
Ultrafine MoO3 nanoparticles embedded in porous carbon nanofibers as anodes for high-performance lithium-ion batteries†
Xiu
Liu‡
a,
Yuan
Liu‡
 b,
Xiaodong
Yan
c,
Jin-Le
Lan
*a,
Yunhua
Yu
a and
Xiaoping
Yang
a
b,
Xiaodong
Yan
c,
Jin-Le
Lan
*a,
Yunhua
Yu
a and
Xiaoping
Yang
a
aState Key Laboratory of Organic−Inorganic Composites, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: lanjl@mail.buct.edu.cn
bState Key Lab of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
cKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China
First published on 2nd November 2018
Abstract
Ultrafine MoO3 nanoparticles uniformly embedded in porous carbon nanofiber (PCNF) anodes synthesized by electrospinning are comprehensively investigated. At a carbonization temperature of 750 °C, PMMA is decomposed to form a worm-like porous carbon matrix, and ultrafine MoO3 nanoparticles (3–5 nm) are uniformly dispersed in carbon nanofibers. Electrochemical tests show that the optimized MoO3/PCNF material displays excellent capacity of 795.8 mA h g−1 after 100 cycles at a current density of 200 mA g−1 and also shows excellent rate performance at a high current density. The excellent performance is observed because MoO3 with ultrafine crystalline particles shortens the transmission path of Li+ and porous carbon nanofibers effectively relieve volume stress caused by Li+ insertion. This study presents a facile and cost-effective approach for the synthesis of high-performance electrode materials through nanostructuring and porous design.
Introduction
Over the past decades, rechargeable lithium-ion batteries (LIBs) have achieved much success in the field of portable electronics and now, they are also used to power electric vehicles (EVs) owing to their high energy and power densities as well as long cycling life.1–4 However, the current anodes of commercial LIBs are graphite materials, which possess low theoretical capacity of 372 mA h g−1; thus, LIBs cannot satisfy the ever-increasing requirements of EVs. Therefore, developing new anode materials with high energy density to replace the graphite anode is now in progress. Some transition metal oxide (TMO) electrode materials that uptake/release lithium based on a conversion reaction mechanism or an alloying reaction mechanism always present larger discharge capacities (sometimes over 1000 mA h g−1), thus receiving much research focus.5–11 Among them, MoO3 has been researched extensively as the LIB anode in recent years due to its high theoretical capacity of 1117 mA h g−1. Nevertheless, the practical application of MoO3 is currently hampered by its large volume change during repeated (de)lithiation processes, which can lead to fast capacity decay of LIBs.To overcome this issue, many efforts have been conducted. In particular, the design of one-dimensional (1D) MoO3/C composite nanostructure has been considered as one of the most ideal approaches. Here, the carbon components not only buffer the volume expansion, but also improve the electron conductivity of the composite as a whole.12,13 Moreover, the one-dimensional nanostructure can provide a shortened diffusion length for Li+ along the radial direction and an efficient electron-transport pathway along the axial orientation.14 For example, 1D MoO3/CNT composites prepared by Ni et al. have shown very enhanced lithium-storage properties over MoO3 due to the structural advantages and inherent excellent electron conductivity provided by CNT.15 Also, other groups have demonstrated that 1D amorphous carbon can be used as a good buffering matrix to improve the structural stability of MoO3.16–20
So far, numerous effective strategies, such as the hydrothermal method and templating methods or electrospinning, have been devoted to designing 1D MoO3/C composites.21–23 Previous researches have shown that electrospinning technology can be considered as a simple and industry-viable method to generate porous carbon nanofibers24–26 and directly control the morphology of 1D MoO3/C for greatly improved lithium-storage properties. For instance, Feng et al. prepared amorphous MoO3/C nanofibers through the electrospinning method, and these 1D composite fibers showed high capacity of 710 mA h g−1 at a low current density of 40 mA g−1 after 100 cycles.27 Furthermore, Ma and co-workers employed the electrospinning technology to synthesize crystalline MoO3/C nanofibers, which showed better capacity at a high rate, corresponding to 623 mA h g−1 at 500 mA g−1 and 502 mA h g−1 at 1000 mA g−1.28 Despite great progress, the high-rate capability of 1D MoO3/C nanofibers is still limited. This is because the Li+ diffusion length from the fiber surface to the core is at least equal to its radius, which is still very long for effective insertion/diffusion of lithium in the electrode.29 Thus, it is necessary to further shorten the Li+ diffusion pathway and make full use of the electrode materials, especially highly embedded MoO3 particles, to achieve improved rate capability.
Herein, ultrafine MoO3 nanoparticles embedded in porous carbon nanofibers (denoted as MoO3/PCNFs) are prepared by co-electrospinning of polyacrylonitrile (PAN)/polymethylacrylate (PMMA)/Mo precursor, followed by controlled pyrolysis. PMMA was used here as a polymer template that can be removed from the PAN-derived carbon nanofiber matrix under high temperature, leading to the formation of many multi-channeled pores along the axial direction of the nanofibers. The pores are very beneficial for easy penetration of the Li+-carrying electrolyte into the fibrous electrode, thus improving the electrolyte/electrode contact area for fast Li+ flux and shortening the diffusion length. Moreover, the relationship between the component composition of MoO3/amorphous carbon and the electrochemical lithium-storage performance was comprehensively studied. The optimum MoO3/PCNF electrode exhibited very high capacity of 795.8 mA h g−1 at 200 mA g−1 and excellent high-rate capability (638.6 mA h g−1 at 2 A g−1), which are superior to those of MoO3/CNF and PCNF electrodes. The outstanding electrochemical performance is ascribed to the synergistic effects of ultrafine MoO3 nanoparticles with high capacity, 1D amorphous carbon buffering matrix and channel-like pores.
