
Pairing 1D/2D-conjugation donors/acceptors towards high-performance organic solar cells†
Jiayu
Wang‡
a,
Yiqun
Xiao‡
b,
Wei
Wang
a,
Cenqi
Yan
a,
Jeromy
Rech
 c,
Mingyu
Zhang
a,
Wei
You
c,
Xinhui
Lu
*b and
Xiaowei
Zhan
*a
c,
Mingyu
Zhang
a,
Wei
You
c,
Xinhui
Lu
*b and
Xiaowei
Zhan
*a
aDepartment of Materials Science and Engineering, College of Engineering, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Peking University, Beijing 100871, China. E-mail: xwzhan@pku.edu.cn
bDepartment of Physics, Chinese University of Hong Kong, New Territories 999077, Hong Kong, China. E-mail: xhlu@phy.cuhk.edu.hk
cUniversity of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA
First published on 11th December 2018
Abstract
Two polymer donors, FTAZ and J71, and two fused-ring electron acceptors, ITIC1 and ITIC2, are used to investigate the effects of conjugation dimension on the performance of organic solar cells (OSCs). FTAZ and J71, and ITIC1 and ITIC2 share the same molecular backbone, respectively, while J71 and ITIC2 possess conjugated thienyl side chains. The addition of conjugated side chains slightly red-shifts the absorption spectra and lowers the bandgap due to the extended 2D conjugation. Conjugated side chains on the acceptor induce the self-aggregation of the acceptors, while conjugated side chains on the donor increase the miscibility of the donors and acceptors, thus optimizing the morphology of the active layers. The blends based on mixed combinations, namely 1D donor/2D acceptor and 2D donor/1D acceptor, show better performance relative to 1D donor/1D acceptor and 2D donor/2D acceptor.
Introduction
Organic solar cells (OSCs) are regarded as a promising alternative to silicon-based photovoltaic technology due to some advantages, such as low cost, light weight, flexibility, semi-transparency, large-area fabrication, and short energy payback times.1–3 A bulk heterojunction (BHJ) is the most widely used architecture of the active layer in OSCs, which consists of a blend of electron donor and acceptor materials.4,5 To achieve high power conversion efficiency (PCE), various donors and acceptors have been developed via various molecular design strategies, among which two dimensional (2D) conjugated (also called side-chain conjugated) materials exhibit good performance and attract considerable attention. Compared to the nonconjugated counterparts, 2D conjugated side chains can extend intramolecular conjugation, which enhances light absorption and photocurrent. Moreover, 2D conjugation facilitates intermolecular interaction and π–π overlap, thus promoting charge transport.6,7 During the past decade, a variety of 2D conjugated donors have been developed, which yielded PCEs over 10% in fullerene-based OSCs.8–10 As for acceptors, Zhan and co-workers introduced the concept of 2D conjugation into fused-ring electron acceptors (FREAs),11 and a high PCE of 13% was achieved.12Recently, nonfullerene acceptors,13–16 especially FREAs,17,18 have attracted much attention and attained high PCEs,19 but to continue achieving high device performance, it is crucial to keep a balanced partnership between donor and acceptor components in terms of absorption, energy level, mobility, miscibility, and morphology.20–24 Considering the conjugation dimension of the donor and acceptor, there are four combinations: 1D donor/1D acceptor,25–32 1D donor/2D acceptor,11 2D donor/1D acceptor,33–53 and 2D donor/2D acceptor.12,54 Although various pairs of 1D/2D donor/acceptor have been studied individually, systematic comparisons of 1D/2D donor/acceptor pairs have rarely been reported. These 1D/2D acceptors usually contain different cores and end-groups, and they are often paired with different donor materials to fabricate OSCs, which exhibit different performance, thus leaving the 1D/2D conjugation effect ambiguous. The approach outlined herein aims to properly understand how the 1D/2D conjugation of donor/acceptor would affect device performance and what 1D/2D donor/acceptor combination would be better to achieve higher performance.
In this work, we report the first example of mapping 1D/2D donor/acceptor combinations; specifically we choose two polymer donors, FTAZ55 and J71,35 and two FREAs, ITIC111 and ITIC211 (Scheme 1), to systematically compare 1D/2D donor/acceptor pairs. These materials are good candidates for this study as they have each demonstrated high performance, thus allowing the differences associated within to be attributed to the 1D/2D conjugation effect. The donor polymers, FTAZ and J71, possess the same molecular backbone, but different side chains on the benzodithiophene units (nonconjugated 3-butylnonyl and conjugated thienyl, respectively). Similarly, the conjugated backbones of acceptors ITIC1 and ITIC2 are the same, and the substituents on the central phenyl varied (hydrogen and conjugated thienyl, respectively). Because of the different molecular structures, FTAZ and ITIC1 are characterized by 1D conjugation, while J71 and ITIC2 have 2D conjugation. With this small library, we are able to systematically probe the effects of the conjugated side-chains on electronic, optical, charge-transport, morphological and photovoltaic properties of the donors and acceptors. This will allow for a better understanding of the effect of 1D/2D conjugation and help provide recommendations for pairing new materials in the future. The mixed combinations, 1D/2D (FTAZ:ITIC2) and 2D/1D (J71:ITIC1), show better performance relative to 1D/1D (FTAZ:ITIC1) and 2D/2D (J71:ITIC2).
