini, A. S. Batsanov, V. Karaluka, T. D. Sheppard, H. S. Rzepa and A. Whiting, Chem. Sci., 2018, 9, 1058 RSC;
(i) K. Wang, Y. Lu and K. Ishihara, Chem. Commun., 2018, 54, 5410 RSC;
(j) Y. Du, T. Barber, S. E. Lim, H. S. Rzepa, I. R. Baxendale and A. Whiting, Chem. Commun., 2019, 55, 2916 RSC.
-
(a) P. Tang, Org. Synth., 2005, 81, 262 CrossRef CAS;
(b) R. Yamashita, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 3654 CrossRef CAS PubMed;
(c) P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320 RSC;
(d) R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512 CrossRef CAS PubMed;
(e) V. Karaluka, R. M. Lanigan, P. M. Murray, M. Badland and T. D. Sheppard, Org. Biomol. Chem., 2015, 13, 10888 RSC;
(f) R. M. Lanigan, V. Karaluka, M. T. Sabatini, P. Starkov, M. Badland, L. Boulton and T. D. Sheppard, Chem. Commun., 2016, 52, 8846 RSC;
(g) M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed;
(h) M. T. Sabatini, V. Karaluka, R. M. Lanigan, L. T. Boulton, M. Badland and T. D. Sheppard, Chem. – Eur. J., 2018, 24, 7033 CrossRef CAS PubMed;
(i) D. N. Sawant, D. B. Bagal, S. Ogawa, K. Selvam and S. Saito, Org. Lett., 2018, 20, 4397 CrossRef CAS PubMed.
-
(a) H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571 CrossRef CAS PubMed;
(b) H. Noda, Y. Asada, M. Shibasaki and N. Kumagai, J. Am. Chem. Soc., 2019, 141, 1546 CrossRef CAS PubMed.
-
(a) H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 2201 CAS;
(b) H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822 CrossRef CAS;
(c) C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Chem. Commun., 2012, 48, 666 RSC;
(d) H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271 CrossRef CAS.
-
(a) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10 CrossRef CAS;
(b) X. Wang, Nat. Catal., 2019, 2, 98 CrossRef;
(c)
B. M. Monks and A. Whiting, Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, Wiley, Hobokon, NJ, USA, 2013, ch. 5 Search PubMed;
(d)
F. Ferdousi and A. Whiting, Green Catalytic Direct Amide Bond Formation in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, ed. L. Summerton, H. F. Sneddon, L. C. Jones and J. H. Clark, RSC Green Chemistry series, Cambridge, 2016, ch. 13 Search PubMed;
(e) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453 CrossRef CAS;
(f) K. Ishihara, Tetrahedron, 2009, 65, 1085 CrossRef CAS;
(g) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714 RSC.
-
(a) J. Limanto, E. R. Ashley, J. Yin, G. L. Beutner, B. T. Grau, A. M. Kassim, M. M. Kim, A. Klapars, Z. Liu, H. R. Strotman and M. D. Truppo, Org. Lett., 2014, 16, 2716 CrossRef CAS PubMed;
(b) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS;
(c) R. M. Bannister, M. H. Brookes, G. R. Evans, R. B. Katz and N. D. Tyrrell, Org. Process Res. Dev., 2000, 4, 467 CrossRef CAS;
(d) R. Kumar Mylavarapu, K. GCM, N. Kolla, R. Veeramalla, P. Koilkonda, A. Bhattacharya and R. Bandichhor, Org. Process Res. Dev., 2007, 11, 1065 CrossRef.
-
(a) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC;
(b) C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC.
- C. E. Coomber, L. Benhamou, D.-K. Bučar, P. D. Smith, M. J. Porter and T. D. Sheppard, J. Org. Chem., 2018, 83, 2495 CrossRef CAS.
- P. M. Murray, F. Bellany, L. Benhamou, D.-K. Bučar, A. B. Tabor and T. D. Sheppard, Org. Biomol. Chem., 2016, 14, 2373 RSC.
-
http://www.homepages.ed.ac.uk/jwp/Chemeng/azeotrope/hetero.html
, Last accessed 11th April 2019; E. C. Wagner, J. Chem. Ed., 1950, 27, 245. The azeotropes contain the following quantities of water (wt%): EtOAc (8); nPrOAc (14); nBuOAc (27); iPrOAc (11); tBuOAc (22); EtCN (24); nPrCN (33).
- J. Cassidy, V. Raina, C. Lewis, L. Adams, M. Soukop, W. G. Rapeport, B. D. Zussman, E. M. Rankin and S. B. Kaye, Br. J. Cancer, 1988, 58, 651 CrossRef CAS PubMed.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and data for all compounds, along with 1H and 13C NMR spectra for all amides. See DOI: 10.1039/c9ob01012b |
| ‡ Analysis of the reaction mixture and Dean–Stark trap by 19F NMR indicated that in tBuOAc, 49% of the CF3CH2OR group remained in the reaction mixture after 24 h (cf. 78% in TAME4g). In contrast, analysis of an amidation performed in nBuOAc showed that only 16% of the CF3CH2OR group remained in the reaction mixture after 24 h. Minor additional signals could be seen in the 19F NMR spectrum of the tBuOAc reaction, whereas in nBuOAc multiple species were present at significant concentration. See ESI† for further details. |
| § We have previously noted that 2-aminopyridine (amide 14) shows low reactivity in stoichiometric B(OCH2CF3)3 amidation reactions.4e Others have recently noted that sterically encumbered (amides 8–10) or electron-deficient (amides 11–13) anilines were particularly challenging substrates for catalytic amidation reactions.5a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Liam T.
Martin
a,
Peter D.
Smith
a,
Liam T.
Martin
a,
Peter D.
Smith