Experimental
Preparation of samples
One g of PAN (average Mw = 150k supplied by Aldrich) and 1 g PMMA (average Mw = 15k supplied by Aldrich) were dissolved in 15 mL N,N-dimethylformamide (DMF) at 70 °C for 12 h under magnetic stirring. Then, a certain amount of molybdenum acetylacetone (C10H14MoO6, 0, 0.25, 0.5, or 1 g) was added into the mixed solution and stirred for another 12 h to obtain the precursor solution for electrospinning. The mixture solution was transferred into a plastic syringe and was stretched by electrostatic force via the high-voltage device. The electrospinning fibers were collected by an Al-foil-enwrapped roller with a rotate speed of 800 rpm. The collector distance was kept at 15 cm, and the roller collector speed was 800 rpm. The obtained electrospinning webs were then stabilized at 220 °C for 3 h in air and subsequently carbonized at 750 °C for 2 hours in an Ar atmosphere to achieve the final products. The obtained products were denoted as 0.25-MoO3/PCNFs (0.25 g), 0.5-MoO3/PCNFs (0.5 g) and 0.75-MoO3/PCNFs (0.75 g). For comparison, MoO3/CNF composite fibers were prepared by the same procedure without the addition of PMMA. Pure PCNFs were obtained in the same way without using molybdenum acetylacetone (C10H14MoO6). In addition, pure CNFs were prepared according to the above procedure without C10H14MoO6 and PMMA.Characterizations
The morphologies and microstructures of these samples were investigated using a field emission scanning electron microscope (FE-SEM, Supra55, Carl Zeiss) and a high-resolution transmission electron microscope (HR-TEM, Tecnai G2 F30 S-TWIN). The crystallite size and composition of the materials were analyzed using X-ray diffraction (XRD), (WAXD, D8 Advance, Bruker, Cu Kα, λ = 0.154 nm). The Raman spectrum was recorded on a Raman spectrometer (Horiba) with an excitation laser wavelength of 632.81 nm. Thermogravimetric analysis (TGA) was conducted on a TGA instrument (TA-Q50, America) at a heating rate of 10 °C min−1 from 25 to 800 °C in air. An X-ray photoelectron spectrometer (XPS, EscaLab 250, Thermo Fisher Scientific) was used to analyze the elemental chemical status of the samples.Electrochemical measurements
The obtained samples were separately mixed with carbon black and poly-(vinylidene difluoride) (PVDF) to form a slurry with a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in N-methyl pyrrolidone (NMP). The working electrode was prepared by casting the slurry onto nickel foil through a doctor-blade method and was then dried in a vacuum oven at 120 °C overnight. The cells were assembled in an Ar-filled glovebox. Lithium metal foil was used as the counter electrode and the Celgard 2300 membrane was used as a separator. The electrolyte was 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v). The mass loading of the active material for each electrode was controlled to be about 1.0 mg cm−2. After assembly, cyclic voltammetry (CV) (0.005–3 V vs. Li+/Li) and electrochemical impedance spectroscopy (EIS) (from 100 kHz to 0.01 Hz) were performed by using an Autolab PGSTAT 302 N (Metrohm) workstation. The galvanostatic charge/discharge measurements were carried out between 0.005 and 3.0 V on Land CT2001A (China).
:1:1 in N-methyl pyrrolidone (NMP). The working electrode was prepared by casting the slurry onto nickel foil through a doctor-blade method and was then dried in a vacuum oven at 120 °C overnight. The cells were assembled in an Ar-filled glovebox. Lithium metal foil was used as the counter electrode and the Celgard 2300 membrane was used as a separator. The electrolyte was 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1 v/v). The mass loading of the active material for each electrode was controlled to be about 1.0 mg cm−2. After assembly, cyclic voltammetry (CV) (0.005–3 V vs. Li+/Li) and electrochemical impedance spectroscopy (EIS) (from 100 kHz to 0.01 Hz) were performed by using an Autolab PGSTAT 302 N (Metrohm) workstation. The galvanostatic charge/discharge measurements were carried out between 0.005 and 3.0 V on Land CT2001A (China).
Results and discussion
The synthetic process for the MoO3/PCNF samples is schematically presented in Fig. 1a. First, a nanofiber network was produced by the electrospinning method. After that, the fiber membrane was carbonized at 750 °C for 2 hours in an Ar atmosphere to obtain MoO3/PCNFs by removing PMMA. This electrospinning process was incorporated with a heat treatment process to form ultrafine MoO3 nanoparticles in a porous CNF matrix. During the decomposition of the PMMA precursor, numerous pores were formed within the CNF matrix.30,31 Here, as-designed PCNFs exhibited many channel-like and open pores with large diameters of 20–30 nm, and they could act as an ion-buffering reservoir, thus offering fast ion-transport pathways similar to those in the liquid electrolyte. Moreover, the carbon matrix with many pores could accommodate the volume change of highly active MoO3 during lithiation/delithiation processes.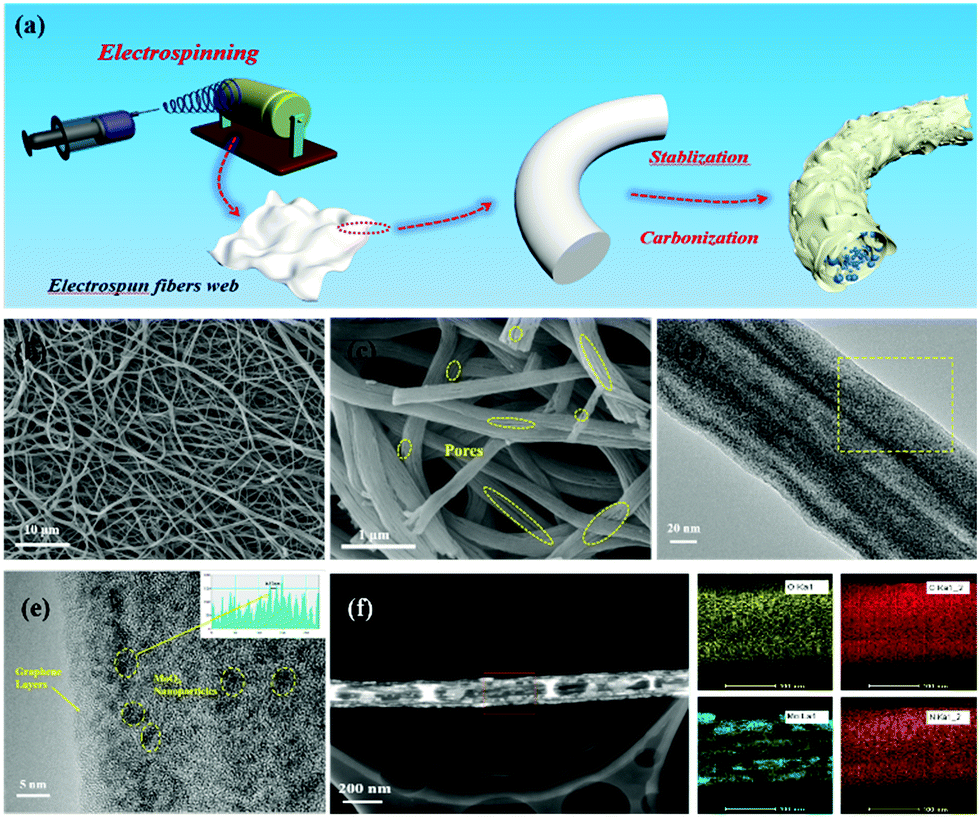 | ||
| Fig. 1 (a) Schematic illustration of the synthetic process of the MoO3/PCNF composite; (b and c) SEM images of 0.5-MoO3/PCNFs; (d and e) TEM image and HRTEM images of 0.5-MoO3/PCNFs; (f) STEM image of 0.5-MoO3/PCNFs and the corresponding elemental mapping images of oxygen, carbon, molybdenum and nitrogen. | ||
The morphologies and microstructures of 0.5-MoO3/PCNFs are shown in Fig. 1b–f. The SEM image of the sample (Fig. 1b) shows that all the nanofibers are bead-free and have a uniform diameter of about 200 nm. From the enlarged SEM image shown in Fig. 1c, it can be seen that there are numerous channel-like pores on the surface of PCNFs. Moreover, the inner porous structure was observed via SEM observation on the cross-section of a single nanofiber. These holes are formed due to the decomposition of PMMA during heat treatment, whereas it is clear that neat CNFs (Fig. S1a, ESI†) and MoO3/CNFs (Fig. S2a, ESI†) are both nonporous and completely solid. The TEM image of the 0.5-MoO3/PCNF sample shows many ultrafine crystalline nanoparticles with a diameter of about 5 nm, and the ultrafine nanoparticles are uniformly dispersed in the porous carbon nanofibers. The lattice spacing of the nanocrystals marked in Fig. 1e is measured to be 0.37 nm, which corresponds to the (101) plane of the orthogonal phase MoO3. Meanwhile, the elemental mapping images in Fig. 1f show that Mo is evenly distributed within the fiber and the results also clearly indicate the presence of pores. Fig. S3 (ESI†) exhibits HRTEM of MoO3/PCNFs with two different molybdenum contents. The 0.25-MoO3/PCNF sample is almost amorphous; it is clear that crystal particles are present in 0.75-MoO3/PCNFs and they unevenly agglomerate together.
Fig. 2a displays the XRD patterns for MoO3/PCNF samples with various MoO3 contents. A broad hump located at about 25° is related to amorphous carbon (JCPDS 13-0148). The significant broadening of XRD peaks confirms the nanocrystalline nature and ultrafine crystallite size of the products. The diffraction peaks can be exclusively indexed to crystalline MoO3 with the monoclinic phase (JCPDS no. 47-1320). The relative intensities of these MoO3 peaks do not show a strong preferential orientation, which may be due to ultrafine MoO3 crystals.32 The Raman spectrum clearly shows D- and G-bands of carbon at 1354 and 1598 cm−1, respectively (Fig. 2b), indicating the presence of disordered turbostratic carbon in the composite, corresponding to an amorphous structure.33I(D)/I(G) is the intensity ratio of the D-peak and the G-peak, which can be used as a parameter for determining the degree of graphitization. The values for 0.25-MoO3/PCNFs, 0.5-MoO3/PCNFs and 0.75-MoO3/PCNFs are 0.931, 0.920 and 0.889, respectively, indicating a relatively lower degree of graphitization. In addition, the Brunauer–Emmett–Teller (BET) surface area of 0.5-MoO3/PCNFs is measured to be 11.41 m2 g−1. The surface area and pore distribution (Fig. S4, ESI†) indicate that the synthesized 0.5-MoO3/PCNF sample possesses many micropores in addition to channel-type pores, which is very suitable for easy penetration of Li+-containing electrolyte into these composite nanofibers.34 Fig. S5a (ESI†) shows the TGA curves of PCNFs and 0.5-MoO3/PCNFs in an air atmosphere. As can be seen from the figure, PCNFs show a very clear weight loss of 30% at 270 °C due to the decomposition of PMMA; the clear weight loss between 400 °C and 500 °C may be due to the gradual decomposition of PAN, finally stabilizing at 670 °C with a residual amount of 2.5%. A rapid weight loss occurs in the temperature range of 330–520 °C in 0.5-MoO3/PCNFs due to PMMA and PAN decomposition, followed by a stable weight. The result in Fig. S5b (ESI†) clearly shows an increase in Mo content remaining in the system with the addition of molybdenum precursor.
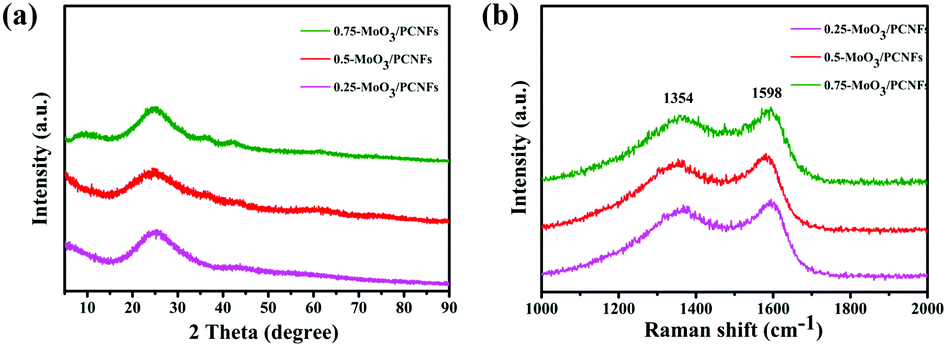 | ||
| Fig. 2 (a) XRD patterns and (b) Raman spectra of the MoO3/PCNF composite according to different precursor contents. | ||
Fig. 3a shows a typical measured XPS spectrum of 0.5-MoO3/PCNFs. The survey spectrum indicates the presence of Mo, C, N and O in the composites. From the high-resolution XPS spectrum of Mo 3d (Fig. 3b), two main peaks are observed at 232.7 eV and 235.8 eV in the spectrum, corresponding to Mo(VI) 3d5/2 and Mo(VI) 3d3/2, indicating the existence of molybdenum oxide.35 In addition, a weak peak at 228.3 eV indicates the presence of a small amount of MoO2.32Fig. 3c shows the high-resolution XPS spectrum of carbon. The C–C bond of the carbon skeleton is observed as the strongest peak at 284.5 eV, whereas those at 286.1 eV and 288.2 eV correspond to the C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups.36 The O 1s spectra from Fig. 3d show three separate peaks (530.7 eV, 532.6 eV, 533.2 eV) belonging to Mo–O bond, CO bond, and C–O bond, respectively.37
O functional groups.36 The O 1s spectra from Fig. 3d show three separate peaks (530.7 eV, 532.6 eV, 533.2 eV) belonging to Mo–O bond, CO bond, and C–O bond, respectively.37
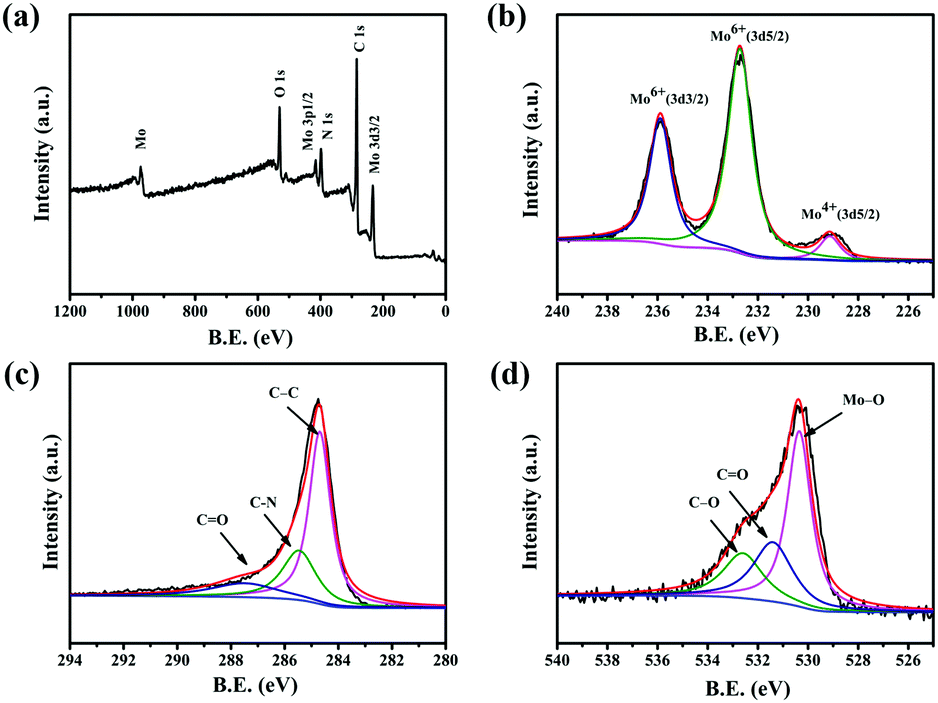 | ||
| Fig. 3 XPS spectra for the 0.5-MoO3/PCNF composite: (a) survey XPS spectrum and high-resolution XPS of (b) Mo 3d; (c) C 1s; and (d) O 1s. | ||
The first three CV curves of 0.5-MoO3/PCNFs are shown in Fig. 4a. The peak visible at 0.75 V corresponds to the formation of the solid electrolyte interface (SEI); it disappears in the subsequent cycles and therefore causes partially observed irreversible capacity during the first cycle.38 According to previous reports, lithium storage within MoO3 is a two-phase process that can be described by following equations:
| MoO3 + xLi+ + xe− → LixMoO3 | (1) |
| LixMoO3 + (6 − x)Li+ + (6 − x)e− → Mo + 3Li2O | (2) |
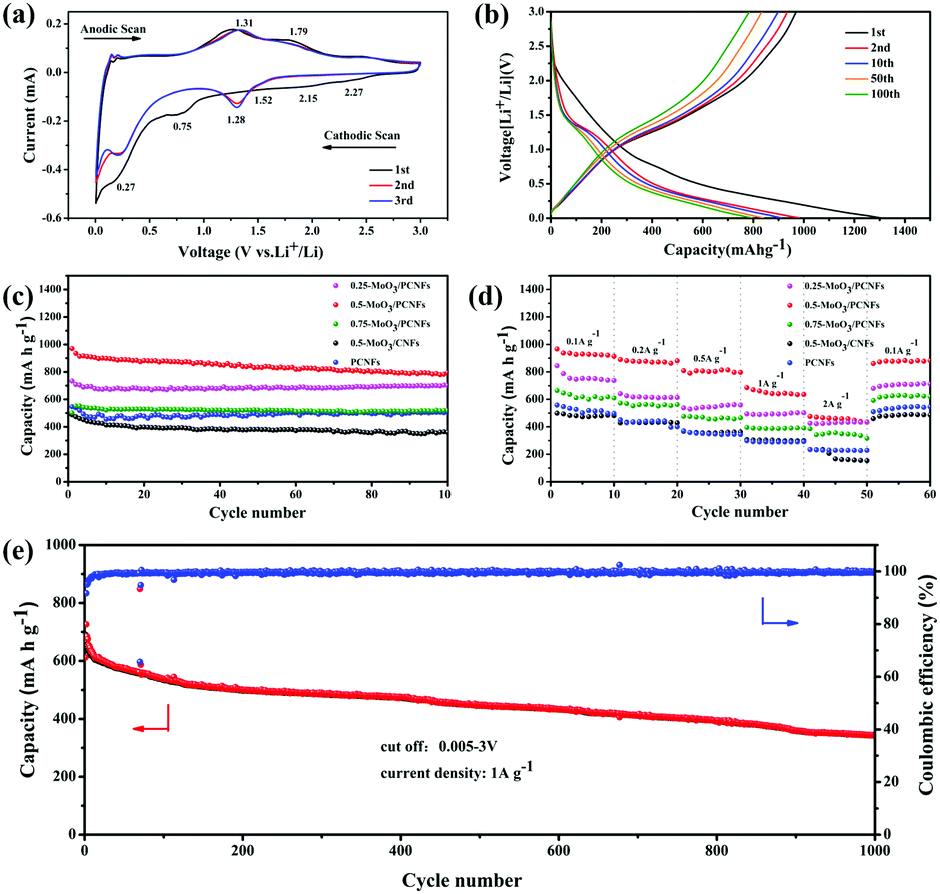 | ||
| Fig. 4 (a) CV curves of the 0.5-MoO3/PCNF electrode and (b) charge/discharge profiles of the 0.5-MoO3/PCNF electrode; (c) cyclic performance at a current density of 0.2 A g−1 and (d) rate performance of MoO3/PCNF composite according to different precursor contents; (e) long cycling performance and coulombic efficiency of 0.5-MoO3/PCNF at a current density of 1 A g−1. | ||
First, two small reduction peaks around 2.27 and 2.15 V only appearing in the first cycle correspond to the multi-step intercalation of Li+ into MoO3 to form a solid solution LixMoO3, as shown in eqn (1).12 During first lithiation, irreversible structural changes from ultrafine MoO3 nanocrystalline materials to non-stoichiometric amorphous LixMoO3 occur in MoO3/PCNFs. The following peak at 0.27 V is ascribed to the reaction of lithium with as-formed LixMoO3, corresponding to eqn (2).8 The peaks of different reversible oxidation/reduction pairs are observed at 1.29/1.31 and 1.52/1.79 V, which can be due to the insertion/extraction of lithium ions from different sites and the phase transition process. Moreover, the third circle and the second circle are highly coincident, indicating that 0.5-MoO3/PCNFs have excellent lithium uptake/release reversibility. Besides, the small polarization between the oxidation peak and the reduction peak reflects rapid Li+ insertion/extraction kinetics, corresponding to excellent rate performance.
The charge–discharge curves of the 0.5-MoO3/PCNF electrode are shown in Fig. 4b. At a current density of 200 mA g−1, the initial discharge capacity was 1304.1 mA h g−1 with good initial coulomb efficiency of 74.4%. As mentioned above, the initial irreversible capacity loss was assigned to the formation of the SEI layer. Furthermore, after the first cycle, the charge curve remained almost changeless, indicating high reversibility of the lithiation/delithiation process and good stability of the electrode structure.
Fig. 4c presents a comparison of cycle performances of different samples. After 100 cycles, the reversible capacity of the 0.5-MoO3/PCNF electrode can still be maintained at 795.8 mA h g−1 at a current density of 200 mA g−1. Compared to the neat PCNF (504.3 mA h g−1) electrode, the presence of ultrafine high-capacity MoO3 nanoparticles in the PCNF matrix increases the capacity by about 37%. On the other hand, the 0.25-MoO3/PCNF electrode delivers an inferior discharge capacity of 700 mA h g−1, which is due to low amount of high-capacity MoO3 in the nanocomposite electrode. Among the MoO3/PCNF electrodes, the 0.75-MoO3/PCNF electrode shows the lowest capacity of only 519.3 mA h g−1 after 100 cycles. This is because the addition of a high amount of Mo precursor can easily lead to the formation of agglomerated clusters of MoO3 nanoparticles in the PCNF matrix, which results in an inferior electron-transport pathway and long Li+-diffusion distance.39 On the whole, all the MoO3/PCNF electrodes show very improved reversible capacity than the MoO3/CNF electrode. For example, the 0.5-MoO3/CNF electrode only delivers capacity of 360.3 mA h g−1 after 100 cycles. These results have demonstrated that the as-designed porous structure can contribute to the realization of an improved electrolyte/electrode contacting area, thus making full use of the dispersed high-capacity MoO3 nanoparticles. As the current density increases, all the electrodes exhibit a gradual decay in ability (Fig. 4d). The 0.5-MoO3/PCNF electrode exhibits the best rate performance, with initial discharge capacities of 1071.7, 925, 888.1, 802.3, and 638.6 mA h g−1 at current densities of 0.1, 0.2, 0.5, 1, and 2 A g−1, respectively. When the current density is restored to 100 mA g−1, the capacity is restored to 896.3 mA h g−1, indicating that the 0.5-MoO3/PCNF electrode has excellent electrochemical reversibility and structural integrity. In addition, the capacity of the 0.5-MoO3/PCNF electrode is gradually stabilized at 345 mA h g−1 after 1000 cycles at the current density of 1 A g−1 (Fig. 4e). Next, the long cyclic performances of 0.5-MoO3/PCNFs and 0.25-MoO3/PCNFs are compared in Fig. S8 (ESI†). Although the cycling stability of 0.5-MoO3/PCNFs is slightly worse than that of 0.25-MoO3/PCNFs, the final capacity is twice as much as that of 0.25-MoO3/PCNFs. The excellent high rate performance of 0.5-MoO3/PCNFs is highly correlated with the channel-like porous nanostructures to shorten the Li+-diffusion length and carbon matrix to guarantee good electronic conductivity. Consequently, the structure can be maintained during the cycle. Due to the above synergistic effect, the 0.5-MoO3/PCNF electrode combines high capacity and high rate of long-term cycle life as an anode material for LIBs. Table 1 shows comparison of the electrochemical performances of our electrodes and other as-reported MoO3/C composites. The capacity of the 0.5-MoO3/PCNF electrode synthesized in the current study is the highest, indicating that the electrode material has great potential as an anode for use in high-performance LIBs.
| Material | Preparation method | Current density (mA g−1) | Cycle number | Specific capacity (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| CNT-wired oxygen-deficient MoO3 nanobelt | Hydrothermal | 200 | 100 | 421 | 22 |
| MoO3/C nanobelts | Hydrothermal | 100 | 50 | 1000 | 12 |
| MoO3@C nanofibers | Electrospinning | 500 | 100 | 623 | 16 |
| MoO3 nanoparticles in C matrix | Ball milling mixture | 149 | 120 | 780 | 40 |
| C-MoO3 NR | Grinding & heating | 250 | 180 | 639 | 41 |
| MoO3/G | Hydrothermal | 50 | 80 | 869 | 42 |
| MoO3/C nanocomposite | One-pot citric-nitrate method | 100 | 100 | 500 | 8 |
| 0.5-MoO 3 /PCNFs | Electrospinning | 200 | 100 | 795.8 | Our work |
To explain the high-rate performance, we further analyzed the kinetics of the 0.5-MoO3/PCNF electrodes (Fig. 5a) to separate the diffusion-controlled capacity and capacitive capacity. CV was tested at different scan rates from 0.2 to 1 mV s−1. As we know, the measured current (i) and scan rate (ν) obey the following power-law relationship:43
| i = aνb, | (3) |
| i (V) = k1ν + k2ν1/2, | (4) |
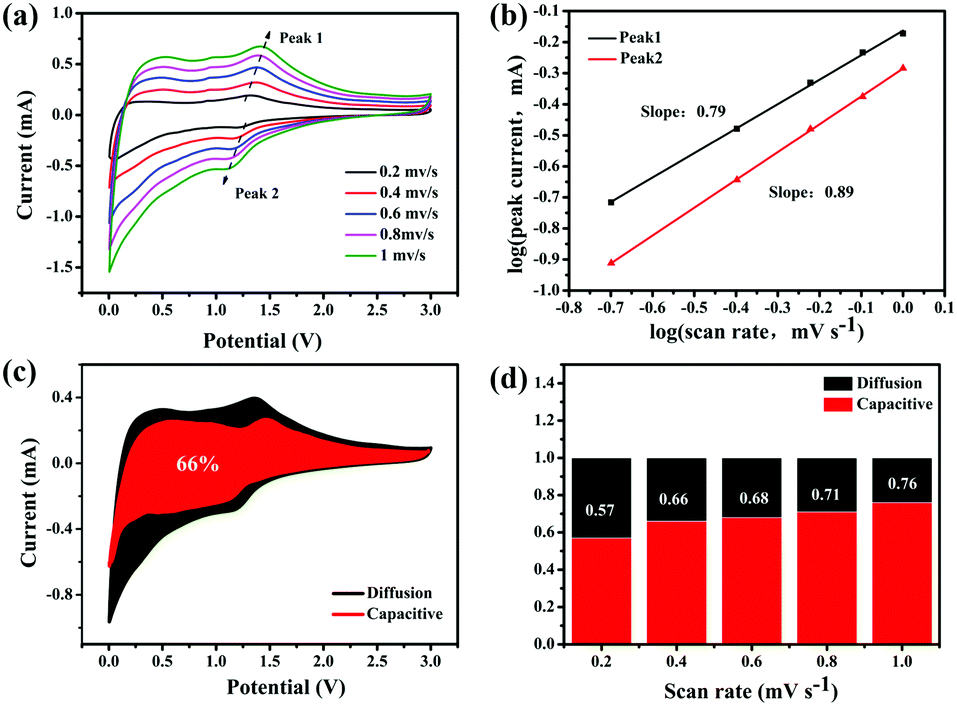 | ||
| Fig. 5 Electrochemical kinetic analysis. (a) CV curves at different scan rates from 0.2 to 1.0 mV s−1, (b) corresponding log(i) versus log(v) plots at specific peak currents, (c) CV curve with the pseudocapacitive fraction shown by the shaded area at a scan rate of 0.4 mV s−1, and (d) bar chart showing the percent of pseudocapacitive contribution at different scan rates. | ||
The galvanostatic intermittence titration technique (GITT) is a reliable technique for determining the chemical diffusion of lithium (DLi+), which is used in the cycling of the two kinds of electrodes. Fig. 6a (details in the Supporting Information) depicts the GITT curve of the first discharge process, corresponding to the intercalation reaction of LixMoO3 formation by MoO3 during the first lithiation process and the subsequent redox reaction.45 Basically, the initial polarization obtained from the difference between the closed-circuit voltage and the open-circuit voltage (inset of Fig. 6a) can determine the enhanced kinetics of the porous structure, shortening the Li+-diffusion distance, which results in an increase in its discharge capacity.46Fig. 6b exhibits the DLi+ values of the two electrodes by discharging from 3 V to 0.001 V. The DLi+ values of the two electrodes are on the same order of magnitude around a similar voltage range, and they are on the order of 10−6 cm2 s−1. In the initial higher voltage range, Li+ diffuses very quickly, indicating that Li+ diffusion is better during the phase transfer of lithium intercalation. The higher Li+ diffusion coefficient of 0.5-MoO3/PCNFs corresponds well with the benefits of ultrafine nanocrystalline domains, which shortens the solid-phase diffusion length, making the size effect of the shorter diffusion path more dominant.47 Furthermore, in the high voltage range of 2 V–3 V, Li+ diffuses rapidly, whereas the value goes down quickly as the Li content in the samples increases. From 2 V to 0.001 V, 0.5-MoO3/PCNFs show slower reduction in diffusion rate than 0.5-MoO3/CNFs.48,49 This can be because the 1D nature of PCNF can be beneficial for decreasing ion transport resistance.31 Besides, the Li+ diffusion coefficient in 0.5-MoO3/PCNFs fluctuates less and is generally higher than that of non-porous 0.5-MoO3/CNFs. This indicates that the porous structure of carbon fibers can provide a way for electrolyte transport, shortening the diffusion path of Li+ and speeding up Li+ diffusion.
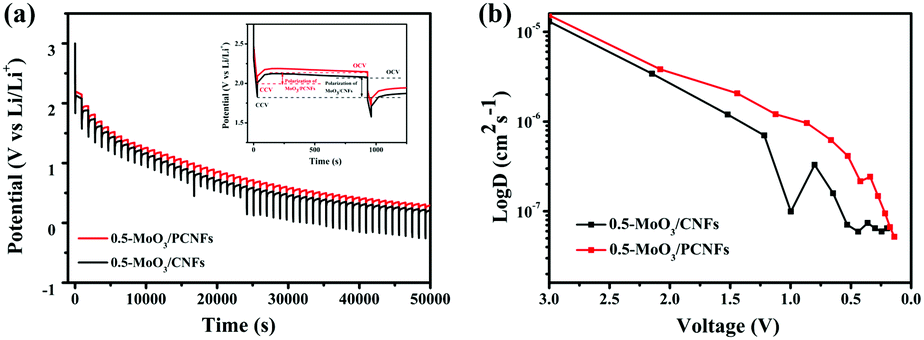 | ||
| Fig. 6 (a) Galvanostatic intermittent titration curves of 0.5-MoO3/PCNFs and 0.5-MoO3/CNFs and (b) variation of DLi+ determined by GITT during discharge cycle for 0.5-MoO3/PCNFs and 0.5-MoO3/CNFs, respectively. | ||
To further determine the reason why the 0.5-MoO3/PCNF electrode exhibits highly improved electrochemical properties, we compared the LIB charge transport kinetics of 0.5-MoO3/PCNFs and 0.5-MoO3/CNFs after 20 cycles using electrochemical impedance spectroscopy (EIS). Fig. 7 shows their corresponding Nyquist plots; each contains two distinct sections: a semicircle in the high and medium frequency ranges and a sloped line in the low frequency range. The semicircle at high frequencies indicates the resistance of Li+ through the interphase (Rf) between the electrode and the electrolyte, whereas the semicircle at the medium frequency reflects the charge transfer reaction (Rct); the oblique line reflects the diffusion of Li+ to the electrode and the impedance of Warburg.50 The intercept at the high frequency of the Z real axis (Z′) corresponds to the internal impedance (Rs) of the electrolyte. The diameter of the semicircle is related to the charge transfer resistance (Rct) from the electrolyte to the electrode, whereas the slope line indicates the Li+ diffusion process. Although the Rs values of the two electrodes are almost similar, the Rct value of 0.5-MoO3/PCNFs is much smaller than that of 0.5-MoO3/CNFs, which shows that the charge transfer process between the 0.5-MoO3/PCNF electrode and the electrolyte is faster during the reaction. It is further proved that the porous structure can provide a larger contact area for the electrode material and the electrolyte, accelerate the transmission speed of electrons and lithium ions, and provide double protection for the volume change of MoO3 during the electrochemical reaction. Most importantly, the electrical conductivity of the entire electrode material is improved during the electrochemical process.
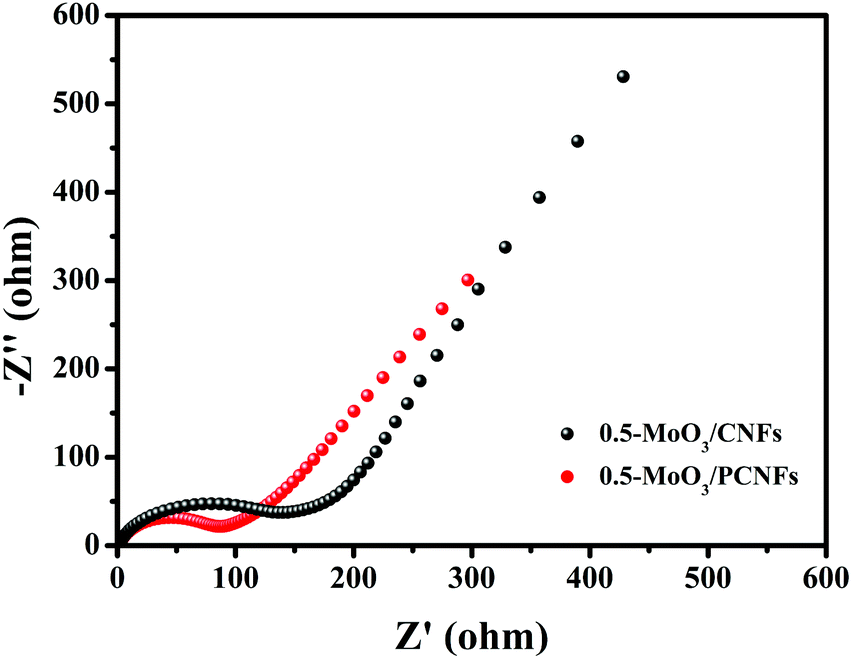 | ||
| Fig. 7 Electrochemical impedance spectra of 0.5-MoO3/PCNFs and 0.5-MoO3/CNFs after 20 cycles in a frequency range of 0.01 Hz–100 kHz and the equivalent electrical circuit (inset). | ||
Conclusions
In this study, we synthesized porous carbon nanofibers containing uniformly dispersed ultrafine MoO3 nanoparticles by a simple one-step electrospinning process. The porous structure not only buffers the mechanical stress caused by lithiation/delithiation of the electrode, but also prevents the aggregation of MoO3 nanoparticles. Moreover, the interconnected pores can facilitate diffusion and transfer of electrons and lithium ions, effectively reducing the transfer path. The ultrafine MoO3 nanoparticles uniformly dispersed in the carbon fiber have very small size (<3 nm), which promotes fast diffusion of the electrolyte. While a certain degree of N-doping enhances the conductivity and increases the reversible capacity of the carbon matrix, synergizing with ultrafine MoO3 nanoparticles can provide additional capacity and buffer volume changes during cycling. In addition, the behavior of the pseudocapacitor and the higher lithium diffusion coefficient exhibited in the 0.5-MoO3/PCNF electrode indicate the rapid electrochemical kinetics and high cycle capacity of the electrode. The 0.5-MoO3/PCNF electrode is stable at 795.8 mA h g−1 after 100 cycles at a current density of 200 mA g−1, thus demonstrating that the energy storage performance of MoO3/PCNFs is significantly improved by introducing a porous structure; therefore, it is an alternative highly viable material that can replace current commercial anodes in the next generation of LIBs.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51772016, 51272021, 51402010 and 51072013) and the Fundamental Research Funds for the Central Universities (No. XK1802-2).References
- J. Y. Li, Q. Xu, G. Li, Y. X. Yin, L. J. Wan and Y. G. Guo, Mater. Chem. Front., 2017, 1, 1691–1708 RSC.
- P. P. R. M. L. Harks, F. M. Mulder and P. H. L. Notten, J. Power Sources, 2015, 288, 92–105 CrossRef CAS.
- D. Zhou, X. Li, L. Z. Fan and Y. Deng, Electrochim. Acta, 2017, 230, 212–221 CrossRef CAS.
- Z. D. Zhou, W. L. Song, X. Li and L. Z. Fan, Electrochim. Acta, 2016, 207, 9–15 CrossRef.
- L. M. Zheng, Y. Liu, J. L. Lan, Y. H. Yu and X. P. Yang, Chem. Eng. J., 2017, 330, 1289–1296 CrossRef CAS.
- Y. Q. Jin, H. C. Yuan, J. L. Lan, Y. H. Yu, Y. H. Lin and X. P. Yang, Nanoscale, 2017, 9, 13298–13304 RSC.
- D. Zhou, W. L. Song, X. Li, L. Z. Fan and Y. Deng, J. Alloys Compd., 2016, 699, 730–737 CrossRef.
- Q. Xia, H. Zhao, Z. Du, J. Wang and T. Zhang, J. Power Sources, 2013, 226, 107–111 CrossRef CAS.
- Y. Guo, L. M. Zheng, J. L. Lan, Y. H. Yu and X. P. Yang, Electrochim. Acta, 2018, 269, 624–631 CrossRef CAS.
- H. C. Yuan, Y. Q. Jin, J. L. Lan, Y. Liu, Y. H. Yu and X. P. Yang, Inorg. Chem. Front., 2018, 5, 932–938 RSC.
- Y. Liu, X. Yan, Y. Yu and X. Yang, J. Mater. Chem. A, 2015, 3, 20880–20885 RSC.
- Q. Xia, H. Zhao, Z. Du, Z. Zeng and C. Gao, Electrochim. Acta, 2015, 180, 947–956 CrossRef CAS.
- Y. Liu, X. Yan, Y. Yu and X. Yang, ACS Sustainable Chem. Eng., 2016, 4, 2951–2959 CrossRef CAS.
- Q. Xia, P. Wang and Q. Tan, Mater. Lett., 2018, 215, 221–224 CrossRef CAS.
- L. Zhou, L. Yang, P. Yuan, J. Zou and Y. Wu, J. Phys. Chem. C, 2010, 114, 21868–21872 CrossRef CAS.
- G. Wang, J. Ni, H. Wang and L. Gao, J. Mater. Chem. A, 2013, 1, 4112–4118 RSC.
- X. Li, J. Xu, L. Mei, Z. Zhang and C. Cui, J. Mater. Chem. A, 2014, 3, 3257–3260 RSC.
- Y. Dong, S. Li, H. Xu, M. Yan and X. Xu, Phys. Chem. Chem. Phys., 2013, 15, 17165–17170 RSC.
- M. F. Hassan, Z. P. Guo, Z. Chen and H. K. Liu, J. Power Sources, 2010, 195, 2372–2376 CrossRef CAS.
- L. C. Yang, W. L. Guo, Y. Shi and Y. P. Wu, J. Alloys Compd., 2010, 501, 218–220 CrossRef CAS.
- M. Xu, J. Tang, H. Wu and G. Zheng, RSC Adv., 2014, 4, 29586–29590 RSC.
- X. Xiao, Z. Peng, C. Chen, C. Zhang and M. Beidaghi, Nano Energy, 2014, 9, 355–363 CrossRef CAS.
- J. Ni, G. Wang, J. Yang, D. Gao and J. Chen, J. Power Sources, 2014, 247, 90–94 CrossRef CAS.
- G. Xia, Q. Gao, D. Sun and X. Yu, Small, 2017, 13, 1701561 CrossRef PubMed.
- G. Xia, L. Zhang, F. Fang, D. Sun, Z. Guo, H. Liu and X. Yu, Adv. Funct. Mater., 2016, 26, 6188–6196 CrossRef CAS.
- L. Zhang, G. Xia, Y. Huang, C. Wei, Y. Yu, D. Sun and X. Yu, Energy Storage Mater., 2018, 10, 160–167 CrossRef.
- D. Yan, X. Luo, H. Zhang, G. Zhu, L. Chen, G. C. Chen, H. R. Xu and A. B. Yue, J. Alloys Compd., 2016, 688, 481–486 CrossRef CAS.
- C. Q. Feng, H. Gao, C. F. Zhang, Z. P. Guo and H. K. Liu, Electrochim. Acta, 2013, 93, 101–106 CrossRef CAS.
- H. He, L. Shi, Y. Fang, X. Li and Q. Song, Small, 2014, 10, 4671–4676 CrossRef CAS PubMed.
- S. Abouali, M. A. Garakani, B. Zhang, H. Luo, Z. L. Xu, J. Q. Huang, J. Huang and J. K. Kim, J. Mater. Chem. A, 2014, 2, 16939–16944 RSC.
- H. He, L. Shi, Y. Fang, X. Li, Q. Song and L. Zhi, Small, 2014, 10, 4671–4676 CrossRef CAS PubMed.
- Y. Sun, X. Hu, W. Luo and Y. Huang, J. Mater. Chem., 2011, 22, 25–431 Search PubMed.
- L. Yang, X. Li, S. He, G. Du, X. Yu, J. W. Liu, Q. S. Gao, R. Z. Hua and M. Zhu, J. Mater. Chem. A, 2016, 4, 10842–10849 RSC.
- Y. Liu, X. Yan, J. Lan, Y. Yu, X. Yang and Y. Lin, Mater. Chem. Front., 2017, 1, 1331–1337 RSC.
- L. C. Yang, W. Sun, Z. W. Zhong, J. W. Liu, Q. S. Gao, R. Z. Hu and M. Zhu, J. Power Sources, 2016, 306, 78–84 CrossRef CAS.
- H. Li, H. Ye, Z. Xu, C. Wang, J. Yin and H. Zhu, Phys. Chem. Chem. Phys., 2016, 19, 2908–2914 RSC.
- Y. Sun, X. Hu, W. Luo and Y. Huang, J. Mater. Chem., 2011, 22, 425–431 RSC.
- F. Xia, X. Hu, Y. Sun, W. Luo and Y. Huang, Nanoscale, 2012, 4, 4707–4711 RSC.
- Y. Sun, J. Wang, B. Zhao, R. Cai, R. Ran and Z. P. Shao, J. Mater. Chem. A, 2013, 1, 4736–4746 RSC.
- M. Karevan, R. V. Pucha, M. A. Bhuiyan and K. Kalaitzidou, Carbon Lett., 2010, 11, 325–331 CrossRef.
- T. Tao, A. M. Glushenkov, C. Zhang, H. Zhang, D. Zhou, Z. P. Guo, H. K. Liu, Q. Y. Chen, H. P. Hu and Y. Chen, J. Mater. Chem., 2011, 21, 9350–9355 RSC.
- J. Ding, S. A. Abbas, C. Hanmandlu, L. Lin, C. S. Lai, P. C. Wang, L. J. Li, C. W. Chu and C. C. Chang, J. Power Sources, 2017, 348, 270–280 CrossRef CAS.
- C. L. Liu, Y. Wang, C. Zhang, X. S. Li and W. S. Dong, Mater. Chem. Phys., 2014, 143, 1111–1118 CrossRef CAS.
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo and J. Hjelm, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- Y. Wu, M. V. Reddy, B. V. R. Chowdari and S. Ramakrishna, ACS Appl. Mater. Interfaces, 2013, 5, 12175–12184 CrossRef CAS PubMed.
- R. J. Mi, Y. S. Jung and Y. M. Kang, Nanoscale, 2012, 4, 6870–6875 RSC.
- G. Fang, Z. Wu, J. Zhou, C. Zhu, X. Cao, T. Q. Lin, Y. M. Chen, C. Wang, A. Q. Pan and S. Q. Liang, Adv. Energy Mater., 2018, 1703155 CrossRef.
- B. Markovsky, M. D. Levi and D. Aurbach, Electrochim. Acta, 1998, 43, 2287–2304 CrossRef CAS.
- J. Huang, J. Yan, J. Li, L. Cao, Z. Xu, J. P. Wu, L. Zhou and Y. J. Luo, J. Alloys Compd., 2016, 688, 588–595 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00497h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |