
V2(PO4)O/C@CNT hollow spheres with a core–shell structure as a high performance anode material for lithium-ion batteries†
Bin
Xiao
,
Wen-hai
Zhang
,
Hai-feng
Xia
,
Zhi-teng
Wang
,
Lin-bo
Tang
,
Chang-sheng
An
,
Zhen-jiang
He
,
Hui
Tong
and
Jun-chao
Zheng
 *
*
School of Metallurgy and Environment, Central South University, Changsha 410083, P. R. China. E-mail: jczheng@csu.edu.cn; Fax: +86-731-88836357; Tel: +86-731-88836357
First published on 9th January 2019
Abstract
V2(PO4)O is a novel promising anode material because of its stable crystal structure, low cost, and environmentally friendliness. However, its low electronic and ionic conductivity result in poor specific capacity and rate performance, hindering its industrial application. In this study, V2(PO4)O/C@CNT hollow spheres (HSs) with a core–shell structure were successfully synthesized by a simple spray drying method. Pluronic® F-127 acted as a carbon source and template, and induced the V2(PO4)O particles to form HSs. The V2(PO4)O/C@CNT HSs were composed of uniform HSs of about 0.4–0.6 μm in diameter and carbon nanotubes (CNTs). The HS structure enhances the electronic conductivity and lithium ion transport rate of V2(PO4)O/C materials and thus helps improve the electrochemical performance. CNTs not only increase the electronic conductivity and lithium ion transport rate, but also hinder the aggregation of the HSs. These activities enhance the electrochemical performance. The lithium ion storage behavior of V2(PO4)O/C@CNT HSs is systematically studied in the potential range 0.01–3.0V. The V2(PO4)O/C@CNT HS anode can achieve a high reversible capacity of 894.9 mA h g−1 at 0.1 A g−1 and can obtain a reversible capacity of 490.4 mA h g−1 at a high rate of 5 A g−1, perhaps the best electrochemical performance demonstrated so far for a V2(PO4)O anode material (specific capacity and rate performance), indicating its promise for application as an anode material in advanced lithium-ion batteries.
1. Introduction
With the continuous growth of energy demand, energy storage has become a very important issue. Among the numerous existing energy storage technologies, rechargeable lithium batteries (LIBs) have been extensively applied to portable electronic devices and electric vehicles, owing to their numerous advantages, such as nonexistent memory effect, long cycling life, and high energy density.1–13 As an important part in the battery, anode materials should be continuously developed and optimized.14 As far as the charge–discharge mechanism is concerned, anode materials for LIBs can be divided into three types: (i) intercalation type anode materials, such as graphite, Ti,15 and V-based oxides;16 (ii) conversion type anode materials, such as transition-metal oxides17 and transition metal sulfides;18 and (iii) alloy/de-alloy type anode materials, including Si19 and Sn.20 Given the advantages of low cost, outstanding electronic conductivity, and good chemical/thermal stability, the typical intercalation type anode material graphite has been applied extensively in commercial LIBs. However, the LIBs possess many weaknesses, such as low theoretical capacity (372 mA h g−1) and weak rate property, which hinder the achievement of market demand for high energy density and high power density.21 Another frequently used commercial anode material, Li4Ti5O12, has good reversibility, but its low theoretical capacity (175 mA h g−1) and low energy density restrict the material's development.22 Conversion type anode materials, such as Co3O4,23 MoS2,24 and MoSe2,25 have high capacity and high energy density,26 but they also have many disadvantages, such as solid electrolyte interphase (SEI) film instability and weak capacity retention. As alloy/de-alloy type anode materials, Si19 and Sn20 have high specific capacity. Nevertheless, the large volume expansion in the charge–discharge process hinders their commercial application.27V-based anode materials, for instance Li3VO4,22 VPO4,11 and VOPO4,10 have been widely investigated by researchers. Li3VO4, an intercalation type anode material, possesses the advantages of small volume, slight structural changes, and rapid lithium ion diffusion rate. However, the material's low theoretical capacity and weak electronic conductivity hinder its further application. VPO4 and VOPO4 have stable crystal structures and high theoretical capacities but poor rate and cycling performances. Nevertheless, even with some weaknesses, V-based anode materials possess enormous potential applications for LIBs due to the various chemical valence states of V.
V2(PO4)O, as a novel V-based anode material, has attracted attention due to its stable crystal structure, low cost, and environmentally friendliness. The crystal structure of V2(PO4)O is composed of [VO6] octahedra and [PO4] tetrahedra, and the linking between [VO6] octahedra is by face contact.28 The arrays shared by the [VO6] octahedra with one face can lead to a short V–V distance, which promotes electron hopping between low- and high-valence V sites. The special structure of V2(PO4)O equips the material with a relatively high intrinsic electronic conductivity (1.8 × 10−15 S cm−1). The rigid [PO4] tetrahedra also afford V2(PO4)O with excellent structural stability. At present, only one researcher has studied the V2(PO4)O material, and a V2(PO4)O/C composite was prepared by a hydrothermal method and calcination steps that achieved an appreciable specific capacity of 541 mA h g−1 at 0.5 A g−1.28 However, the obtained V2(PO4)O/C composite only delivered a specific capacity of 345.6 mA h g−1 at 5 A g−1, which could not meet the needs of industrial applications. And the V2(PO4)O/C was composed of nanoscale particles with a particle size between 10 and 30 nm, which can shorten the distance of lithium ion diffusion, but the nanoscale particles had many shortcomings, such as the agglomeration and pulverization of nanoscale particles. This problem may be solved by synthesizing hollow spheres (HSs) of about 0.4–0.6 μm diameter, which can offer a high specific surface area and specific capacity, shorten the lithium ion diffusion path, and provide an enlarged space for volume expansion.29–31 Meanwhile, the agglomeration of HSs can be inhibited by adding carbon nanotubes (CNTs), which also enhance the electronic conductivity of the V2(PO4)O material. In the current study, V2(PO4)O/C@CNT HSs were synthesized through a facile spray dryer approach with Pluronic® F-127 acting as a template and carbon (C) source. Pluronic® F-127 can adsorb VO3−, PO43−, and NH4+, and then Pluronic® F-127 with absorbed ions was assembled into hollow spheres. The properties of the V2(PO4)O/C@CNT HSs were investigated.
2. Experimental
2.1 Material synthesis
NH4VO3 (Aladdin >99.9%) and NH4H2PO4 (>99.0%) were purchased from Aladdin Co., Ltd. Pluronic® F-127 (Sigma-Aldrich >99.0%) was acquired from Sigma-Aldrich Co., Ltd CNTs (>98%) were obtained from the Chinese Academy of Science, Chengdu Organic Chemistry Co., Ltd. All chemical reagents besides CNTs used in this study were not further purified.The CNTs used in this study were purified for improving the hydrophilicity of CNTs. 100 mg CNTs was added into 500 mL concentrated HNO3 solution (68% w/w) and the mixture were sonicated at room temperate for 6 h. After that, the CNTs were washed with deionized water several times and dried by vacuum freeze drying for 16 h.
V2(PO4)O/C@CNT HSs were synthesized by the following steps: NH4VO3 (117.0 mg), NH4H2PO4 (121.0 mg), and Pluronic® F-127 (1000 mg) were mixed in 100 mL of deionized water under continuous stirring at 80 °C for 2 h. CNTs (100 mg) were added to 100 mL of deionized water and dispersed by ultrasonication for 4 h. Then, the two liquids were mixed, and the resultant solution was dried through a spray dryer with an inlet temperature of 260 °C at a flow rate of 600 mL h−1. Finally, the V2(PO4)O/C@CNT precursor was heated at 850 °C for 10 h under a 10% H2/Ar atmosphere to acquire V2(PO4)O/C@CNT HSs. By contrast, V2(PO4)O/C HSs were fabricated by the same steps without CNTs. The process of preparing V2(PO4)O/C@CNT HSs is shown in Fig. 1.
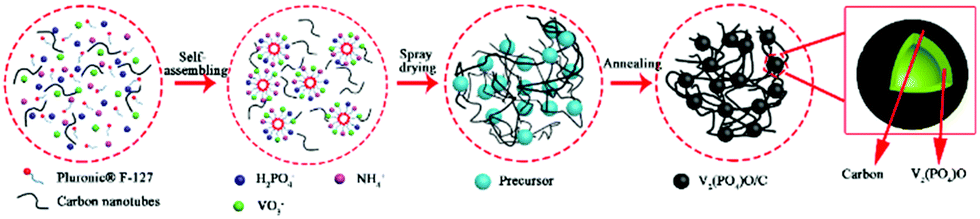 | ||
| Fig. 1 Schematic diagram of the synthesis process for V2(PO4)O/CNT HSs. | ||
2.2 Material characterization
Cu Kα radiation X-ray diffraction (XRD, Rint-2000, Rigaku) was applied to test the structural and crystalline phase analyses of the as-prepared samples. A simultaneous thermal analyzer (STA 449F3, NETZSCH, Germany) was used to measure the C content of the as-prepared samples at a heating rate of 5 °C min−1 at an air flow of 100 mL min−1. The Brunauer–Emmett–Teller (BET) method was employed to examine the specific surface area through a micromeritics surface area and porosity analyzer (ASAP 2020 HD88, USA). An Invia Raman spectrometer was employed to examine the Raman spectra of both samples. An IRAffinity-1 FTIR spectrometer (SHIMADZU) was applied to obtain Fourier transform infrared (FTIR) spectra. The morphology of the as-synthesized samples was obtained by field-emission scanning electron microscopy (FESEM; JEOL, JSM-7001F) coupled with energy-dispersive X-ray spectroscopy (EDX) and transmission electron microscopy (TEM; JEOL, JEM-3000F). X-ray photoelectron spectroscopy (XPS; VG ESCALAB MK II) was used to test the surface chemical valences of both samples.2.3 Electrochemical measurements
The electrochemical characterization of V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs was examined by a coin-type cell (CR2025). The typical electrode loading was in the range of 3–3.5 mg cm−2. The working electrodes in the coin-type cell were prepared through the following steps. The active materials, polyvinylidene fluoride adhesive, and acetylene black were mixed at a mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2. Then, a finite amount of N-methyl-2-pyrrolidone (NMP) was dropped into the above mixture; after grinding, the slurry was cast onto a Cu foil. The coated Cu foil was placed in a vacuum drying oven at 120 °C for 8 h and punched into disks with diameters of 12 mm. A glove box under dry Ar was applied to assemble the coin-type cell, in which the Cu foil acted as the counter electrode and the polypropylene film served as the separator. The electrolyte used in the coin-type cell was 1 mol L−1 LiPF6 dissolved in a 1:1 volume mixture of ethylene carbonate/dimethyl carbonate. The rate and cycling performances of the cells were examined by a LAND battery cycler between 0.01 and 3.0 V versus Li/Li+ at 25 °C. A CHI660D electrochemical analyzer (Shanghai, Chenhua) was applied to achieve cyclic voltammograms in the potential range of 0.01–3.0 V and electrochemical impedance spectroscopy (EIS) between 0.1 Hz and 100.00 kHz at 25 °C.
:1:2. Then, a finite amount of N-methyl-2-pyrrolidone (NMP) was dropped into the above mixture; after grinding, the slurry was cast onto a Cu foil. The coated Cu foil was placed in a vacuum drying oven at 120 °C for 8 h and punched into disks with diameters of 12 mm. A glove box under dry Ar was applied to assemble the coin-type cell, in which the Cu foil acted as the counter electrode and the polypropylene film served as the separator. The electrolyte used in the coin-type cell was 1 mol L−1 LiPF6 dissolved in a 1:1 volume mixture of ethylene carbonate/dimethyl carbonate. The rate and cycling performances of the cells were examined by a LAND battery cycler between 0.01 and 3.0 V versus Li/Li+ at 25 °C. A CHI660D electrochemical analyzer (Shanghai, Chenhua) was applied to achieve cyclic voltammograms in the potential range of 0.01–3.0 V and electrochemical impedance spectroscopy (EIS) between 0.1 Hz and 100.00 kHz at 25 °C.
3. Results and discussion
The preparation of the V2(PO4)O/C@CNT HS composite involves two steps. At first, a suspension solution containing VO3−, PO43−, NH4+, Pluronic® F-127 and CNTs was dried by spray drying, and the V2(PO4)O/C@CNT HS precursor was obtained. Pluronic® F-127 is an amphiphilic triblock copolymer, which can form macromolecule micelles consisting of a hydrophilic shell and lipophilic core after dissolving in water.32 After the formation of spherical micelle templates, the ions of VO3−, PO43−, and NH4+ were adsorbed on the surface of the templates. During the annealing process, Pluronic® F-127 (H(OCH2CH2)x(OCH2CHCH3)y(OCH2CH2)2OH) and NH4VO3 reacted with NH4H2PO4 in a high temperature environment (850 °C, 10 hours) to form V2(PO4)O and carbon. The reaction equation for this process should be writen as the following:The process of synthesizing V2(PO4)O/C@CNT HSs is shown in Fig. 1.
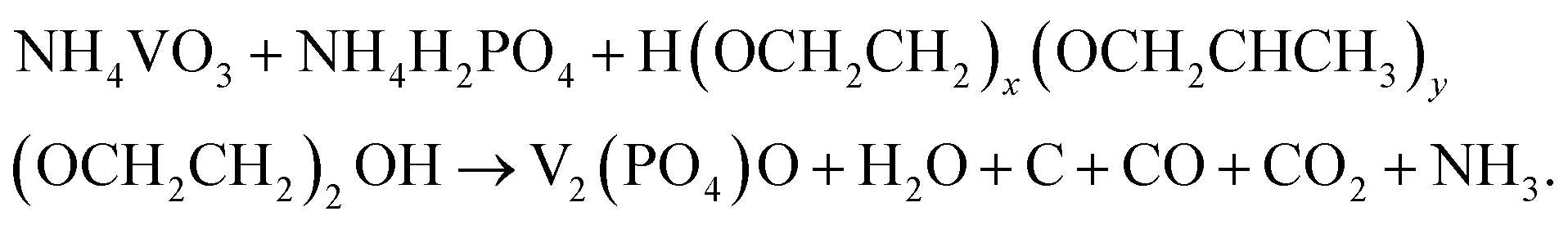
The crystallographic structure of the synthesized V2(PO4)O/C@CNT HSs and V2(PO4)O/C HS composite was examined through XRD (Fig. 2). As shown in the XRD patterns (Fig. 2a), the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HS composite were well indexed to the tetragonal structure (space group I41/amd (141), a = 5.362 Å, b = 5.362 Å, c = 12.378 Å; JCPDS No. 83-0585), which indicates the high crystallinity and purity of V2(PO4)O.28 Comparing the XRD results of the CNTs and V2(PO4)O/C@CNT HS composite, we showed the CNT diffraction peak in the XRD of V2(PO4)O/C@CNT HSs. This finding suggests the existence of CNTs in the V2(PO4)O/C@CNT HS composite. The C content of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HS composite was tested by thermogravimetric analysis (Fig. 2b) tested with a heating rate of 5 °C min−1 under an air flow of 100 mL min−1. The weight of V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs decreased between 400 °C and 570 °C because of C oxidation reaction. The weight loss of the V2(PO4)O/C HS composite was estimated to be 31.79%, and that of the V2(PO4)O/C@CNT HS composite was 36.36%. These values suggest that the CNT content of the V2(PO4)O/C@CNT HS composite was approximately equal to 4.57%.
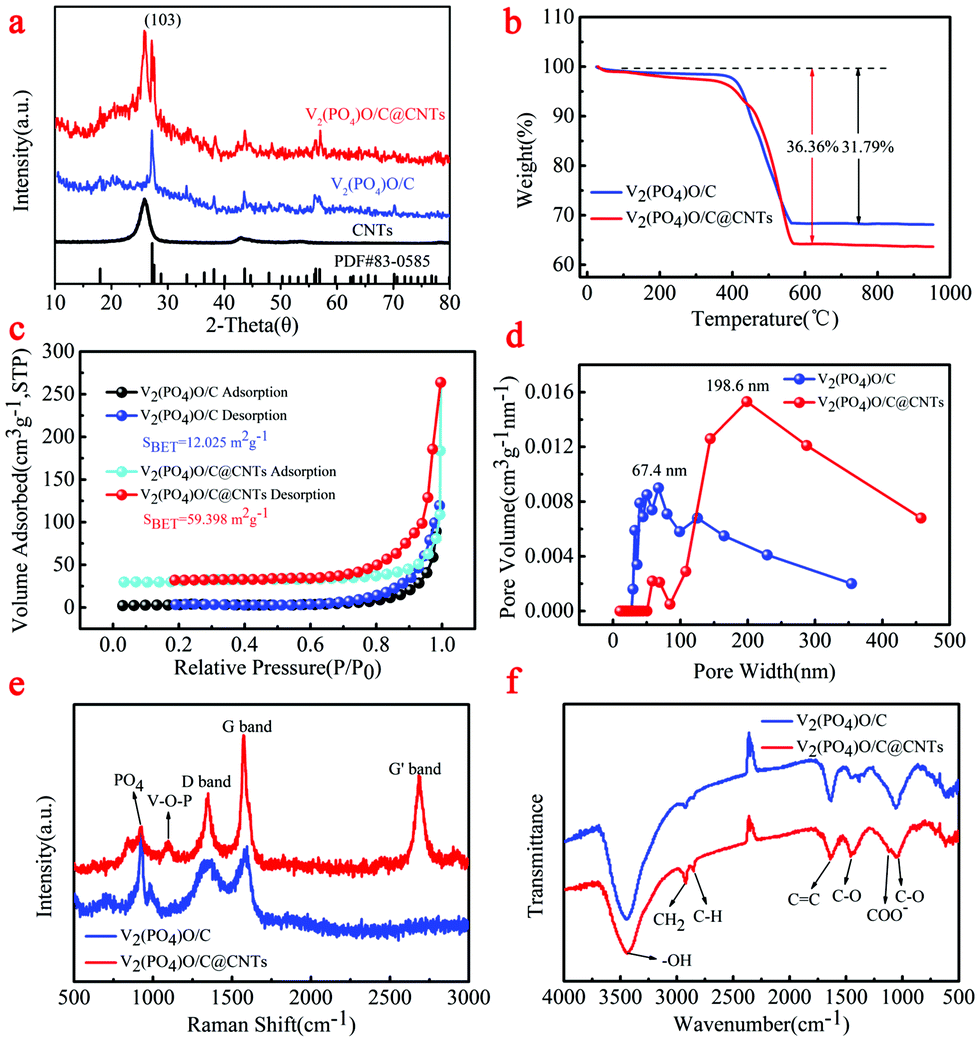 | ||
| Fig. 2 (a) XRD of V2(PO4)O/CNT HSs and V2(PO4)O/C HSs; (b) TG curves of V2(PO4)O/CNT HSs and V2(PO4)O/C HSs; (c) N2 adsorption–desorption isotherms of V2(PO4)O/CNT HSs and V2(PO4)O/C HSs; (d) corresponding pore size distribution performed by the BJH method; (e) Raman spectrum of V2(PO4)O/CNT HSs and V2(PO4)O/C HSs; (f) FTIR of V2(PO4)O/CNT HSs and V2(PO4)O/C HSs. | ||
To characterize the porosity properties of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HS composite, N2 absorption–desorption isotherms were examined. In Fig. 2c, the specific surface area of the V2(PO4)O/C@CNT HS composite was 59.398 m2 g−1, which was higher than that of the V2(PO4)O/C HS composite (12.025 m2 g−1). Such an augmented specific surface area for the V2(PO4)O/C@CNT HS composite increased the area of contact between the active material and the electrolyte. This expanded area helped shorten the lithium diffusion distance. The pore size distributions of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs were calculated from the desorption branch (Fig. 2d). The pore size distribution of the V2(PO4)O/C@CNT HSs was in the range of 58.3–457.5 nm, and a wide peak was located at about 198.6 nm. Meanwhile, the pore size distribution of the V2(PO4)O/C HSs was in the range of 29.5–354.3 nm, and a wide peak was located at about 67.4 nm. Voids in the particle not only provide access to electrolyte but also provide space for material volume expansion. Both factors can improve effectively the long cycling stability of materials.
The Raman spectra of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs are shown in Fig. 2e. Two strong peaks are located at 1347 and 1575 cm−1 in the spectra of both samples; these peaks are assigned to the disorder-induced phonon (D band) and G-line tangential modes (G band), respectively, of C materials.33 A strong peak also situated at 2686 cm−1 in the spectrum of the V2(PO4)O/C@CNT HSs corresponds to the G′ mode (G′ band) of the CNTs. Another two peaks at 925 and 1093 cm−1 in the spectrum of the V2(PO4)O/C@CNT HSs are indexed to the totally symmetric PO4 “breathing” vibration and the V–O–P stretch, respectively.34 To obtain further information on both samples, we performed FTIR spectroscopy in Fig. 2f. Three peaks were observed at 3439, 2925, and 2849 cm−1 at 2500–3500 cm−1 and assigned to the –OH mode and CH2 and C–H bands, respectively.35–37 In addition, the signals at 1634, 1448, 1388, and 1055 cm−1 can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups, CO band, COO– groups, and C–O band, respectively.38
C groups, CO band, COO– groups, and C–O band, respectively.38
To observe the morphology of the as-synthesized samples, we conducted FESEM (Fig. 3). Clearly, the as-synthesized V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs were composed of uniform spheres of 0.4–0.6 μm diameter (Fig. 3a, b, d, and e). The agglomeration phenomenon of V2(PO4)O/C spheres appeared in the V2(PO4)O/C HS samples. However, V2(PO4)O/C spheres in the V2(PO4)O/C@CNT HS sample were uniformly dispersed in the CNT network and became beneficial for improving the specific surface area of the V2(PO4)O/C@CNT HS sample. Enlarging the FESEM image of the V2(PO4)O/C@CNT HS sample, it can be observed that some CNTs penetrated into the V2(PO4)O/C@CNT HSs in Fig. S3 (ESI†), which can be proved by the following TEM image. Improving the specific surface area of the V2(PO4)O/C@CNT HS sample can increase the contact area between the active material and the electrolyte, which is in favor of shortening the lithium diffusion distance.39,40 This CNT network can efficiently increase the electron transport rate between the V2(PO4)O/C spheres, thereby improving the electronic conductivity of V2(PO4)O/C@CNT spheres. The results can be proved by the following EIS findings. The high specific surface area and enhanced electronic conductivity of the V2(PO4)O/C@CNT spheres are beneficial for enhancing the electrochemical performances of V2(PO4)O/C, especially the rate capability and long cycle performance. To clarify the spatial distribution of V, phosphorus (P), and C in the V2(PO4)O/C HSs and V2(PO4)O/C@CNT HSs, we performed EDX elemental mapping. The mapping images in Fig. 3c and f demonstrate that the V, P, and C elements in both samples are distributed uniformly on the surface and suggest the presence of a thin C layer on the surface of the V2(PO4)O/C spheres. This phenomenon can be observed in the following TEM images.
 | ||
| Fig. 3 (a and b) SEM images of V2(PO4)O/C HSs; (d and e) SEM images of V2(PO4)O/C@CNT HSs; (c) EDX elemental mapping of V2(PO4)O/C HSs; (d) EDX elemental mapping of V2(PO4)O/C@CNT HSs. | ||
To illustrate the internal structure of the as-prepared samples, we examined high-magnification TEM and high-resolution TEM (HRTEM) images. As shown in Fig. 4a and b, the V2(PO4)O/C spheres were hollow structures, and the diameters of both samples lay within 0.4–0.6 nm, which matched the result of the SEM images. The V2(PO4)O/C spheres in the V2(PO4)O/C@CNT HS sample were wrapped with CNTs. Furthermore, a uniform C-coating layer was present on the surface of the nanoparticles forming a core–shell structure, and the thickness of the C layer was about ∼18 nm. As shown in the TEM image at low magnification, it can be clearly observed that a complete spherical carbon shell coated the V2(PO4)O/C sphere in Fig. S2 (ESI†). The carbon shell not only promotes the electron transfer but also inhibits the volume expansion of the V2(PO4)O spheres in the charge–discharge process. In addition, it can be seen that CNTs are not simply attached to the surface of V2(PO4)O/C HSs, but penetrated into them, as shown in Fig. S4 (ESI†). Clear lattice fringes are displayed in the HRTEM images in Fig. 4e and f. The observed interplane spaces were 0.329 and 0.331 nm, which were well indexed to the (103) plane of tetragonal V2(PO4)O (JCPDS No. 83-0585). The selected area electron diffraction patterns of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs are shown in Fig. 4g and h, which can be indexed to the tetragonal structure of V2(PO4)O (JCPDS No. 83-0585). The diffraction rings in Fig. 4g can be indexed to (200), (206), and (208), and those in Fig. 4h can be indexed to (103), (200), (220), (107), and (224), respectively.
 | ||
| Fig. 4 (a and c) TEM images of V2(PO4)O/C HSs; (b and d) TEM images of V2(PO4)O/C@CNT HSs; (e) HRTEM image of V2(PO4)O/C HSs; (f) HRTEM image of V2(PO4)O/C@CNT HSs; (g) SAED patterns of V2(PO4)O/C HSs; (h) SAED patterns of V2(PO4)O/C@CNT HSs. | ||
To confirm the surface chemical valences of both samples, we conducted XPS. Indeed, the survey spectra (Fig. S1a and b, ESI†) of the V2(PO4)O/C@CNT HS and V2(PO4)O/C HS samples clearly revealed the existence of the V, P, O, and C elements. This result agreed with the above EDX elemental mapping results. As shown in Fig. 5a, the binding energy centered at 517.0 and 524.5 eV in the V 2p spectrum of V2(PO4)O/C@CNT HSs can be attributed to V 2p3/2 and V 2p1/2 of V2+ and V3+, which were different from that of the V2(PO4)O/C HSs. The V 2p spectrum of the V2(PO4)O/C HSs can be indexed to V 2p3/2 and V 2p1/2 and confirms the tervalence of V (Fig. S1c, ESI†). The V 2p3/2 peak of the V2(PO4)O/C HS sample deconvoluted into three subpeaks centered at 516, 517.2, and 517.5 eV (Fig. S1c, ESI†), which correspond to V4+, V3+, and V5+, respectively. Due to the poor air tightness of the furnace, there may be air entering the furnace in the annealing process. In the synthetic reaction of V2(PO4)O, water was formed at high temperature. CNTs reacted with water and produced CO and H2; the reaction equation for this process was the following: C + H2O → CO + H2. CO and H2 both are reductive gases, which can avoid vanadium oxidation. The P 2p XPS spectrum of the V2(PO4)O/C@CNT HSs in Fig. 5b can be fitted to three subpeaks centered at 132.8, 133.5, and 134.4 eV, which correspond to P–C, P–O, and P–O bonds, respectively, whereas that of V2(PO4)O/C HSs (Fig. S1d, ESI†) was divided into two components assigned to the P–O bond (133.1 and 133.8 eV).34 The O 1s spectrum of V2(PO4)O/C@CNT HSs in Fig. 5c was divided into two subpeaks corresponding to the P–O (134.0 eV) and C–O (133.1 eV) bonds, respectively, whereas that of V2(PO4)O/C HSs was split into four peaks (Fig. S1e, ESI†) corresponding to V–O (530.2 and 531.4 eV), P–O (531.4 and 532 eV), and C–O (533 eV), respectively. Furthermore, the P–C bond (285.1 eV) was further proved by the C 1s spectrum of V2(PO4)O/C@CNT HSs (Fig. 5d), which cannot be found in the C 1s spectrum of V2(PO4)O/C HSs. The reason for the formation of the P–C bond may be the high-temperature reaction between CNTs and (NH4)H2PO4, thereby demonstrating that the V2(PO4)O/C HSs are chemically bonded to the CNTs via P–C bonds. The results revealed the successful improvement of electrical conductivity of the materials and alleviation of the volume expansion. The other two divided peaks of V2(PO4)O/C@CNT HSs at 284.7 and 286 eV were assigned to the C–O bond, whereas those of V2(PO4)O/C HSs (Fig. S1f, ESI†) were fitted to C–O (284.7 and 286.2 eV) and C–C (285.7 eV), respectively.
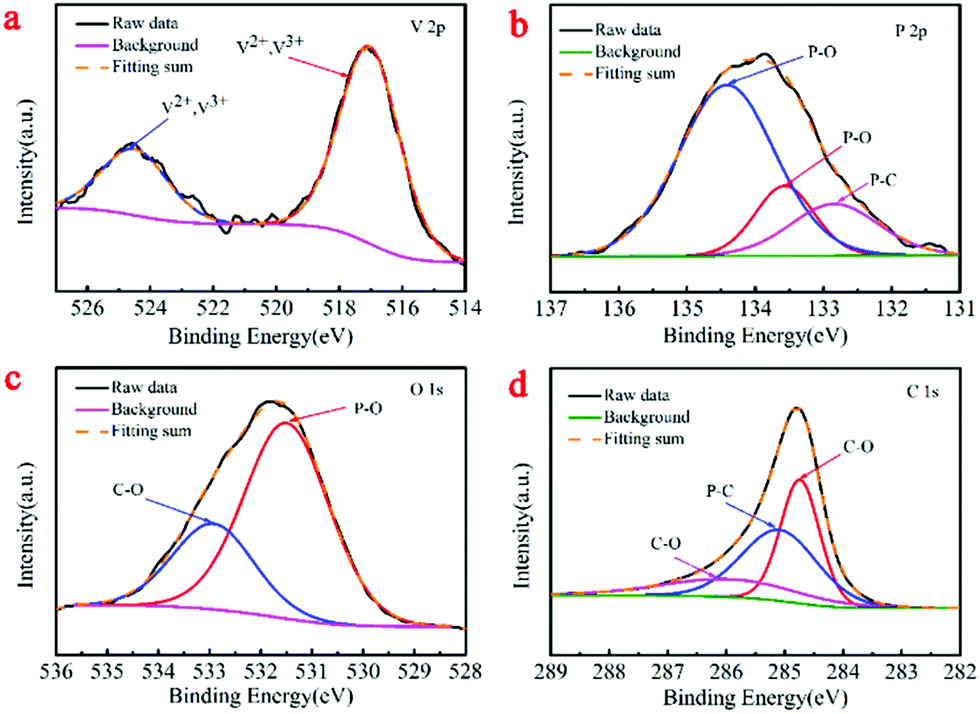 | ||
| Fig. 5 (a) XPS core level of V 2p in V2(PO4)O/C@CNT HSs; (b) XPS core level of P 2p in V2(PO4)O/C@CNT HSs; (c) XPS core level of O 1s in V2(PO4)O/C@CNT HSs; (d) XPS core level of C 1s in V2(PO4)O/C@CNT HSs. | ||
The electrochemical performances of V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs as LIB anodes were tested. The cyclic voltammetry (CV) curves of the V2(PO4)O/C@CNT HS composite are shown in Fig. 6a when these were tested in the potential range 0.01–3.0 V at a scanning rate of 0.1 mV s−1. The charge–discharge curves of the V2(PO4)O/C@CNT HS and V2(PO4)O/C HS samples at various rates, from 0.1 A g−1 to 5.0 A g−1 between 0.01 and 3.00 V, are shown in Fig. 6b and c, respectively. We observed the existence of three reduction peaks located at 0.77, 1.7, and 2.52 V and two oxidation peaks at 1.8 and 2.65 V in the first cycle of the CV curves (Fig. 6a). The obvious reduction peak centered at 0.77 V in the first cathodic cycle that disappeared in the following cycles was assigned to the formation of an SEI layer, which corresponded to the plateau at 0.77 V of the first discharge curves of the V2(PO4)O/C@CNT HS sample in Fig. 6b. The other two reduction peaks at 1.7 and 2.52 V were assigned to the insertion reaction between V2(PO4)O and Li+. In the anodic process of the first cycle, the two oxidation peaks at 1.8 and 2.65 V were attributed to the Li+ deintercalation reaction. In the following two cycles, the CV curves of the V2(PO4)O/C@CNT HS composite maintain high coincidence and demonstrate the stability of the electrochemical reaction.
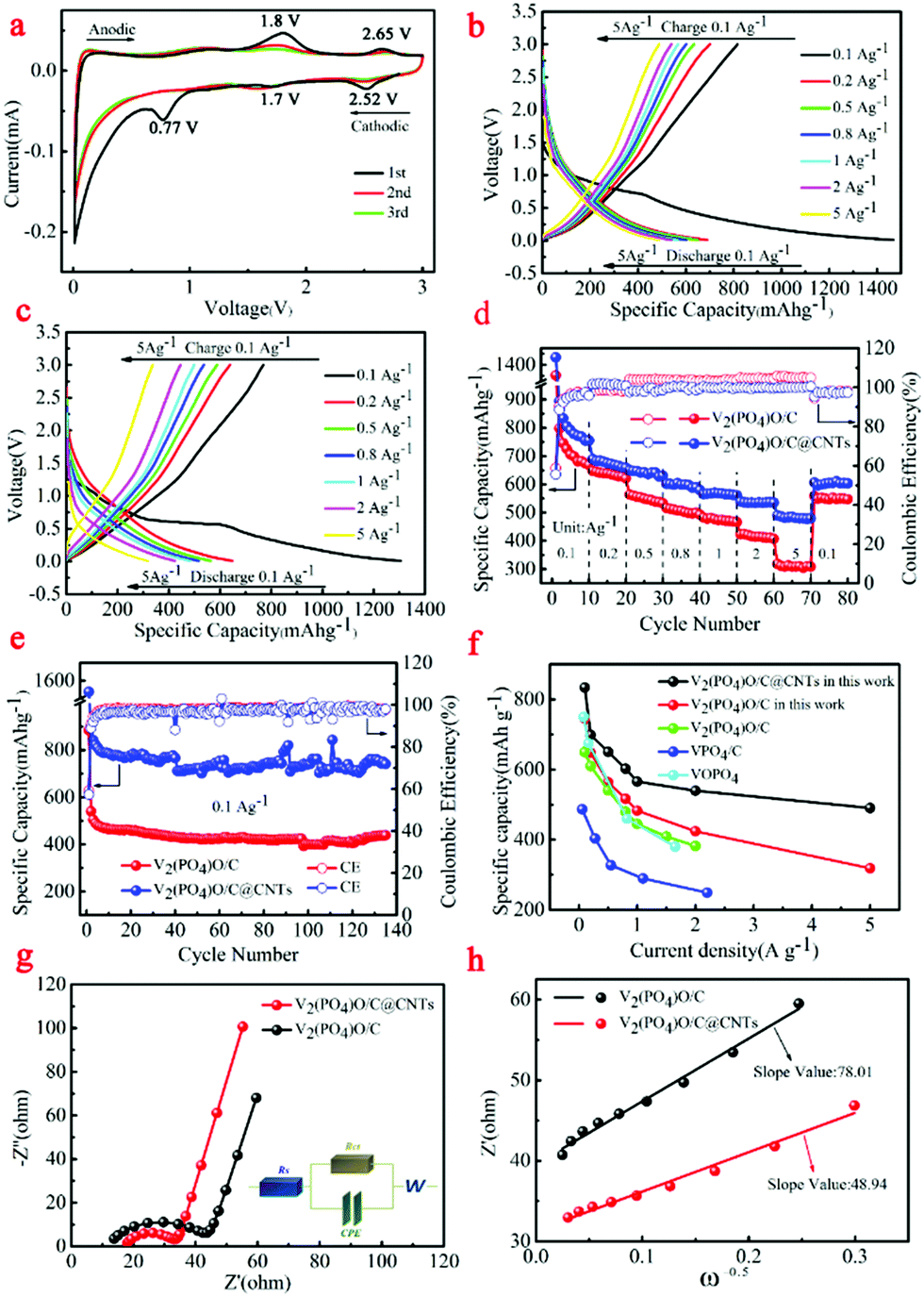 | ||
| Fig. 6 (a) The CV curves of V2(PO4)O/C@CNT HSs; (b) the first charge–discharge profiles of the V2(PO4)O/C@CNT HS sample from 0.1 to 5.0 A g−1; (c) the first charge–discharge profiles of the V2(PO4)O/C HS sample from 0.1 to 5.0 A g−1; (d) rate performance of V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs; (e) cycling performance of V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs; (f) the rate performance of V2(PO4)O/C@CNT HSs compared to other materials (V2(PO4)O/C,28 VPO4/C,5 VOPO410); (g) the Nyquist plots and the corresponding fitted curves of V V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs; (h) plots showing the relationship between Z′ and ω−0.5 at low frequencies, calculated from the EIS data. | ||
In the discharge/charge profiles of the V2(PO4)O/C@CNT HS sample (Fig. 6b), two pairs of weak discharge/charge plateaus of 1.7/1.8 V and 2.52/2.65 V in the first cycle at 0.1 A g−1 between 0.01 and 3.00 V were assigned to the related peaks of CV curves. The V2(PO4)O/C@CNT HS sample can achieve a discharge capacity of 1476.7 mA h g−1 and a charge capacity of 846.9 mA h g−1, whereas the coulombic efficiency was 57.35%. With regards the rate performance, the V2(PO4)O/C@CNT HS sample can achieve a high reversible capacity of 894.9, 689.8, 650.4, 602.6, 565.8, and 539.9 mA h g−1 at the different current densities of 0.1, 0.2, 0.5, 0.8, 1.0, and 2.0 A g−1, respectively, which are all higher than that of the V2(PO4)O/C HS sample at various densities (Fig. 6c and d). In particular, the sample can even obtain a reversible capacity of 490.4 mA h g−1 at the high rate of 5.0 A g−1. When the current density returns to 0.1 A g−1, it achieved a reversible capacity of 606.9 mA h g−1 with a capacity retention of 97.4% (Fig. 6d), which suggested satisfactory rate tolerance of the V2(PO4)O/C@CNT HS sample. Moreover, the long-term cycling performances of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs are shown in Fig. 6e. The V2(PO4)O/C@CNT HS sample delivered a reversible capacity of 741.5 mA h g−1 after 135 cycles at 0.1 A g−1, which was higher than that of the V2(PO4)O/C HS sample. Compared with other anode materials, the rate performance of the V2(PO4)O/C@CNT HS sample was much better.28 The major causes of this result were the higher theoretical specific capacity, higher electronic conductivity, and good Li+ ion coefficient.
To determine the source of the good electrochemical properties, we examined the EIS results in the range of 0.1 Hz to 100 kHz (Fig. 6g). To simulate Nyquist plots, we adopted a simplified equivalent circuit (Fig. 6g). The first intercept of the semicircle at the Z′ axis in the high-frequency region in Fig. 6g was relevant to the resistance Rs, which was the combined Ohmic resistance of the separator, electrolyte, and metal electrode. The charge transfer resistance was represented by Rct, which corresponded to the distance between the two intercepts of the semicircle on the Z′ axis. The fitting results of the Nyquist plots for both samples are listed in Table 1. The Rs values of the V2(PO4)O/C@CNT HSs and V2(PO4)O/C HSs were 17.37 and 11.74 Ω, respectively, which were higher than that of the V2(PO4)O/C HSs in the literature.28 However, the Rct of the V2(PO4)O/C@CNT HSs was 13.5 Ω, which was far lower than that of the V2(PO4)O/C HSs (29.47 Ω); this result suggested that CNTs reduced the charge resistance and improve electronic conductivity. Moreover, the Rct values of both samples were lower than those of the literature and indicated that the HS structure was conducive to the transmission of electrons.28 The electrochemical reaction is dominated by ion diffusion when a threshold of the electronic conductivity is exceeded. The lithium ion diffusion coefficient DLi+ was calculated using EIS data on the basis of the following equations:41
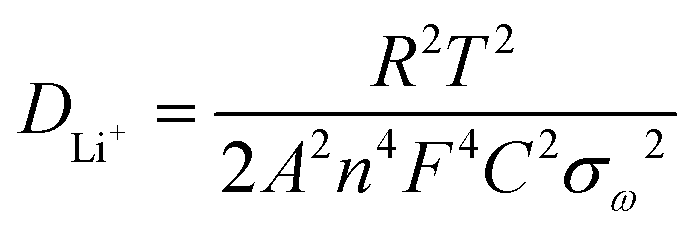 | (1) |
| Z′ = Re + Rct + σωω−0.5 | (2) |
| Sample | R s (Ω) | R ct (Ω) | Slope value | D Li+ (cm2 s−1) |
|---|---|---|---|---|
| V2(PO4)O/C | 11.74 | 29.47 | 78.01 | 7.28 × 10−15 |
| V2(PO4)O/C@CNTs | 17.37 | 13.5 | 48.94 | 1.85 × 10−14 |
| V2(PO4)O/C28 | 3 | 48 | 198.6 | 5.75 × 10−15 |
4. Conclusions
In summary, V2(PO4)O/C@CNT HSs with a core–shell structure were successfully synthesized via a facile spray drying method. The V2(PO4)O/C@CNT HS composite exhibited a high specific capacity and good rate performance. The V2(PO4)O/C@CNT HSs delivered a high reversible capacity of 894.9, 689.8, 650.4, 602.6, 565.8, and 539.9 mA h g−1 at the current densities of 0.1, 0.2, 0.5, 0.8, 1.0, and 2.0 A g−1, respectively. In particular, the V2(PO4)O/C@CNT HSs obtained a reversible capacity of 490.4 mA h g−1 at the high rate of 5.0 A g−1. The good electrochemical performances of the V2(PO4)O/C@CNT HSs benefitted from the material's unique HS structure, which enhanced the electronic conductivity and lithium ion transport rate of the V2(PO4)O/C HS materials. The CNTs also played an important role in this enhancement. The addition not only increased the electronic conductivity and lithium ion transport rate but also hindered the aggregation of the HSs. Thus, the V2(PO4)O/C@CNT HSs, as a potential novel anode material can be used in advanced lithium ion batteries in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 51572300) and the Graduate Innovation Project of Central South University (No. 2018zzts136).Notes and references
- C. Shen, B. Zhang, J. F. Zhang, J. C. Zheng, Y. D. Han and H. Li, RSC Adv., 2015, 5, 7208–7214 RSC.
- J. C. Zheng, Y. D. Han, B. Zhang, C. Shen, L. Ming and J. F. Zhang, RSC Adv., 2014, 4, 41076–41080 RSC.
- J. C. Zheng, Y. D. Han, B. Zhang, C. Shen, L. Ming and J. F. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 13520–13526 CrossRef CAS PubMed.
- C. Shen, J. C. Zheng, B. Zhang, Y. D. Han, J. F. Zhang, L. Ming, H. Li and X. B. Yuan, RSC Adv., 2014, 4, 40912–40916 RSC.
- J. C. Zheng, Y. D. Han, B. Zhang, C. Shen, L. Ming, X. Ou and J. F. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 6223–6226 CrossRef CAS PubMed.
- J. C. Zheng, Y. D. Han, D. Sun, B. Zhang and E. J. Cairns, Energy Storage Mater., 2017, 7, 48–55 CrossRef.
- B. Xiao, B. Zhang, J. C. Zheng, L. B. Tang, C. S. An, Z. J. He, H. Tong and W. J. Yu, Ceram. Int., 2018, 44, 13113–13121 CrossRef CAS.
- B. Zhang, Y. D. Han, J. C. Zheng, C. Shen, L. Ming and J. F. Zhang, J. Power Sources, 2014, 264, 123–127 CrossRef CAS.
- B. Xiao, B. Zhang, L. B. Tang, C. S. An, Z. J. He, H. Tong, W. J. Yu and J. C. Zheng, Ceram. Int., 2018, 44, 15044–15049 CrossRef CAS.
- B. Zhang, Y. D. Han, J. C. Zheng, J. F. Zhang, C. Shen, L. Ming, X. B. Yuan and H. Li, Chem. Commun., 2014, 50, 11132–11134 RSC.
- L. B. Tang, B. Xiao, C. S. An, H. Li, Z. J. He and J. C. Zheng, Ceram. Int., 2018, 44, 14432–14438 CrossRef CAS.
- H. Chen, M. Ling, L. Hencz, H. Y. Ling, G. Li, Z. Lin, G. Liu and S. Zhang, Chem. Rev., 2018, 118, 8936–8982 CrossRef CAS PubMed.
- D. Adekoya, X. Gu, M. Rudge, W. Wen, C. Lai, M. Hankel and S. Zhang, Adv. Funct. Mater., 2018, 1803972, DOI:10.1002/adfm.201803972.
- M. Zhang, T. Wang and G. Cao, Int. Mater. Rev., 2015, 60, 330–352 CrossRef CAS.
- L. Shen, E. Uchaker, X. Zhang and G. Cao, Adv. Mater., 2012, 24, 6502–6506 CrossRef CAS PubMed.
- X. Wang, Y. Huang, D. Jia, W. K. Pang, Z. Guo, Y. Du, X. Tang and Y. Cao, Inorg. Chem., 2015, 54, 11799–11806 CrossRef CAS PubMed.
- M. Liu, C. Yan and Y. Zhang, Sci. Rep., 2015, 5, 8326 CrossRef PubMed.
- L. Zhang, Q. Wei, D. Sun, N. Li, H. Ju, J. Feng, J. Zhu, L. Mai, E. J. Cairns and J. Guo, Nano Energy, 2018, 51, 391–399 CrossRef CAS.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed.
- D. Kong, H. He, Q. Song, B. Wang, W. Lv, Q.-H. Yang and L. Zhi, Energy Environ. Sci., 2014, 7, 3320–3325 RSC.
- C. Zhang, H. Song, C. Liu, Y. Liu, C. Zhang, X. Nan and G. Cao, Adv. Funct. Mater., 2015, 25, 3497–3504 CrossRef CAS.
- J. Liu, H. Xia, L. Lu and D. Xue, J. Mater. Chem., 2010, 20, 1506 RSC.
- K. Chang, W. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J. Y. Lee, J. Mater. Chem., 2011, 21, 6251 RSC.
- Y. Liu, M. Zhu and D. Chen, J. Mater. Chem. A, 2015, 3, 11857–11862 RSC.
- J. Liu and D. Xue, Nanoscale Res. Lett., 2010, 5, 1525–1534 CrossRef CAS PubMed.
- M. Holzapfel, H. Buqa, W. Scheifele, P. Novak and F. M. Petrat, Chem. Commun., 2005, 1566–1568, 10.1039/b417492e.
- X. Nan, C. Liu, C. Zhang, W. Ma, K. Wang, Z. Li and G. Cao, J. Mater. Chem. A, 2016, 4, 9789–9796 RSC.
- D. Mao, J. Wan, J. Wang and D. Wang, Adv. Mater., 2018, e1802874, DOI:10.1002/adma.201802874.
- J. Wang, Y. Cui and D. Wang, Adv. Mater., 2018, e1801993, DOI:10.1002/adma.201801993.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- C. M. Papadakis, R. Ivanova, K. Lüdtke, K. Mortensen, P. K. Pranzas and R. Jordan, J. Appl. Crystallogr., 2010, 40, s361–s362 CrossRef.
- L. Tzounis, S. Debnath, S. Rooj, D. Fischer, E. Mäder, A. Das, M. Stamm and G. Heinrich, Mater. Des., 2014, 58, 1–11 CrossRef CAS.
- D. Zhao, T. Meng, J. Qin, W. Wang, Z. Yin and M. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 1437–1445 CrossRef CAS PubMed.
- D. Kumar, G. Kumar and V. Agrawal, Parasitol. Res., 2018, 117, 377–389 CrossRef PubMed.
- O. Carnevali, C. Conti, P. Ferraris, M. G. Garavaglia, G. Gioacchini, E. Giorgini, C. Rubini, S. Sabbatini and G. Tosi, J. Mol. Struct., 2009, 938, 207–213 CrossRef CAS.
- S. Orefuwa, M. E. Naggar, I. Shehadi, M. M. Chehimi and A. A. Mohamed, J. Nanosci. Nanotechnol., 2017, 17, 4063–4068 CrossRef CAS.
- J. T. Kloprogge, L. Hickey and R. L. Frost, J. Raman Spectrosc., 2004, 35, 967–974 CrossRef CAS.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414–430 RSC.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang and Y. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- J. C. Zheng, Z. Yang, Z. J. He, H. Tong, W. J. Yu and J. F. Zhang, Nano Energy, 2018, 53, 613–621 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00619a |
| This journal is © the Partner Organisations 2019 |