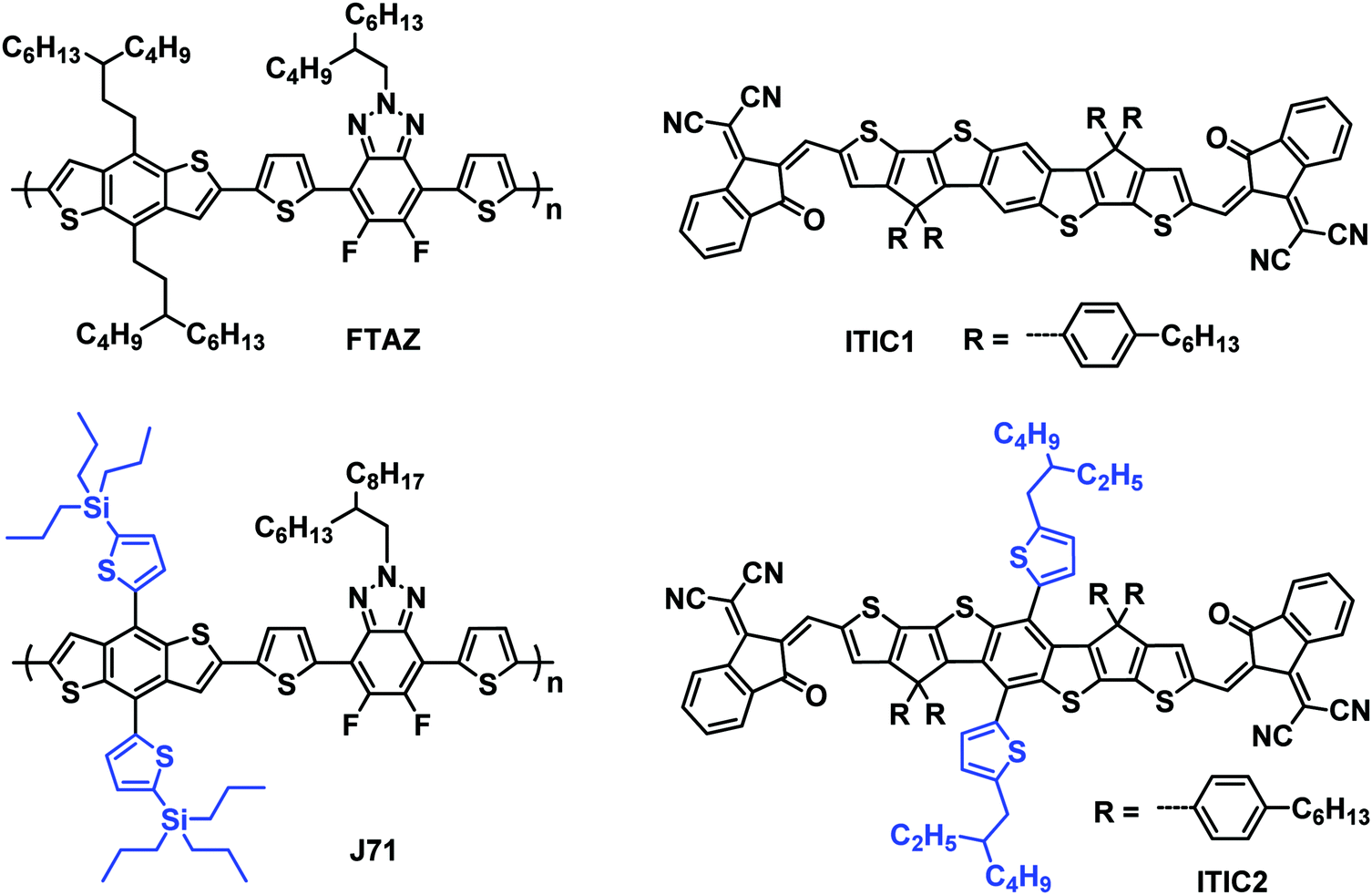 | ||
| Scheme 1 Chemical structures of FTAZ, J71, ITIC1, and ITIC2. | ||
Results and discussion
Optical and electronic properties
The optical impact of 1D vs. 2D conjugation was first explored with UV-vis absorption. The normalized optical absorption spectra of FTAZ, J71, ITIC1, and ITIC2 in chloroform and in thin films are shown in Fig. S1a (ESI†) and Fig. 1a, respectively. In solution, the 1D conjugated FTAZ shows two peaks at 534 and 572 nm, while 2D conjugated J71 shows a slightly red-shifted spectrum with two peaks at 536 and 578 nm. The absorption maximum of 1D conjugated acceptor ITIC1 locates at 702 nm, while that of 2D conjugated ITIC2 red-shifts to 714 nm. Relative to those in solution, the absorption spectra of FTAZ and J71 in films do not differ much, while those of ITIC1 and ITIC2 red-shift. The absorption maxima of 1D conjugated FTAZ and ITIC1 are 533 and 734 nm, respectively; while those of 2D conjugated J71 and ITIC2 slightly red-shift to 540 and 738 nm, respectively. The increase in absorption maxima in the 2D conjugated systems is attributed to the extended intramolecular conjugation from the side chains. The optical bandgaps of FTAZ, J71, ITIC1, and ITIC2 are estimated from the absorption edge of the thin film: 2.00, 1.96, 1.55, and 1.53 eV, respectively. This information is also found in Table 1. The wide-bandgap polymer donors and low-bandgap acceptors exhibit complementary absorption, which is beneficial for harvesting panchromatic light and improving short-circuit current density (JSC).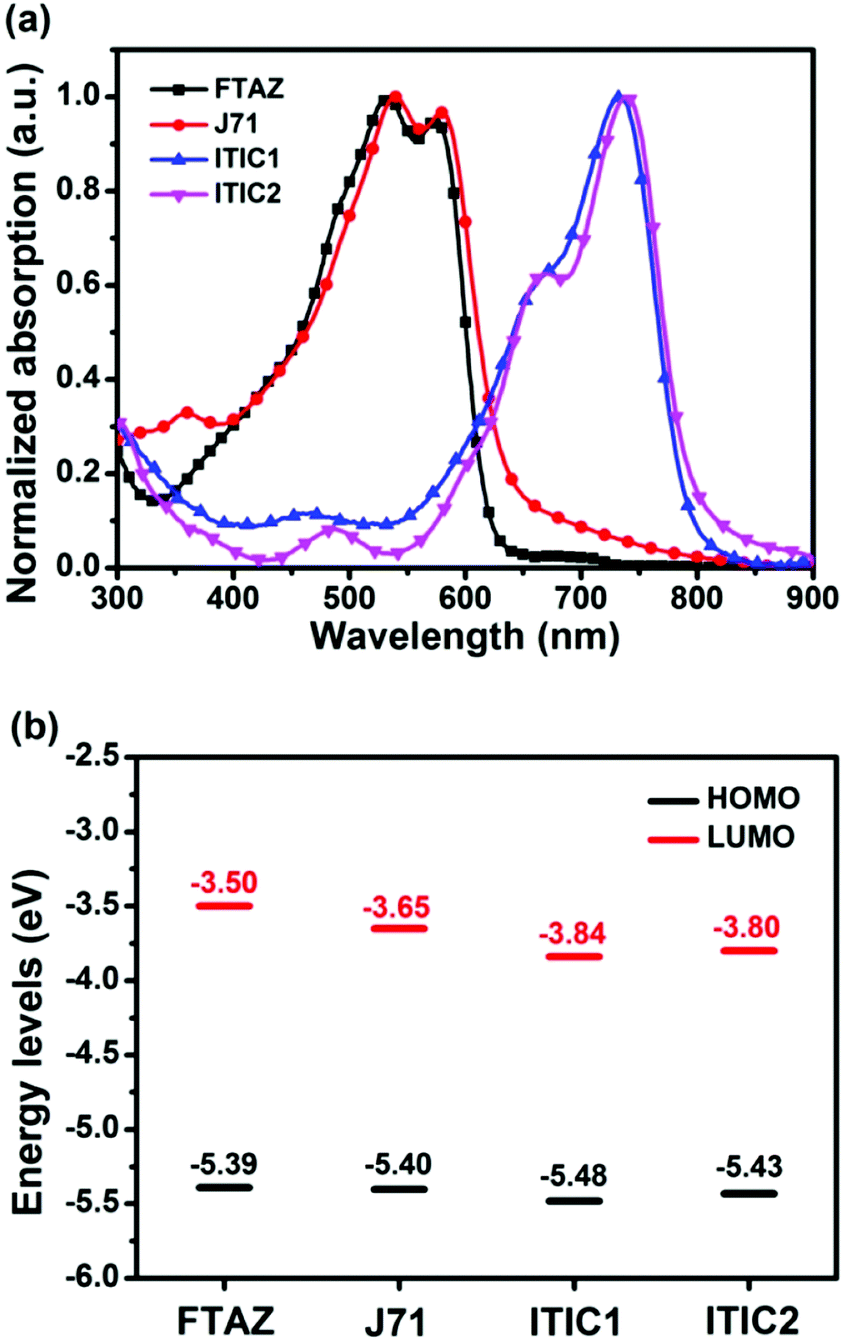 | ||
| Fig. 1 (a) Thin film UV-vis absorption spectra and (b) energy level diagram from CV of FTAZ, J71, ITIC1, and ITIC2. | ||
| Material | λ abssolution (nm) | λ absfilm (nm) | E g (eV) | HOMO (eV) | LUMO (eV) | μ (10−3 cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| a Estimated from the absorption edge in film. | ||||||
| FTAZ | 534, 572 | 533, 574 | 2.00 | −5.39 | −3.50 | 4.4 |
| J71 | 536, 578 | 540, 580 | 1.96 | −5.40 | −3.65 | 3.6 |
| ITIC1 | 702 | 734 | 1.55 | −5.48 | −3.84 | 1.0 |
| ITIC2 | 714 | 738 | 1.53 | −5.43 | −3.80 | 1.3 |
Next, the electrochemical properties of FTAZ, J71, ITIC1, and ITIC2 were investigated by a cyclic voltammetry (CV) method with films on a glassy carbon working electrode in 0.1 M [nBu4N]+[PF6]− CH3CN solution at a potential scan rate of 100 mV s−1. Each of these molecules exhibits irreversible oxidation and reduction waves (Fig. S1b, ESI†). Additionally, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels (Fig. 1b and Table 1) were estimated from the onset oxidation and reduction potentials, respectively, assuming the absolute energy level of FeCp2+/0 to be 4.8 eV below vacuum56 (the oxidation potential of FeCp2+/0versus Ag/AgCl was measured to be 0.45 V). The HOMO energy levels of FTAZ (−5.39 eV) and J71 (−5.40 eV) are similar; while J71 shows a lower LUMO energy level of −3.65 eV relative to FTAZ (−3.50 eV), due to the σ inductive effect of silicon atom.57 The HOMO and LUMO energy levels of ITIC1 are −5.48 and −3.84 eV, respectively. ITIC2 shows slightly higher HOMO and LUMO energy levels of −5.43 and −3.80 eV, respectively, owing to the electron-donating property of thiophene units.
Next, the charge transport properties of these materials are explored. The hole mobilities of FTAZ and J71, and electron mobilities of ITIC1 and ITIC2 in neat films were measured using the space charge limited current (SCLC) method in hole-only (ITO/PEDOT:PSS/FTAZ or J71/Au) or electron-only (Al/ITIC1 or ITIC2/Al) devices (Fig. S2 (ESI†) and Table 1).58 The hole mobilities of FTAZ and J71 are 4.4 × 10−3 and 3.6 × 10−3 cm2 V−1 s−1, respectively; the electron mobilities of ITIC1 and ITIC2 are 1.0 × 10−3 and 1.3 × 10−3 cm2 V−1 s−1, respectively.
Photovoltaic properties
To further explore the effect of 1D vs. 2D conjugation of these materials, BHJ OSCs with the structure of ITO/ZnO/donor:acceptor/MoOx/Ag were fabricated using FTAZ or J71 as donor and ITIC1 or ITIC2 as acceptor. Each of the 4 pairings of donor:acceptor were carefully optimized and the results are summarized in Fig. 2a and Table 2. The optimized FTAZ:ITIC1-based devices (i.e. 1D/1D) show an open-circuit voltage (VOC) of 0.921 V, JSC of 16.45 mA cm−2, fill factor (FF) of 0.564, and PCE of 8.54%. When switching to a 2D acceptor, FTAZ:ITIC2-based devices (1D/2D) exhibit a higher VOC of 0.925 V, higher JSC of 18.88 mA cm−2, higher FF of 0.630, and an overall higher PCE of 11.0%. However, when switching to a 2D donor, J71:ITIC2-based devices (2D/2D) exhibit a lower PCE of 9.55% with VOC of 0.940 V, JSC of 16.55 mA cm−2, and FF of 0.614. When moving back to the 1D acceptor, J71:ITIC1-based devices (2D/1D) show higher JSC, FF and PCE of 17.90 mA cm−2, 0.653, and 10.6%, respectively. The higher VOC of ITIC2-based devices is due to the higher LUMO energy level of ITIC2 (−3.80 eV compared to −3.84 eV of ITIC1). As the HOMO energy levels of FTAZ and J71 are similar, the difference in VOC between FTAZ and J71-based devices may originate from morphology-induced VOC loss.59,60 The JSC and FF of mixed combinations 1D/2D and 2D/1D (FTAZ:ITIC2 and J71:ITIC1) are higher than those of 1D/1D (FTAZ:ITIC1) and 2D/2D (J71:ITIC2) combinations, which leads to higher PCEs.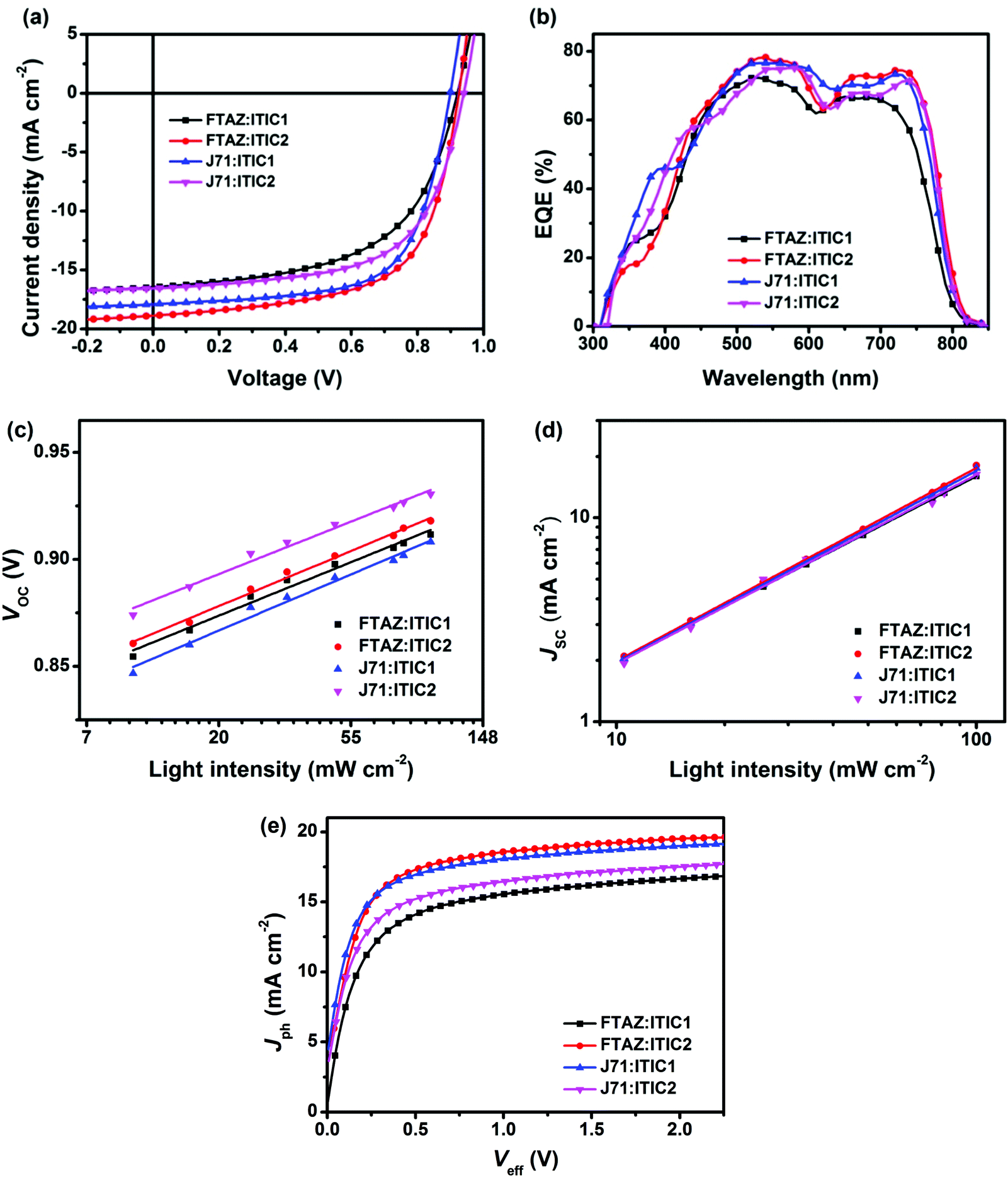 | ||
| Fig. 2 (a) J–V curves, (b) EQE spectra, (c) VOCversus light intensity, (d) JSCversus light intensity, and (e) Jphversus Veff of the optimized devices with the structure of ITO/ZnO/active layer/MoOx/Ag. | ||
| Active layer | V OC (V) | J SC (mA cm−2) | FFa | PCEa (%) | Calculated JSC (mA cm−2) | μ h (10−4 cm2 V−1 s−1) | μ e (10−4 cm2 V−1 s−1) | μ h/μe |
|---|---|---|---|---|---|---|---|---|
| a Average values with standard deviation were obtained from 20 devices, the values in parentheses are the parameters of the best device. | ||||||||
| FTAZ:ITIC1 | 0.922 ± 0.003 (0.921) | 16.06 ± 0.36 (16.45) | 0.562 ± 0.004 (0.564) | 8.32 ± 0.19 (8.54) | 15.84 | 25 | 2.0 | 12.5 |
| FTAZ:ITIC2 | 0.922 ± 0.003 (0.925) | 18.63 ± 0.26 (18.88) | 0.620 ± 0.006 (0.630) | 10.6 ± 0.2 (11.0) | 18.13 | 12 | 4.1 | 2.92 |
| J71:ITIC1 | 0.908 ± 0.005 (0.911) | 17.55 ± 0.23 (17.90) | 0.645 ± 0.015 (0.653) | 10.2 ± 0.2 (10.6) | 17.59 | 10 | 3.5 | 2.85 |
| J71:ITIC2 | 0.935 ± 0.003 (0.940) | 16.29 ± 0.29 (16.55) | 0.598 ± 0.014 (0.614) | 9.11 ± 0.23 (9.55) | 16.50 | 12 | 3.7 | 3.24 |
The external quantum efficiency (EQE) spectra of the optimized devices are shown in Fig. 2b. All of the four EQE spectra exhibit a slight valley at around 600 nm, arising from the separated absorption of donors (400–600 nm) and acceptors (600–800 nm); thus, the donors and acceptors contribute to the photoresponse in the region of 400–600 nm and 600–800 nm, respectively. The JSC of the optimized FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends calculated from integration of EQE spectra with the AM 1.5G reference spectrum are 15.84, 18.13, 17.59, and 16.50 mA cm−2, respectively, consistent with JSC values measured from J–V (the error is <5%, Table 2).
Next, to better understand the differences between the various 1D and 2D pairings, charge recombination in the devices was investigated by measuring VOC (Fig. 2c) and JSC (Fig. 2d) under different incident light intensities (P). The relationship between VOC and P is described by the formula of VOC ∝ ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P,61 where a slope of 1 kBT/q (kB: Boltzmann constant, T: temperature, and q: elementary charge) indicates that bimolecular recombination dominates in the device, a slope of 2 kBT/q indicates that geminate or Shockley–Read–Hall recombination dominates,62 while a slope of 0.5 kBT/q indicates that surface recombination dominates.63,64 The slope for FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends is 0.99 kBT/q, 1.02 kBT/q, 1.04 kBT/q, and 0.97 kBT/q, respectively, suggesting that bimolecular recombination dominates in all 4 blends. The relationship between JSC and P is described by the formula of JSC ∝ PS, where a value of S = 1 indicates that all free carriers are swept out and collected at electrodes before recombination, and S < 1 indicates some extent of bimolecular recombination.65 The S values of FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends are 0.92, 0.95, 0.94 and 0.93, respectively, indicating relatively weaker bimolecular recombination in mixed combinations 1D/2D and 2D/1D (FTAZ:ITIC2 and J71:ITIC1), which is beneficial for higher FF.
P,61 where a slope of 1 kBT/q (kB: Boltzmann constant, T: temperature, and q: elementary charge) indicates that bimolecular recombination dominates in the device, a slope of 2 kBT/q indicates that geminate or Shockley–Read–Hall recombination dominates,62 while a slope of 0.5 kBT/q indicates that surface recombination dominates.63,64 The slope for FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends is 0.99 kBT/q, 1.02 kBT/q, 1.04 kBT/q, and 0.97 kBT/q, respectively, suggesting that bimolecular recombination dominates in all 4 blends. The relationship between JSC and P is described by the formula of JSC ∝ PS, where a value of S = 1 indicates that all free carriers are swept out and collected at electrodes before recombination, and S < 1 indicates some extent of bimolecular recombination.65 The S values of FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends are 0.92, 0.95, 0.94 and 0.93, respectively, indicating relatively weaker bimolecular recombination in mixed combinations 1D/2D and 2D/1D (FTAZ:ITIC2 and J71:ITIC1), which is beneficial for higher FF.
Charge transfer in blended films was investigated by photoluminescence (PL) quenching (Fig. S3, ESI†). FTAZ and J71 are excited at 466 nm and emit at 631 and 636 nm (Fig. S3a, ESI†), respectively; while ITIC1 and ITIC2 are excited at 687 nm and emit at 784 and 776 nm (Fig. S3b, ESI†), respectively. According to the absorption spectra of the four materials, excitation at 466 nm mainly excites donors while that at 687 nm mainly excites acceptors. When excited at 466 nm, all of the four blends show over 99% PL quenching, suggesting highly efficient charge transfer from donor to acceptor. When excited at 687 nm, the PL quenching of 1D/2D and 2D/1D, FTAZ:ITIC2 and J71:ITIC1, are 98% and 97%, respectively, slightly higher than that of 1D/1D FTAZ:ITIC1 (92%) and 2D/2D J71/ITIC2 (96%), indicating more efficient charge transfer from acceptor to donor, which is beneficial for higher JSC.
Charge generation and extraction efficiencies of these devices were also investigated by measuring the photocurrent density (Jph) versus the effective voltage (Veff) (Fig. 2e).66 At high Veff (>2 V), all excitons are dissociated into free charge carriers and collected by electrodes, the Jph becomes saturated photocurrent density (Jsat), and JSC/Jsat characterizes the charge generation and extraction under short-circuit conditions. The JSC/Jsat values of mixed combinations 1D/2D and 2D/1D, FTAZ:ITIC2 and J71:ITIC1, are both 94%, higher than that of 1D/1D FTAZ:ITIC1 (92%) and 2D/2D J71/ITIC2 (93%). Therefore, the pairings of mixed 1D and 2D materials, compared to 1D/1D or 2D/2D, show more efficient charge generation and extraction, which is beneficial to higher JSC and FF.
Finally, the hole (μh) and electron mobilities (μe) of the blended films were measured by the SCLC method with device structures of ITO/PEDOT:PSS/active layer/Au for holes (Fig. S4a (ESI†) and Table 2) and Al/active layer/Al for electrons (Fig. S4b (ESI†) and Table 2). The μh/μe of FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends are 12.5, 2.92, 2.85, and 3.24, respectively. The 2D/1D system of J71:ITIC1 shows the most balanced hole/electron transport, which can help explain the highest FF. The unbalanced hole/electron transport in the 1D/1D system of FTAZ:ITIC1 is responsible for the lowest FF. Both high mobilities and balanced charge transport are needed for high JSC and FF.
Film morphology
While the photovoltaic properties illustrated some of the impact from the various pairings of 1D and 2D conjugated materials, the morphology of each of these blends is also important in elucidating the 1D/2D conjugation effect. Transmission electron microscopy (TEM) was first used to characterize the bulk morphology of the active layers (Fig. S5, ESI†). All of the four blends show relatively uniform morphology without pinholes or large aggregates, which prevents severe recombination. Grazing incidence wide-angle X-ray scattering (GIWAXS) was used to gain more molecular level morphology information.67 The 2D GIWAXS patterns and the corresponding intensity profiles of the neat donor and acceptor films in the in-plane (qr) and out-of-plane (qz) directions are shown in Fig. S6 (ESI†). All four of these materials exhibit a favored face-on orientation in neat films. The lamellar peaks of FTAZ, J71, ITIC1, and ITIC2 locate at qr = 0.321, 0.292, 0.333, and 0.299 Å−1, corresponding to d-spacings of 19.6, 21.5, 18.9, and 21.0 Å, respectively. The 2D conjugated materials of J71 and ITIC2 exhibit larger lamellar d-spacings relative to the 1D conjugated counterparts of FTAZ and ITIC1. The π–π stacking peaks of FTAZ, J71, ITIC1, and ITIC2 locate at qz = 1.66, 1.66, 1.79, and 1.85 Å−1, corresponding to d-spacings of 3.79, 3.79, 3.51, and 3.40 Å, respectively. While the π–π stacking peaks of FTAZ and J71 are both at 3.79 Å, FTAZ shows a larger π–π crystallite coherence length (CCL) of 20.6 Å relative to J71 (16.2 Å). A larger CCL is beneficial to high mobility, and this difference can help explain the differences in the hole mobilities for the two materials. Additionally, ITIC1 (13.3 Å) and ITIC2 (13.2 Å) exhibit similar π–π CCLs, leading to similar electron mobilities in neat films.Next, the optimized blended films were also investigated, and the 2D GIWAXS patterns and the corresponding intensity profiles of the blended films are presented in Fig. 3. Much like the neat materials, all of the four blended films show a preferential face-on orientation. The lamellar packing peaks of FTAZ:acceptor films locate at qr ∼ 0.330 Å−1 (d = 19.0 Å), while those of J71:acceptor blends locate at qr ∼ 0.310 Å−1 (d = 20.3 Å). J71:acceptor blends exhibit larger lamellar d-spacings relative to FTAZ:acceptor blends, resembling the trend in the neat films of J71 (2D) and FTAZ (1D). The π–π stacking peaks of FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 blends locate at qz =1.70, 1.67, 1.69, and 1.66 Å−1, respectively, with CCLs of 7.13, 12.7, 10.5, and 10.1 Å, respectively. The 1D/2D mixed blends show slightly larger CCLs than the 1D/1D and 2D/2D blends, which might contribute to the relatively better device performance results.
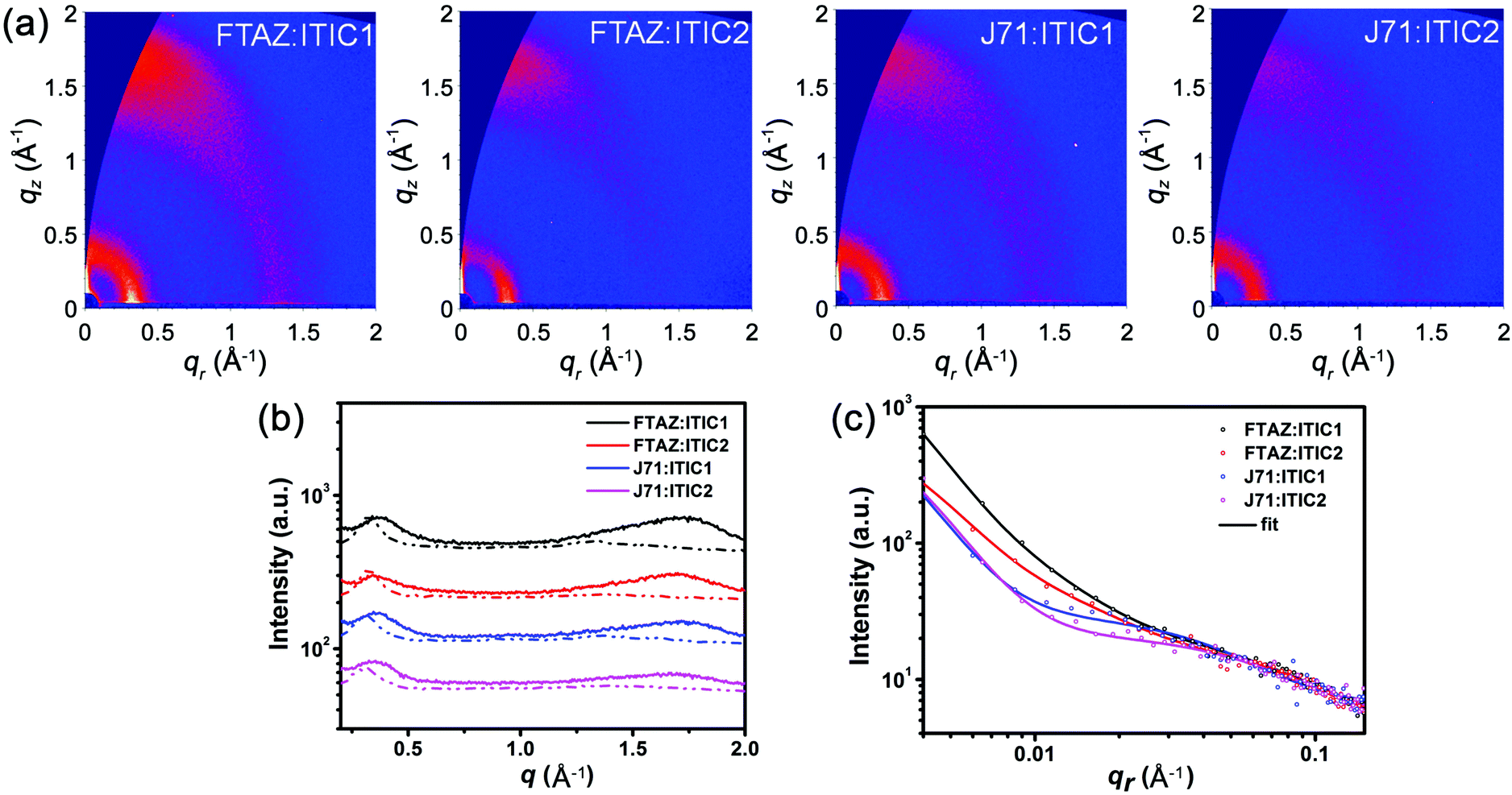 | ||
| Fig. 3 (a) 2D GIWAXS patterns, (b) GIWAXS intensity profiles along the in-plane (scattered line) and out-of-plane (solid line) directions, and (c) GISAXS intensity profiles and best fittings along the in-plane direction of the optimized active layers. | ||
Finally, grazing incidence small-angle X-ray scattering (GISAXS) measurements were performed to understand the phase separation information for the various blends. Phase separation, which originates from the miscibility of donor and acceptor, significantly affects the device performance.68,69 Low miscibility leads to formation of large pure domains, while high miscibility leads to formation of large intermixing domains. Large pure domain is beneficial to charge transport but unfavorable to exciton splitting, while large intermixing domain facilitates exciton splitting but restrains charge transport. Thus it is crucial to solve this paradox by precisely tuning the miscibility of donor and acceptor to reach a balance between exciton splitting and charge transport. The in-plane intensity profiles and the 2D GISAXS patterns of the neat and blended films are presented in Fig. 3c and Fig. S7 (ESI†), respectively. For FTAZ:acceptor blended films, we adopt the Debye–Anderson–Brumberger (DAB) model, a polydispersed hard sphere model and a fractal-like network model to account for the scattering contribution from intermixing amorphous phases, FTAZ domains and acceptor domains, respectively.70 For J71:acceptor blended films, we adopt the DAB model and a fractal-like network model70 since the scattering of J71 is very weak (Fig. S7, ESI†). The FTAZ domains retain similar sizes for FTAZ:acceptor blends (4.2–4.5 nm). The intermixing domain sizes of FTAZ:ITIC1, FTAZ:ITIC2, J71:ITIC1, and J71:ITIC2 films are 26.2, 24.4, 30.7, and 36.3 nm, respectively, while the acceptor domain sizes are 21.0, 22.0, 5.74, and 7.14 nm, respectively (Table 3). The acceptor domain sizes of the ITIC2-based (2D) films are larger than those of ITIC1-based (1D) films, suggesting that the conjugated side chains on the acceptor facilitate molecular packing and growth of acceptor domains, which is beneficial for higher electron mobility in blended films. The intermixing domain sizes of J71-based (2D) films are also larger than those of FTAZ-based (1D) films, indicating that the conjugated side chains on the donor increase the miscibility of the donor and acceptor. However, when mixing the 2D donor of J71 with the 2D acceptor of ITIC2, the film exhibits the largest intermixing domain size, suggesting that the increased miscibility may originate from the interactions between the side chains of the donor and acceptor. Compared to the FTAZ:ITIC1 (1D/1D) system, FTAZ:ITIC2 (1D/2D) shows relatively larger acceptor domains and a smaller intermixing domain, contributing to the higher electron mobility and less recombination, which improves JSC, FF, and PCE. J71 and ITIC1 show better miscibility relative to FTAZ and ITIC1, which is beneficial to efficient charge generation and balanced charge transport, and thus higher JSC and FF. Finally, the extra miscibility between J71 and ITIC2 leads to the largest intermixing domain size and thus more recombination and lower performance.
| Intermixing (nm) | Acceptor (nm) | FTAZ (nm) | |
|---|---|---|---|
| FTAZ:ITIC1 | 26.2 | 21.0 | 4.26 |
| FTAZ:ITIC2 | 24.4 | 22.0 | 4.48 |
| J71:ITIC1 | 30.7 | 5.74 | — |
| J71:ITIC2 | 36.3 | 7.14 | — |
Conclusions
In summary, we choose two polymer donors with the same molecular backbone, FTAZ and J71, and two nonfullerene acceptors with the same molecular backbone, ITIC1 and ITIC2, to investigate the effects of conjugation dimension (1D and 2D) on the performance of OSCs. FTAZ and ITIC1 have 1D conjugation (i.e. only along backbone), while J71 and ITIC2 have 2D conjugation due to conjugated thienyl side chains. By pairing each of the different donors and acceptors together, a better understanding of the 1D/2D conjugation effect can be illustrated. First, 2D conjugated side chains slightly red-shift the absorption spectra and lower the bandgap for both the donor and acceptor materials. J71 (2D) exhibits lower LUMO energy levels relative to FTAZ (1D) due to the σ inductive effect of silicon atoms on thienyl substituents, while ITIC2 (2D) shows higher LUMO energy levels relative to ITIC1 (1D) owing to the electron-donating property of thienyl side chains. Furthermore, 2D conjugated side chains on the acceptor induce self-aggregation of the small molecule acceptors, leading to larger acceptor domain size. Conjugated side chains on the polymer donor improve the miscibility of the donor and acceptor, and thus increase the intermixing domain size. Large pure domain facilitates charge transport but restrains exciton splitting, while large intermixing domain is beneficial to exciton splitting but unfavorable to charge transport. Additionally, the 2D conjugated side chain can effectively adjust the crystallinity and miscibility of donors and acceptors simultaneously, thus reaching a balance between charge transport and exciton dissociation, and finally achieving better performance. In this work, the combinations of 1D donor/2D acceptor (FTAZ/ITIC2) and 2D donor/1D acceptor (J71/ITIC1) achieve balance relative to the 1D/1D and 2D/2D blends, thus showing higher PCEs. Therefore, this work suggests that pairing mixed conjugation systems (i.e. 1D and 2D) might be a technique to achieve higher efficiency in OSCs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
X. Z. thanks the National Natural Science Foundation of China (Grant No. 51761165023 and 21734001). X. L. thanks NSFC/RGC Joint Research Scheme No. N_CUHK418/17 and the financial support from Research Grant Council of Hong Kong (General Research Fund No. 14314216 and Theme-based Research Scheme No. T23-407/13-N). J. R. and W. Y. were supported by a NSF grant (CBET-1639429).Notes and references
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS
.
- F. C. Krebs, N. Espinosa, M. Hösel, R. R. Søndergaard and M. Jørgensen, Adv. Mater., 2014, 26, 29–39 CrossRef CAS
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS
- Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS
- K. Feng, G. Yang, X. Xu, G. Zhang, H. Yan, O. Awartani, L. Ye, H. Ade, Y. Li and Q. Peng, Adv. Energy Mater., 2018, 8, 1602773 CrossRef
- J. Wang, W. Wang, X. Wang, Y. Wu, Q. Zhang, C. Yan, W. Ma, W. You and X. Zhan, Adv. Mater., 2017, 29, 1702125 CrossRef
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed
- Y. Lin and X. Zhan, Adv. Energy Mater., 2015, 5, 1501063 CrossRef
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed
- Y. Lin, F. Zhao, Y. Wu, K. Chen, Y. Xia, G. Li, S. K. K. Prasad, J. Zhu, L. Huo, H. Bin, Z.-G. Zhang, X. Guo, M. Zhang, Y. Sun, F. Gao, Z. Wei, W. Ma, C. Wang, J. Hodgkiss, Z. Bo, O. Inganäs, Y. Li and X. Zhan, Adv. Mater., 2017, 29, 1604155 CrossRef PubMed
- S. Chen, Y. An, G. K. Dutta, Y. Kim, Z.-G. Zhang, Y. Li and C. Yang, Adv. Funct. Mater., 2017, 27, 1603564 CrossRef
- S. Chen, H. J. Cho, J. Lee, Y. Yang, Z.-G. Zhang, Y. Li and C. Yang, Adv. Energy Mater., 2017, 7, 1701125 CrossRef
- M. Jeong, S. Chen, S. M. Lee, Z. Wang, Y. Yang, Z.-G. Zhang, C. Zhang, M. Xiao, Y. Li and C. Yang, Adv. Energy Mater., 2017, 8, 1702166 CrossRef
- S. Chen, S. M. Lee, J. Xu, J. Lee, K. Lee, T. Hou, Y. Yang, M. Jeong, B. Lee, Y. Cho, S. Jung, J. Oh, Z.-G. Zhang, C. Zhang, M. Xiao, Y. Li and C. Yang, Energy Environ. Sci., 2018, 11, 2569–2580 RSC
- Y. Qin, M. A. Uddin, Y. Chen, B. Jang, K. Zhao, Z. Zheng, R. Yu, T. J. Shin, H. Y. Woo and J. Hou, Adv. Mater., 2016, 28, 9416–9422 CrossRef CAS
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef
- J. Zhu, Z. Ke, Q. Zhang, J. Wang, S. Dai, Y. Wu, Y. Xu, Y. Lin, W. Ma, W. You and X. Zhan, Adv. Mater., 2018, 30, 1704713 CrossRef
- J.-D. Chen, Y.-Q. Li, J. Zhu, Q. Zhang, R.-P. Xu, C. Li, Y.-X. Zhang, J.-S. Huang, X. Zhan, W. You and J.-X. Tang, Adv. Mater., 2018, 30, 1706083 CrossRef PubMed
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed
- Y. Lin, F. Zhao, S. K. K. Prasad, J.-D. Chen, W. Cai, Q. Zhang, K. Chen, Y. Wu, W. Ma, F. Gao, J.-X. Tang, C. Wang, W. You, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2018, 30, 1706363 CrossRef PubMed
- P. Cheng, J. Wang, Q. Zhang, W. Huang, J. Zhu, R. Wang, S. Y. Chang, P. Sun, L. Meng, H. Zhao, H. W. Cheng, T. Huang, Y. Liu, C. Wang, C. Zhu, W. You, X. Zhan and Y. Yang, Adv. Mater., 2018, 30, 1801501 CrossRef PubMed
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS PubMed
- B. Kan, H. Feng, X. Wan, F. Liu, X. Ke, Y. Wang, Y. Wang, H. Zhang, C. Li, J. Hou and Y. Chen, J. Am. Chem. Soc., 2017, 139, 4929–4934 CrossRef CAS PubMed
- W. Wang, C. Yan, T.-K. Lau, J. Wang, K. Liu, Y. Fan, X. Lu and X. Zhan, Adv. Mater., 2017, 29, 1701308 CrossRef PubMed
- L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z.-G. Zhang, B. Qiu, X. Li, C. Sun, L. Gao, J. Yao, X. Chen, Y. Yang, M. Xiao and Y. Li, Adv. Mater., 2017, 29, 1703344 CrossRef PubMed
- S. Xu, Z. Zhou, W. Liu, Z. Zhang, F. Liu, H. Yan and X. Zhu, Adv. Mater., 2017, 29, 1704510 CrossRef PubMed
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS PubMed
- Z. Xiao, X. Jia, D. Li, S. Wang, X. Geng, F. Liu, J. Chen, S. Yang, T. P. Russell and L. Ding, Sci. Bull., 2017, 62, 1494–1496 CrossRef CAS
- T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed
- J. Zhang, C. Yan, W. Wang, Y. Xiao, X. Lu, S. Barlow, T. C. Parker, X. Zhan and S. R. Marder, Chem. Mater., 2018, 30, 309–313 CrossRef CAS
- J. Zhu, Y. Xiao, J. Wang, K. Liu, H. Jiang, Y. Lin, X. Lu and X. Zhan, Chem. Mater., 2018, 30, 4150–4156 CrossRef CAS
- W. Liu, J. Zhang, Z. Zhou, D. Zhang, Y. Zhang, S. Xu and X. Zhu, Adv. Mater., 2018, 30, 1800403 CrossRef PubMed
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, J. Am. Chem. Soc., 2018, 140, 7159–7167 CrossRef CAS PubMed
- Z. Luo, H. Bin, T. Liu, Z.-G. Zhang, Y. Yang, C. Zhong, B. Qiu, G. Li, W. Gao, D. Xie, K. Wu, Y. Sun, F. Liu, Y. Li and C. Yang, Adv. Mater., 2018, 30, 1706124 CrossRef PubMed
- Y. Xie, F. Yang, Y. Li, M. A. Uddin, P. Bi, B. Fan, Y. Cai, X. Hao, H. Y. Woo, W. Li, F. Liu and Y. Sun, Adv. Mater., 2018, 30, 1803045 CrossRef PubMed
- Y. Zhang, B. Kan, Y. Sun, Y. Wang, R. Xia, X. Ke, Y. Q. Q. Yi, C. Li, H. L. Yip, X. Wan, Y. Cao and Y. Chen, Adv. Mater., 2018, 30, 1707508 CrossRef PubMed
- S. Dai, T. Li, W. Wang, Y. Xiao, T.-K. Lau, Z. Li, K. Liu, X. Lu and X. Zhan, Adv. Mater., 2018, 30, 1706571 CrossRef PubMed
- X. Xu, Z. Li, Z. Bi, T. Yu, W. Ma, K. Feng, Y. Li and Q. Peng, Adv. Mater., 2018, 30, 1800737 CrossRef PubMed
- Z. Li, X. Xu, G. Zhang, T. Yu, Y. Li and Q. Peng, Sol. RRL, 2018, 2, 1800186 CrossRef
- Q. An, W. Gao, F. Zhang, J. Wang, M. Zhang, K. Wu, X. Ma, Z. Hu, C. Jiao and C. Yang, J. Mater. Chem. A, 2018, 6, 2468–2475 RSC
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bassler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS
- J. Ohshita, Macromol. Chem. Phys., 2009, 210, 1360–1370 CrossRef CAS
- G. G. Malliaras, J. R. Salem, P. J. Brock and C. Scott, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13411–13414 CrossRef
- B. Ray, M. S. Lundstrom and M. A. Alam, Appl. Phys. Lett., 2012, 100, 013307 CrossRef
- Z. Tang, J. Wang, A. Melianas, Y. Wu, R. Kroon, W. Li, W. Ma, M. R. Andersson, Z. Ma, W. Cai, W. Tress and O. Inganäs, J. Mater. Chem. A, 2018, 6, 12574–12581 RSC
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef
- S. Wheeler, F. Deledalle, N. Tokmoldin, T. Kirchartz, J. Nelson and J. R. Durrant, Phys. Rev. Appl., 2015, 4, 024020 CrossRef
- S. Solak, P. W. M. Blom and G. A. H. Wetzelaer, Appl. Phys. Lett., 2016, 109, 053302 CrossRef
- I. Riedel, J. Parisi, V. Dyakonov, L. Lutsen, D. Vanderzande and J. C. Hummelen, Adv. Funct. Mater., 2004, 14, 38–44 CrossRef CAS
- M. A. Faist, S. Shoaee, S. Tuladhar, G. F. A. Dibb, S. Foster, W. Gong, T. Kirchartz, D. D. C. Bradley, J. R. Durrant and J. Nelson, Adv. Energy Mater., 2013, 3, 744–752 CrossRef CAS
- J. Mai, Y. Xiao, G. Zhou, J. Wang, J. Zhu, N. Zhao, X. Zhan and X. Lu, Adv. Mater., 2018, 30, 1802888 CrossRef PubMed
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J.-H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J.-L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed
- J. Mai, T.-K. Lau, J. Li, S.-H. Peng, C.-S. Hsu, U. S. Jeng, J. Zeng, N. Zhao, X. Xiao and X. Lu, Chem. Mater., 2016, 28, 6186–6195 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00512e |
| ‡ Contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |