
Self-assembly of positively charged polymer patchy micelles in organic solutions and the reversible ultrasound responsivity of the assemblies†
Ya
Zhao‡
,
Weijing
Fan‡
and
Hanying
Zhao
 *
*
Key Laboratory of Functional Polymer Materials, Ministry of Education, College of Chemistry, Nankai University, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China. E-mail: hyzhao@nankai.edu.cn
First published on 30th January 2019
Abstract
Self-assembly of asymmetric structures has been attractive for applications in the fabrication of hierarchical structures. However, the role of the electrostatic interaction in the self-assembly of charged asymmetric particles in organic solvents has not been fully understood. In this research, self-assembly of positively charged patchy micelles in organic solutions and the reversible ultrasound responsivity of the assemblies are studied. Surface micelles (s-micelles) with cross-linked/quaternized poly(2-(dimethylamino)ethyl methacrylate) (q-PDMAEMA) cores and poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEGMA) coronae were prepared on the surfaces of silica particles. Positively charged patchy micelles were synthesized upon cleavage of the s-micelles. In THF, the patchy micelles self-assemble into vesicles with positively charged q-PDMAEMA in the walls and POEGMA in the coronae. The average size of the vesicles is dependent on POEGMA chain length. The repelling electrostatic interaction between the positively charged q-PDMAEMA patches in the walls is unfavorable for the achievement of low free energy of the assemblies, but it is energetically favorable for the vesicles to overcome energy barriers in the dissociation of the structures under ultrasound irradiation. The vesicle structures present reversible ultrasound responsivity. Under sonication the vesicles formed by patchy micelles dissociate into transient nanostructures, including aggregates and isolated patchy micelles. When the ultrasound is switched off, the transient nanostructures self-assemble into vesicles. By contrast, crosslinked vesicles do not show ultrasound responsivity. This research provides a new approach to the fabrication of responsive assemblies based on asymmetric structures, and the assemblies could find potential applications in medicine and bioengineering.
Introduction
Synthesis and self-assembly of asymmetric particles have received much attention during the last two decades because of their scientific significance and their potential applications in the fabrication of new advanced materials.1–14 Asymmetric particles refer to particles with diverse anisotropy in shape, surface properties and chemical composition.15–21 Among the asymmetric particles, Janus particles and patchy particles are the most widely studied asymmetric particles. Janus particles refer to particles having two hemispheres with different physical properties or chemical compositions.19–25 Similar to small molecular surfactant and amphiphilic block copolymer chains, amphiphilic Janus particles present very interesting surfactant properties and self-assembly behaviors.26–30 Patchy particles are a class of asymmetric particles with different patches on their surfaces.9 Chemically distinct patches on the particles provide directional interaction, and allow for the formation of directional bonds.31 Based on the interaction of patches, patchy particles can be used as the elementary building blocks for the fabrication of nano- or microscale self-assemblies, including clusters,32 chain-like structures,33,34 nanorods,35 and other structures.36–40It is well known that block copolymers are able to self-assemble into micelles in selective solvents. In polymer micelles, the insoluble blocks form the cores and the soluble blocks form the coronae. In the past decades, many different stimuli-responsive polymer micelles have been fabricated, including thermal-responsive, pH-responsive, salt-responsive and enzyme-responsive micelles.41,42 Stimuli-responsive micelles have found applications in nanoscience and medicine.43–45 Compared with the widely studied stimuli-responsive polymer micelles, the study on the stimuli-responsive self-assemblies formed by patchy particles is quite rare.
In this paper a study on the self-assembly of positively charged patchy micelles in organic solutions and the reversible ultrasound responsivity of the assemblies are reported. Ultrasound refers to sound waves with frequencies from 20 kHz up to several gigahertz. It is widely used in cleaning, mixing and chemical reactions. Ultrasound energy was also previously used in self-assembly research studies. The ultrasound responsivity of liposomes and ultrasound-assisted control release of drugs from the assemblies were reported previously.46 Ultrasound energy was also applied in the reorganization of supramolecular nanostructures.47 The ultrasound responsivity of spherical superparticles formed by tadpole-like single chain nanoparticles (SCNPs) was reported by Chen and coworkers.48 After a gentle ultrasonic treatment the superparticles are dissociated into individual SCNPs due to the absence of chain entanglement in the hydrophobic domains. Herein, nanosized patchy micelles composed of crosslinked/quaternized poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) cores partly covered with poly(oligo(ethylene glycol) monomethyl ether methacrylate) (POEGMA) patches were prepared by selective quaternization of PDMAEMA blocks of PDMAEMA-b-POEGMA block copolymer brushes on silica particles and followed by cleavage of the surface micelles (s-micelles) from the surfaces of the silica particles.49 In a previous study, co-assembly of protein molecules and patchy micelles prepared from cleaved s-micelles in aqueous solution was reported.49 In this research, self-assembly of positively charged patchy micelles in organic solution and the ultrasound responsivity of the assemblies are studied. The hydrophilic patchy micelles were dissolved in water and a 10-fold volume of THF was added. THF is a good solvent for POEGMA, but a precipitant for quaternized PDMAEMA (q-PDMAEMA). In order to avoid unfavorable interaction between q-PDMAEMA and THF, the patchy micelles self-assemble into vesicles with q-PDMAEMA in the walls and POEGMA in the coronae. Because of the electrostatic repulsion between the positively charged q-PEMAEMA patches in the walls, it is energetically favourable for the vesicles to dissociate into small aggregates and isolated particles under sonication. Upon cessation of the sonication, the small aggregates and isolated particles re-assemble into vesicles, the original equilibrium state. This research provides a method to control the morphology and size of the assemblies formed by patchy particles. The self-assembly process and the reversible ultrasound responsivity of the patchy micelles are illustrated in Scheme 1.
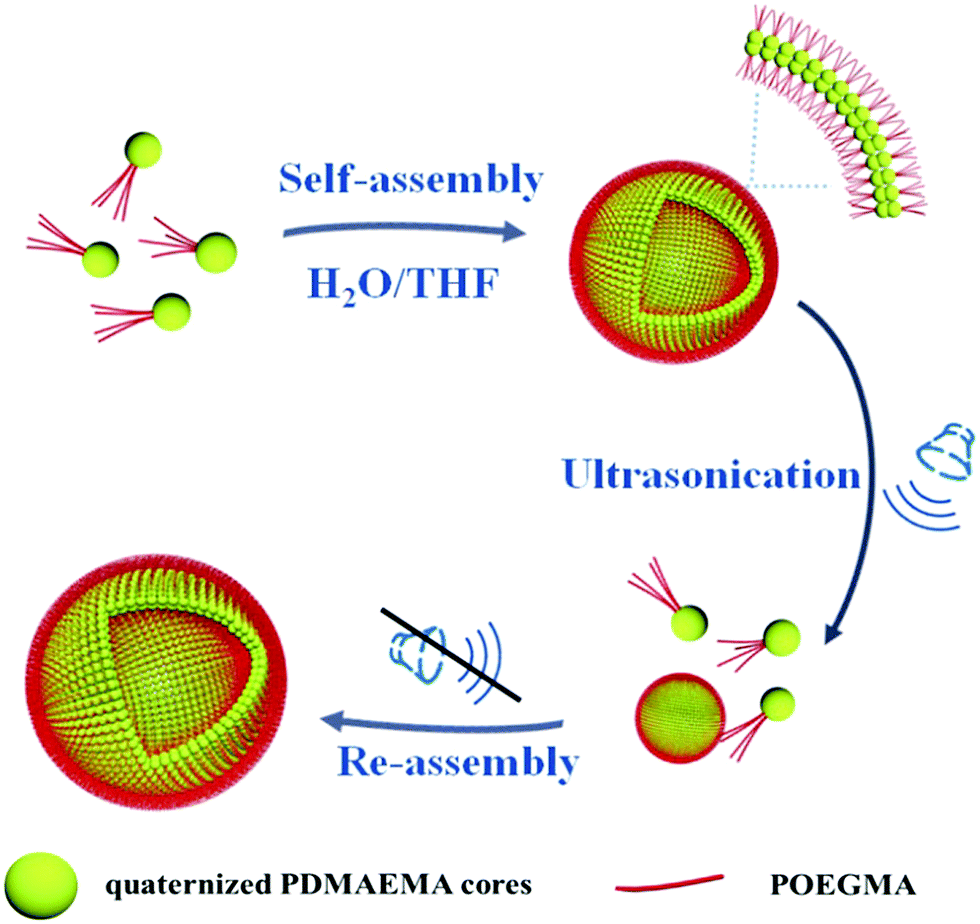 | ||
| Scheme 1 Schematic illustration for the self-assembly and reversible ultrasound responsivity of patchy micelles with quaternized PDMAEMA cores and POEGMA patches on the surfaces. | ||
Experimental
Materials and instruments
Tetraethyl orthosilicate (TEOS, 98%) was purchased from Sigma-Aldrich. Before use it was distilled under reduced pressure. DMAEMA (99%, Acros) was purified by passing it through a basic alumina column, and distilling under reduced pressure. 4,4′-Azobis(4-cyanopentanoic acid) (ABCPA, 97%, Sigma-Aldrich) was recrystallized from methanol and dried in a vacuum oven under reduced pressure at room temperature. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was synthesized in our laboratory. Oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA, M = 500 g mol−1) was purchased from Sigma-Aldrich and it was purified by passing it through a basic alumina column before use. n-Hexylamine (Aladdin, 99%), 2,2′-dipyridyl disulfide (Heowns, 98%), 1,4-diiodobutane (DIB, Alfa Aesar, 99%), benzoyl peroxide (BPO, Alfa Aesar, 97%), 1,3,6,8-pyrenetetrasulfonic acid tetrasodium salt (PTSA, 98%, Sigma-Aldrich), 2′,4′,5′,7′-tetraiodofluorescein disodium salt (erythrosin B, 90%, Sigma-Aldrich) and (3-mercaptopropyl)trimethoxysilane (MPTMS, Alfa Aesar, 95%) were used as received.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 volume ratios were performed in all the measurements. For the light scattering detector at a scattering angle of 90°, both the excitation and emission wavelengths were adjusted to 450 nm with 2 mm slits. All the measurements were performed at room temperature.
:10 volume ratios were performed in all the measurements. For the light scattering detector at a scattering angle of 90°, both the excitation and emission wavelengths were adjusted to 450 nm with 2 mm slits. All the measurements were performed at room temperature.
Synthesis of thiol-modified silica particles (SiO2-SH)
Ethanol (250 mL), distilled water (5 mL) and ammonia solution (20 mL, 25%) were added into a 500 mL flask. Under stirring at 40 °C, 15 mL of TEOS was added to the solution and the mixture was stirred at 40 °C overnight. SiO2 particles were collected by centrifugation (8000 rpm min−1, 10 min) and washed repeatedly with ethanol and water.In a 100 mL three-necked flask, SiO2 particles (1 g) were dispersed in 30 mL of toluene. MPTMS (1 mL, 5.3 mmol) and pyridine (1 mL, 12.3 mmol) were added dropwise. The suspension was refluxed at 115 °C for 24 h under an argon atmosphere. SiO2-SH particles were collected by centrifugation (8000 rpm min−1, 10 min) and washed with methanol for five times. The particles were dried in a vacuum oven at reduced pressure.
Synthesis of pyridyl disulfide-modified silica particles (SiO2-SS-Py)
The SiO2-SH particles (1 g) were ultrasonically dispersed in 10 mL of methanol, and 2,2′-dithiodipyridine (1.12 g) was added into the solution under an argon atmosphere. After three freeze–pump–thaw cycles, thiol–disulfide exchange reaction was performed at room temperature overnight. After the reaction, a colour change of the solution from milk-white to yellow was observed. SiO2-SS-Py particles were collected by centrifugation (10000 rpm, 10 min), and washed repeatedly with methanol.
The surface density of pyridyl disulfide groups on SiO2-SS-Py particles was determined by UV-vis. Pyridyl-SS-SiO2 (10 mg) was ultrasonically dispersed in DMF (1 mL), and tributylphosphine (20 μL) was added into the solution after two freeze–pump–thaw cycles. The solution was stirred at room temperature for 12 h. After centrifugation (10000 rpm, 10 min), the supernatant was diluted to a volume of 10 mL for UV-vis measurement. Pyridine-2-thione produced in the thiol–disulfide exchange reaction has absorption at 375 nm in DMF. The pyridine-2-thione content in the supernatant and the surface density of pyridyl disulfide groups on SiO2-SS-Py particles were calculated based on the UV-vis result. Our calculation result indicated that the surface density was about 1.22 molecules per nm2.
Synthesis of PDMAEMA-b-POEGMA block copolymers by reversible addition–fragmentation chain transfer (RAFT) polymerization
In the first step, a PDMAEMA macromolecular chain transfer agent (macro-CTA) was synthesized. DMAEMA (5.00 g, 31.8 mmol), ABCPA (17.8 mg, 0.0636 mmol) and the RAFT chain transfer agent CPADB (88.9 mg, 0.318 mmol) were dissolved in 15 mL of 1,4-dioxane in a 50 mL Schlenk flask. After three freeze–pump–thaw cycles, RAFT polymerization of DMAEMA was performed at 70 °C for 10 h. The polymerization was stopped by quenching the Schlenk flask in ice water. PDMAEMA was precipitated in n-hexane and pink polymer was obtained after drying in a vacuum oven under reduced pressure.PDMAEMA-b-POEGMA block copolymers with different POEGMA chain lengths were synthesized by RAFT polymerization using PDMAEMA as the macro-CTA. A typical RAFT polymerization is as follows. OEGMA (4.00 g, 8.00 mmol), ABCPA (2.80 mg, 0.01 mmol), and the PDMAEMA macro-CTA (0.854 g) were dissolved in 5 mL of 1,4-dioxane in a 25 mL Schlenk flask. After three freeze–pump–thaw cycles, RAFT polymerization of OEGMA was performed at 65 °C for 6 h. After the RAFT polymerization, a PDMAEMA-b-POEGMA block copolymer was precipitated in an n-hexane/ether mixture (3/2, v/v).
Preparation of PDMAEMA-b-POEGMA polymer brushes on the surfaces of silica particles
PDMAEMA-b-POEGMA block copolymers (180 mg) and SiO2-SS-Py particles (200 mg) were ultrasonically dissolved/dispersed in 3 mL of THF in a 10 mL Schlenk flask. After two freeze–pump–thaw cycles, n-hexylamine (40 μL, 0.30 mmol) was added into the solution and the solution was stirred at room temperature for 24 h. The dithiocarbonate moieties at the ends of the block copolymer chains were reduced to thiol groups by n-hexylamine, which results in a clear colour change of the solution from pink to milky white. Block copolymer brushes on silica particles were prepared by thiol–disulfide exchange reaction. The grafting density of block polymer brushes on silica particles can be calculated according to the amount of pyridine-2-thione produced in the exchange reaction.After the thiol–disulfide reaction, silica particles with block copolymer brushes were collected by centrifugation (10000 rpm, 10 min), and washed repeatedly with THF to remove excess linear block copolymer chains.
Preparation of surface micelles (s-micelles) on the surfaces of silica particles
Silica nanoparticles with PDMAEMA-b-POEGMA brushes (200 mg) were ultrasonically dispersed in 50 mL of THF, and the solution was stirred at room temperature for 2 h. Methyl iodide (0.40 μL) in 50 mL of THF was slowly added to the particle solution and the solution was stirred at 40 °C for 2 h in the dark, and then 1,4-diiodobutane (0.50 μL) in 50 mL of THF and methyl iodide (1.50 μL) in 50 mL of THF were successively added to the solution and the solution was stirred at 40 °C for 24 h. After the reaction, silica particles with s-micelles on the surfaces were collected by centrifugation (10000 rpm, 10 min), and dried at reduced pressure.
Cleavage of the s-micelles from the surfaces of the silica particles
Silica particles with s-micelles on the surfaces (100 mg) were dispersed in 8.0 mL of THF, and 1.0 mg of BPO was added to the solution. After three freeze–pump–thaw cycles, the cleavage reaction was carried out at 65 °C for 10 h. After the reaction, THF was removed on an evaporator and the solid was dispersed in water. After centrifugation (12000 rpm, 10 min), the cleaved s-micelles were left in the supernatant and the silica nanoparticles were at the bottom of the centrifuge tube. After freeze drying, solid s-micelles were obtained.
Self-assembly of patchy micelles in THF
Cleaved s-micelles (or patchy micelles, 15 mg) were dissolved in 1 mL of water, and 10 mL of THF was added into the solution very slowly. It took about 2 h to add all THF into the patchy micellar solution.Cross-linking of the self-assemblies was performed by adding 1 mg of erythrosin B (2′,4′,5′,7′-tetraiodofluorescein disodium salt) into 1 mL of the above solution. The solution was stirred at room temperature overnight, and dialyzed against water using dialysis tubing (MWCO = 5 kDa) to remove THF and excess erythrosin B.
The study on the reversible ultrasound responsivity of the self-assemblies formed by patchy micelles was performed in an ultrasonic bath (power 200 W) operated at a frequency of 40 kHz.
Results and discussion
PDMAEMA-b-POEGMA block copolymers were synthesized by two-step RAFT polymerization. In this research three block copolymers with the same PDMAEMA chain length and different POEGMA block lengths were synthesized. A summary of molecular parameters including the number-average polymerization degrees, apparent molecular weights and dispersities of the block copolymers is provided in Table 1. SEC curves of the block polymers and PDMAEMA precursor are presented in Fig. S1 (ESI†).| Sample | DPPDMAEMAa | DPPOEGMAa | M n (× 103 g mol−1) | PDIb |
|---|---|---|---|---|
| a The number-average degrees of polymerization of PDMAEMA and POEGMA blocks were determined by 1H NMR. b The number-average molecular weights (Mns) and molecular weight dispersities (PDIs) of PDMAEMA and block copolymers were measured by SEC on PMMA standards. | ||||
| PDMAEMA81 | 81 | 11 | 1.26 | |
| PDMAEMA81-b-POEGMA13 | 81 | 13 | 18 | 1.18 |
| PDMAEMA81-b-POEGMA37 | 81 | 37 | 30 | 1.16 |
| PDMAEMA81-b-POEGMA92 | 81 | 92 | 45 | 1.16 |
Silica particles with an average size of 175 nm were prepared by the Stöber method.50 Pyridyl disulfide-modified silica particles were prepared by using a method reported previously.49,51 The grafting density of pyridyl disulfide groups on silica particles was determined to be about 1.22 molecules per nm2.49 The dithiocarbonate moieties at the ends of the PDMAEMA-b-POEGMA block copolymer chains were treated with hexylamine, and thiol-terminated PDMAEMA-b-POEGMA block copolymers were prepared. The thiol-terminated PDMAEMA-b-POEGMA block copolymers were reacted with pyridyl disulfide groups on silica particles and PDMAEMA-b-POEGMA block copolymer brushes on silica particles were prepared. The grafting densities of PDMAEMA-b-POEGMA block copolymer brushes were determined by measuring the absorption of pyridine-2-thione produced in the thiol–disulfide exchange reactions. The UV-vis spectra and the calibration curve of pyridine-2-thione are shown in Fig. S2 (ESI†). Based on the UV-vis results, the grafting densities of PDMAEMA81-b-POEGMA13, PDMAEMA81-b-POEGMA37 and PDMAEMA81-b-POEGMA92 on silica particles were calculated to be 0.15, 0.14, and 0.12 molecule per nm2, respectively. Thermogravimetric analysis (TGA) curves of PDMAEMA81-b-POEGMA13, PDMAEMA81-b-POEGMA37 and PDMAEMA81-b-POEGMA92 brushes are shown in Fig. S3 (ESI†). Based on the TGA results, the grafting densities of the three block copolymer brushes are 0.18, 0.15, and 0.12 molecule per nm2. The grafting densities calculated by using the two different methods are quite close.
Silica particles with PDMAEMA-b-POEGMA block copolymer brushes were dispersed in THF solution, and methyl iodide was added into the solution successively. Upon addition of methyl iodide into the solution, some DMAEMA units on the block copolymer brushes were quaternized. Because THF is a precipitant for the q-PDMAEMA, the block copolymer brushes associate into s-micelles with the solvophobic q-PDMAEMA cores and the solvophilic POEGMA coronae.52–54 Further addition of 1,4-diiodobutane and methyl iodide results in cross-linking and full quaternization of the s-micellar cores. Fig. 1a and b show TEM and SEM images of silica particles with s-micelles formed by quaternization of PDMAEMA81-b-POEGMA13 brushes in THF. In the TEM image, the contrast between the s-micelles and the silica particle is produced by methyl iodide and 1,4-diiodobutane used for quaternization of PDMAEMA blocks, so the dark dots represent the cores of the s-micelles. The TEM result indicates that s-micelles with an average size of around 5 nm are formed on the silica particle (Fig. 1a). The SEM result reveals that s-micelles with an average size of 9 nm are produced (Fig. 1b). Only the cores of the s-micelles are observed by TEM, so the average size of the s-micelles measured by TEM is smaller than the value measured by SEM. SEM images of silica particles with s-micelles formed by PDMAEMA81-b-POEGMA37 and PDMAEMA81-b-POEGMA92 are shown in Fig. S4 (ESI†).
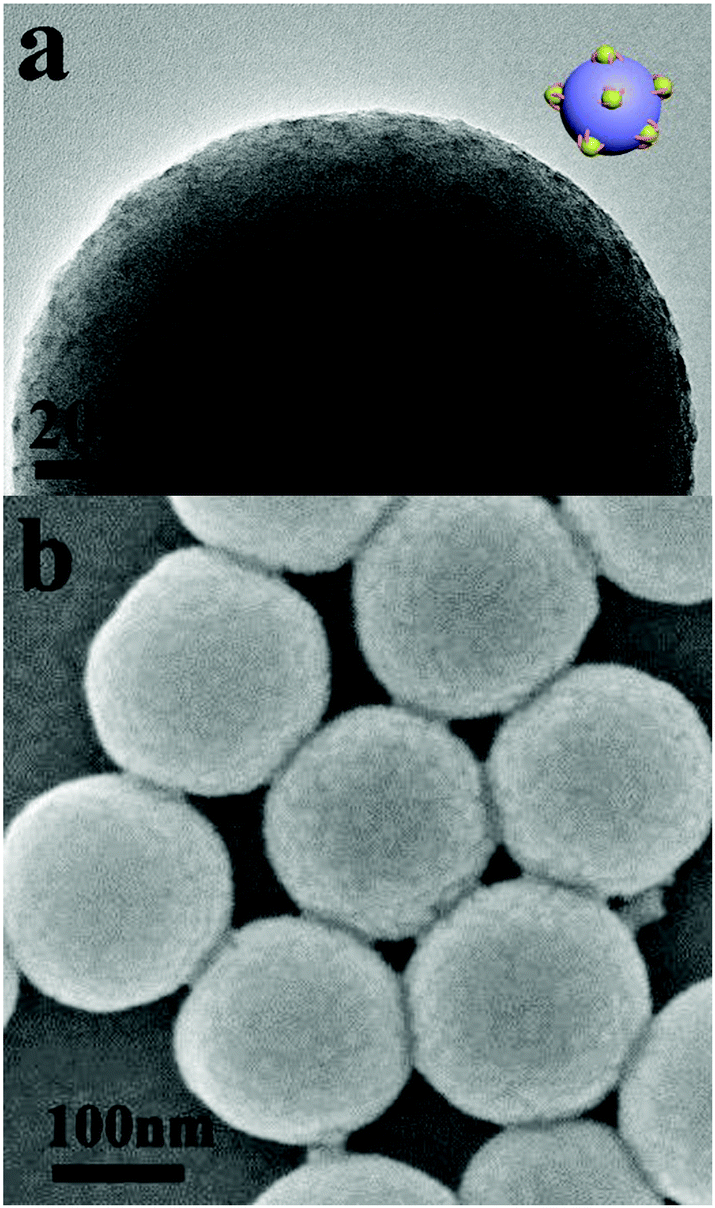 | ||
| Fig. 1 (a) TEM and (b) SEM images of silica particles with s-micelles on their surfaces. The s-micelles are formed by quaternization of PDMAEMA81-b-POEGMA13 brushes in THF solution. A cartoon picture showing the structure of a silica particle with s-micelles on its surface is shown in the inset of (a). | ||
The s-micelles are anchored to the surfaces of the silica particles through disulfide bonds. Upon cleavage of the disulfide bonds, cleaved s-micelles are obtained.55 In this research, disulfide bonds were cleaved under oxidative conditions by using benzoyl peroxide in THF. After the cleavage reaction, THF was removed and water was added into the system. Cleaved s-micelles were collected after removal of silica particles by centrifugation. Fig. 2a shows a TEM image of the cleaved s-micelles formed by quaternized PDMAEMA81-b-POEGMA13. The spherical particles with an average size of 5 nm are observed in the image. The average number of block copolymer chains in a cleaved s-micelle was calculated to be about 3. More TEM images of the s-micelles prepared from quaternized PDMAEMA81-b-POEGMA37, and PDMAEMA81-b-POEGMA92 brushes are shown in Fig. S5 (ESI†), where nano-sized spherical particles are observed. In this paper, the cleaved s-micelles prepared from quaternized PDMAEMA81-b-POEGMA13, PDMAEMA81-b-POEGMA37, and PDMAEMA81-b-POEGMA92 are denoted as PM1, PM2 and PM3, respectively. A cartoon picture of the structure of a cleaved s-micelle is shown in the inset of Fig. 2a. The ζ-potential values of PM1, PM2 and PM3 were measured to be 44.5, 43.9 and 43.2 mV, respectively. Because the three block copolymers have the same PDMAEMA chain length, the ζ-potentials of the cleaved s-micelles are very close.
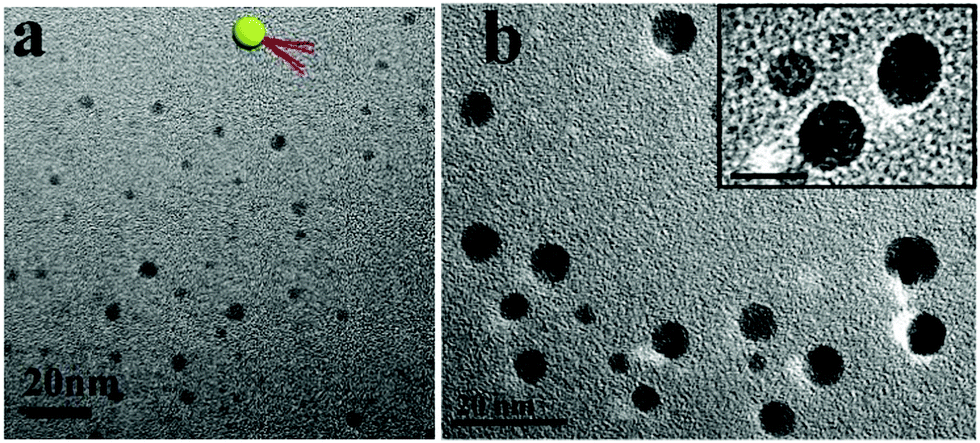 | ||
| Fig. 2 (a) TEM image of the cleaved PM1 s-micelles, and (b) TEM image of the cleaved PM1 s-micelles stained with HAuCl4. A cartoon picture of the structure of a cleaved s-micelle is shown in the inset of (a). A magnified TEM image of stained s-micelles is shown in the inset of (b), where the scale bar represents 10 nm. | ||
To demonstrate the patchy structures of the cleaved s-micelles, traces of HAuCl4 were added to aqueous solutions of the cleaved s-micelles. The negatively charged AuCl4− ions were adsorbed onto the positively charged patches and the structures were stained. Fig. 2b shows a TEM image of stained PM1. The dark nano-sized domains on the particles represent the positively charged patches, demonstrating the synthesis of patchy micelles based on a polymer brush approach. A magnified TEM image of typical patchy micelles is shown in the inset of Fig. 2b, where patchy structures are observed.
THF is a good solvent for POEGMA chains and a precipitant for the positively charged PDMAEMA patches. To prepare the self-assemblies of patchy micelles, a 10-fold volume of THF was added into aqueous solutions of patchy micelles. Fig. 3a–c show TEM images of assemblies formed by PM1, PM2 and PM3, respectively. The TEM results demonstrate that the patchy micelles are able to self-assemble into vesicles. In a vesicle structure, the positively charged PDMAEMA patches aggregate into the wall to avoid the unfavorable interaction with the organic solvent, and the solvophilic POEGMA chains stay in the coronae to stabilize the structure. The repelling electrostatic interaction between the positively charged PDMAEMA patches in the walls is unfavorable for the achievement of low free energy of the assemblies, but it is energetically favourable for the vesicles to overcome energy barriers in the dissociation of the structures under ultrasound irradiation.
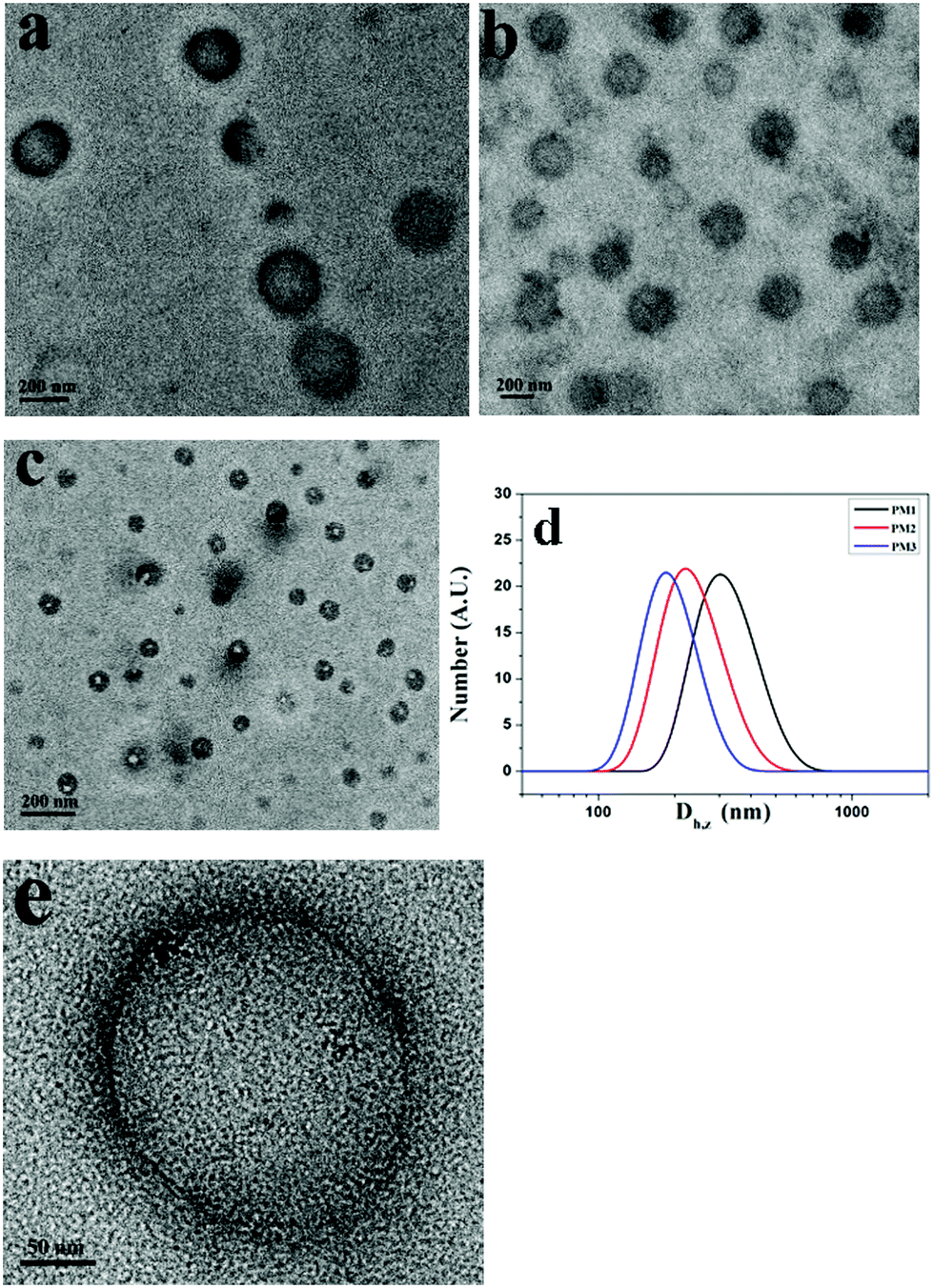 | ||
| Fig. 3 TEM images of vesicles self-assembled from (a) PM1 (b) PM2 and (c) PM3 patchy micelles; (d) DLS curves of the assemblies formed by PM1, PM2 and PM3 patchy micelles, and (e) magnified TEM image of a specific vesicle formed by PM1. | ||
Based on the TEM results, the average sizes of the vesicles formed by PM1, PM2 and PM3 are 250, 218, and 120 nm, respectively, which indicates that the average size of the assemblies is inversely proportional to the POEGMA chain length. This is also confirmed by DLS results. Fig. 3d shows DLS curves of the vesicles self-assembled from PM1, PM2 and PM3, respectively. The Z-average sizes (Dh,z) of the vesicles are 300, 220, and 180 nm, respectively. In the self-assembly of block copolymers in selective solvents, the free energy is determined by the stretching of the blocks in the cores, the interfacial energy between the blocks in the cores and the solvent in the medium, and the repulsive interactions among corona chains.56,57 In this research, the surface charges of the three different patchy micelles are very close, and the structural difference of the patchy micelles is the POEGMA chain length. PM3 patchy micelles with the longest POEGMA chains tend to self-assemble into structures with the highest curvatures, so that the repulsive interactions among POEGMA chains can be relieved. As a result, the smallest vesicles are obtained by self-assembly of PM3 and the biggest ones are fabricated from PM1. A magnified TEM image of a specific vesicle formed by PM1 is shown in Fig. 3e, where patchy micelles located at the membrane can be observed, demonstrating the patchy micelles are used as building blocks in the fabrication of vesicles.
Fig. 4a shows UV-vis spectra of PTSA in a solution of PM1 assemblies at different times. Because of its low solubility in water/THF (1/10, v/v), PTSA salt has broad and low absorbance in the solution. By contrast, in the presence of PM1 assemblies the absorbance of PTSA increases with time and absorption peaks at 342, 360 and 379 nm are observed. In the solution of PM1 assemblies, PTSA salt has electrostatic interaction with positively charged PDMAEMA patches in the vesicle walls, which improves the solubility of PTSA in the solution. With the diffusion of PTSA into the walls, more and more PTSA molecules are dissolved in the solution, so the UV absorbance of PTSA increases with time. Fig. 4b shows fluorescence emission spectra of PTSA in the solution of PM1 assemblies measured at different times. The emission intensity of PTSA increases with time due to the diffusion of the PTSA molecules into the walls of the vesicles.
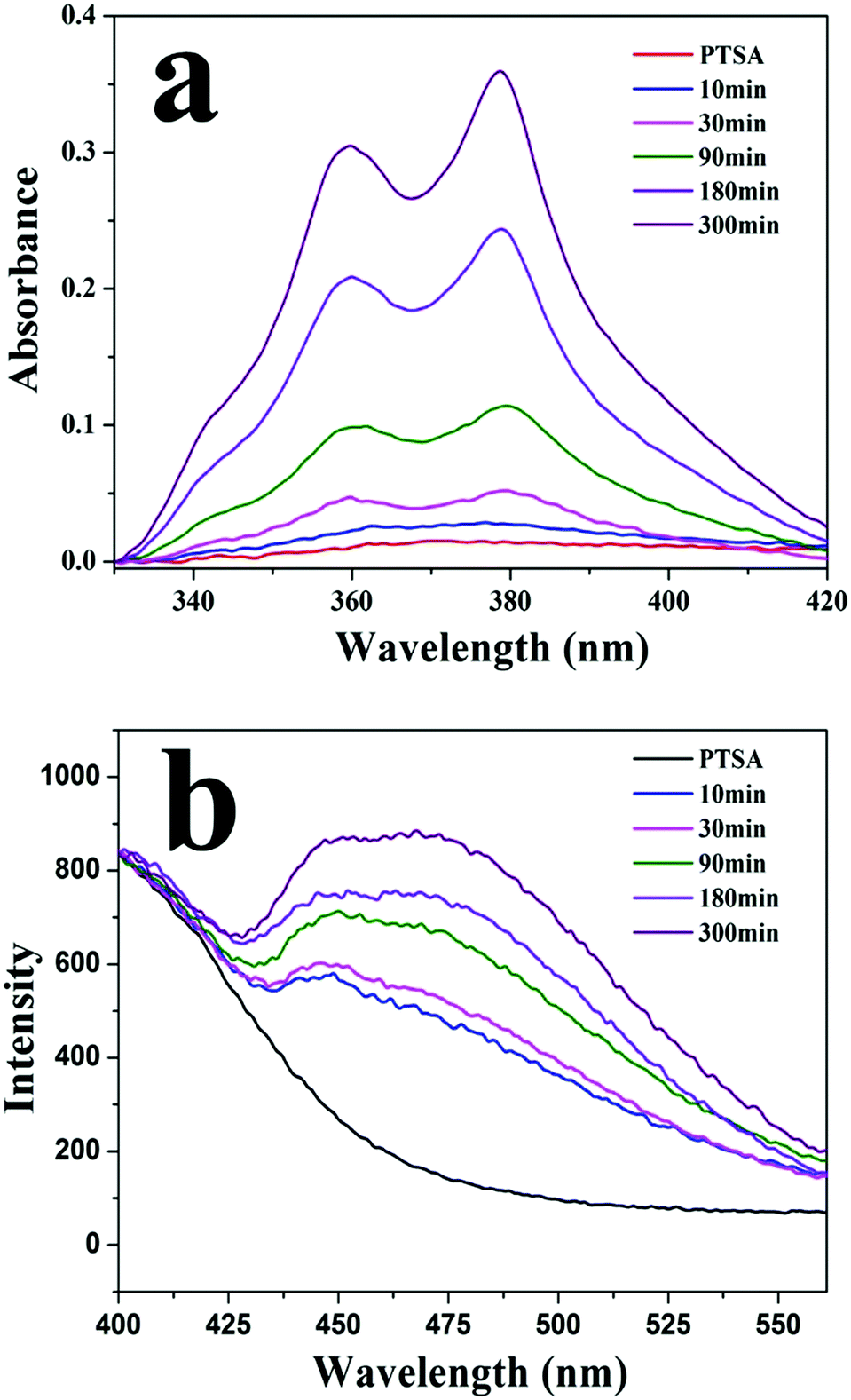 | ||
| Fig. 4 (a) UV-vis and (b) fluorescence spectra of 1,3,6,8-pyrenetetrasulfonic acid tetrasodium salt (PTSA) in a water/THF (1/10, v/v) solution of PM1 assemblies measured at different times. | ||
To explore the self-assembly behaviors of the patchy micelles, the stopped-flow technique was used to study the kinetics of the self-assembly of PM1 patchy micelles. An aqueous solution of PM1 was mixed with THF at a 1:10 volume ratio and the self-assembly process was monitored on a stopped-flow apparatus. The self-assembly process of PM1 was compared with that of the precursor linear polymer, quaternized PDMAEMA81-b-POEGMA13 block copolymer prepared by quaternization of PDMAEMA81-b-POEGMA13 with methyl iodide. Fig. 5a shows the time dependence of the intensities of scattered light upon mixing an aqueous solution of PM1 at 15 mg mL−1 or an aqueous solution of quaternized PDMAEMA81-b-POEGMA13 at the same concentration with a ten-fold volume of THF. The curves in Fig. 5a were fitted with double-exponential functions,
| (I∞ − It)/I∞ = a1e−t/τ1 + a2e−t/τ2 | (1) |
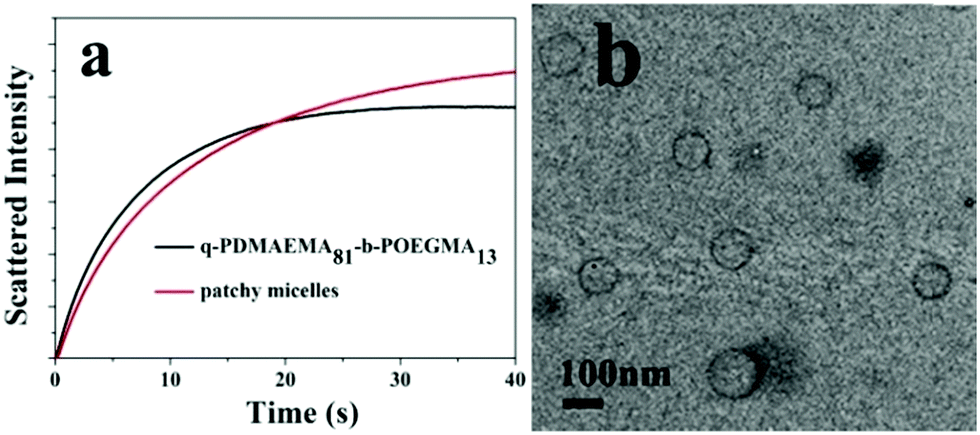 | ||
| Fig. 5 (a) Time-dependent light scattering intensities recorded upon stopped-flow mixing of an aqueous solution of PM1 (or quaternized PDMAEMA81-b-POEGMA13) with a ten-fold volume of THF, and (b) TEM image of self-assemblies of quaternized PDMAEMA81-b-POEGMA13 in a water/THF solution at a 1:10 volume ratio. | ||
Fig. 5b shows a TEM image of assemblies formed by the quaternized PDMAEMA81-b-POEGMA13 linear block copolymer in a water/THF solution. The block copolymer chains self-assemble into vesicles with quaternized PDMAEMA in the walls and POEGMA in the coronae. Although both PM1 and the linear block copolymer self-assemble into vesicles, the average size of the vesicles formed by PM1 (Fig. 3a) is bigger than the average size of the assemblies formed by the linear block copolymer. So the scattering intensity of the PM1 vesicles at equilibrium is higher than that of the block copolymer vesicles (Fig. 5a).
Under sonication self-assemblies formed by patchy micelles experience a transition from an equilibrium state to a non-equilibrium state due to the energy input to the system, and transient nanostructures are formed. The transient nanostructures formed in the non-equilibrium state go back to the equilibrium state when the energy supply is stopped, and vesicle structures are formed. The repelling electrostatic interaction between the positively charged PDMAEMA patches in the walls decreases the energy barriers associated with the dissociation of vesicles under sonication, so the vesicles formed by positively charged PM are supposed to have high ultrasound responsivity. Fig. 6a shows a TEM image of assemblies formed by PM1 after 15 min sonication. In the image aggregates with sizes in the range of 13–25 nm, as well as isolated PM particles with an average size of around 5 nm, are observed, which demonstrates that the positively charged patchy micelles in organic solution can make transient reorganization under ultrasound irradiation, resulting in a morphological change from big-sized vesicles to small-sized aggregates and isolated particles. A typical aggregate and isolated PM particles are indicated by a red and a yellow circle in Fig. 6a, respectively. The reorganization process is shown in Scheme 1. Fig. 6b and c represent TEM images of assemblies formed by PM2 and PM3 after 15 min sonication. In the TEM images transient nanosized aggregates and isolated PM particles are observed. Panels d–f in Fig. 6 show TEM images of PM1, PM2 and PM3 assemblies prepared from solutions standing for 12 h after the ultrasound is switched off. In the TEM images vesicle structures are observed, which demonstrate that the transient nanostructures revert to the original state when the energy input is stopped.
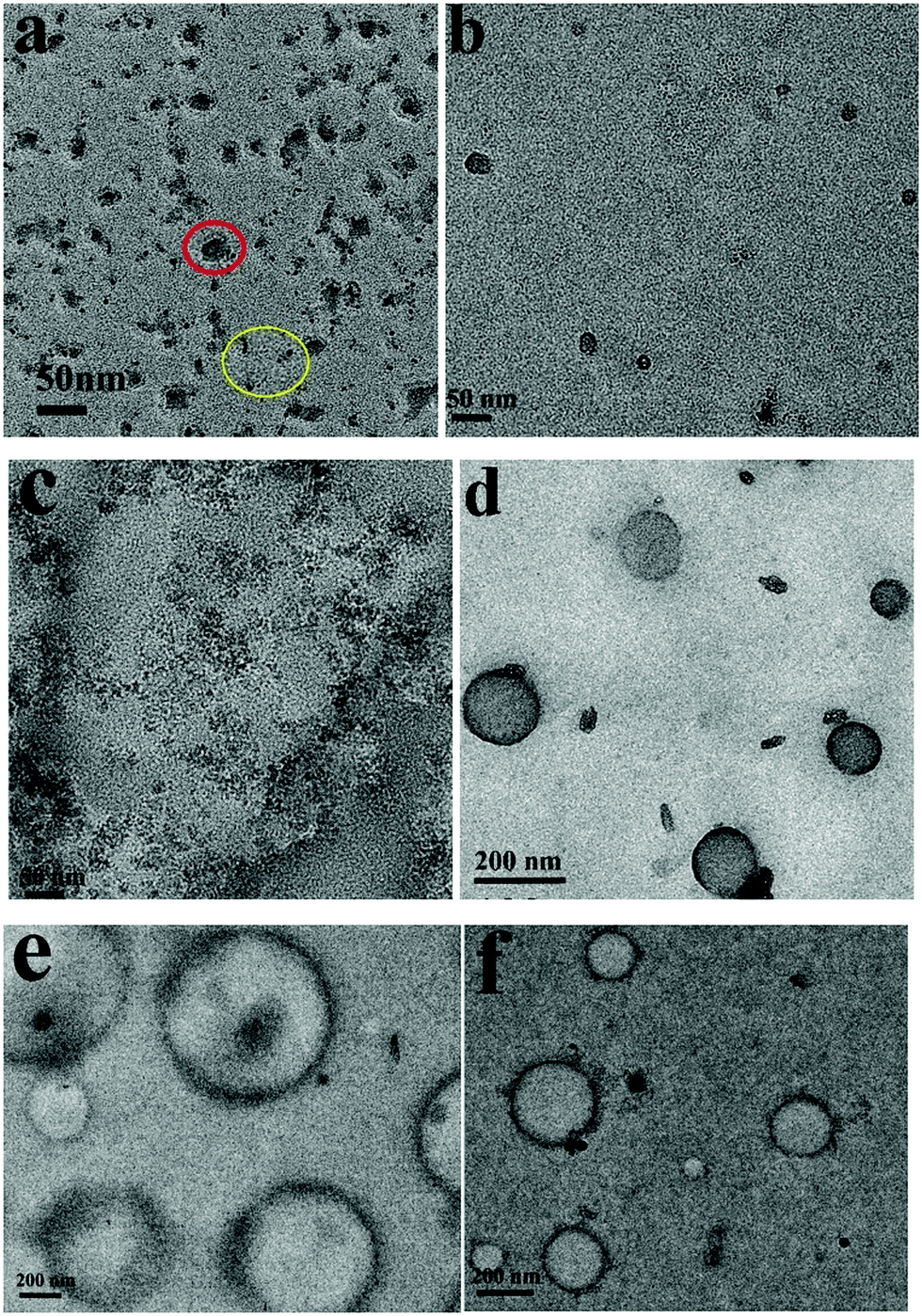 | ||
| Fig. 6 TEM images of assemblies formed by (a and d) PM1, (b and e) PM2 and (c and f) PM3 after 15 minutes ultrasound exposure and when the ultrasound is off. | ||
The reversible ultrasound responsivity of PM particles in a water/THF (1/10, v/v) solution was also confirmed by DLS. Curves 1 and 2 in Fig. 7a represent the DLS curves of PM1 assemblies before and after 15 min ultrasound exposure. The Z-average hydrodynamic diameter of the assemblies decreases from 270 to 45 nm upon ultrasound exposure, indicating the dissociation of the assemblies by ultrasound energy. When the ultrasound is switched off, the average size goes back to 300 nm (curve 3) due to the reorganization of the transient nanostructures into the assemblies. DLS curves of PM2 and PM3 assemblies before and after 15 min ultrasound exposure are shown in Fig. S6a and b (ESI†), and the same phenomena are observed. By contrast, vesicles formed by the quaternized PDMAEMA81-b-POEGMA13 block copolymer do not show ultrasound responsivity due to the chain entanglement in the walls of the vesicles (Fig. S6c, ESI†).
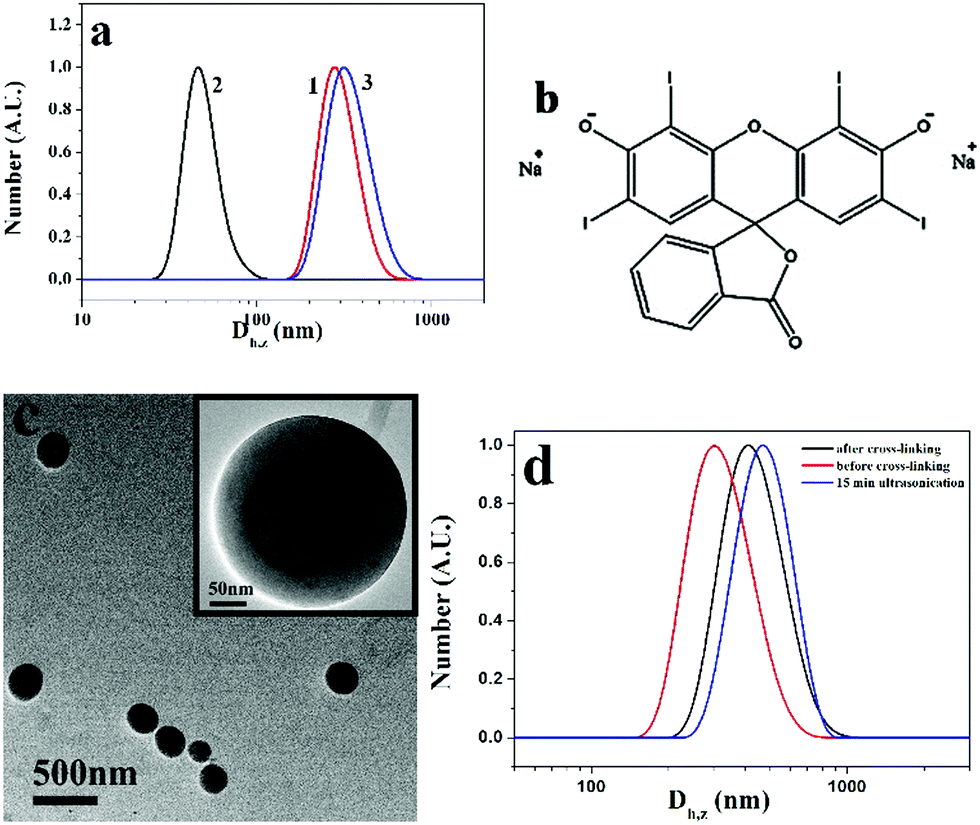 | ||
| Fig. 7 (a) DLS curves of assemblies formed by PM1 particles before (curve 1) and after (curve 2) 15 minutes ultrasound exposure and when the ultrasound is off (curve 3), (b) chemical structure of erythrosin B, (c) TEM image of PM1 vesicles in aqueous solution cross-linked with erythrosin B, and (d) DLS curves of PM1 vesicles before and after cross-linking and the cross-linked vesicles after 15 min ultrasonication treatment. | ||
In the formation of PM vesicles, the positively charged q-PDMAEMA patches are in the walls and the neutral POEGMA chains are in the coronae, so the walls of the vesicles can be cross-linked by negatively charged compounds. Herein, erythrosin B (2′,4′,5′,7′-tetraiodofluorescein disodium salt) was used to cross-link the structures. The chemical structure of the compound is shown in Fig. 7b. After addition of erythrosin B, the solution of PM1 assemblies was dialyzed against water to remove THF. Fig. 7c shows a TEM image of cross-linked PM1 vesicles in aqueous solution. Before cross-linking, PM particles are soluble in water, and no assemblies are formed in water. Upon addition of erythrosin B, the vesicles are cross-linked through electrostatic interaction between negatively charged erythrosin B and positively charged q-PDMAEMA patches in the walls and the vesicle structures are fixed, so the assembled structures are maintained in aqueous solution. It is noted that the assemblies in aqueous solution are bigger than those in water/THF due to the swelling of the membrane in the aqueous solution. The size change was also confirmed by DLS results. In comparison with the vesicles in the water/THF solution, the cross-linked vesicles in the aqueous solution have bigger hydrodynamic diameters (Fig. 7d). Because the structures are fixed with erythrosin B, the cross-linked vesicles do not present ultrasound responsivity. DLS results indicate that the vesicles do not dissociate after 15 min ultrasonication treatment (curve c in Fig. 7d), so no cross-linking structure is necessary in the achievement of reversible ultrasound responsivity for the PM assemblies.
Conclusions
In conclusion, patchy micelles with positively charged PDMAEMA cores partly covered with POEGMA patches were prepared by selective quaternization of PDMAEMA-b-POEGMA block copolymer brushes and followed by cleavage of the s-micelles from the surfaces of the silica particles. In a water/THF mixture (1/10, v/v), the patchy micelles are able to self-assemble into vesicles, where q-PDMAEMA patches are in the walls and POEGMA chains are in the coronae. The average size of the vesicles is strongly dependent on the POEGMA chain length. The average size of the vesicles decreases with an increase in the POEGMA chain length. The vesicles formed by patchy micelles show reversible ultrasound responsivity. Under ultrasound irradiation, vesicles dissociate into small-sized aggregates and isolated particles. When the ultrasound is switched off, the transient nanostructures go back to the vesicle structures. This research provides a new approach for the synthesis of patchy particle assemblies with reversible ultrasound responsivity, and the self-assemblies may find application as a platform for drug/protein delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (NSFC, 51673098 and 51473079) and the National Basic Research Program of China (973 Program, 2012CB821500).Notes and references
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- J. Hu, S. Zhou, Y. Sun, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 4356–4378 RSC.
- S. Jiang, Q. Chen, M. Tripathy, E. Luijten, K. S. Schweizer and S. Granick, Adv. Mater., 2010, 22, 1060–1071 CrossRef CAS PubMed.
- J. Du and R. K. O’Reilly, Chem. Soc. Rev., 2011, 40, 2402–2416 RSC.
- S. C. Glotzer and A. M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- G. Loget and A. Kuhn, J. Mater. Chem., 2012, 22, 15457–15474 RSC.
- A. Perro, S. Reculusa, S. Ravaine, E. Bourgeat-Lami and E. Duguet, J. Mater. Chem., 2005, 15, 3745–3760 RSC.
- T. Zhou, B. Dong, H. Qi, S. Mei and C. Y. Li, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1620–1640 CrossRef CAS.
- I. Jo, S. Lee, J. Zhu, T. S. Shim and G. Yi, Curr. Opin. Colloid Interface Sci., 2017, 30, 97–105 CrossRef CAS.
- R. Deng, F. Liang, J. Zhu and Z. Yang, Mater. Chem. Front., 2017, 1, 431–443 RSC.
- Y. Zhang and H. Zhao, Langmuir, 2016, 32, 3567–3579 CrossRef CAS PubMed.
- A. B. Pawar and I. Kretzschmar, Macromol. Rapid Commun., 2010, 31, 150–168 CrossRef CAS PubMed.
- F. Li, D. P. Josephson and A. Stein, Angew. Chem., Int. Ed., 2011, 50, 360–388 CrossRef CAS PubMed.
- J. He, Y. Liu, T. C. Hood, P. Zhang, J. Gong and Z. Nie, Nanoscale, 2013, 5, 5151–5166 RSC.
- S. Liu, S. Bai, Y. Zheng, K. W. Shah and M. Han, ChemCatChem, 2012, 4, 1462–1484 CrossRef CAS.
- S. Park, J. H. Lim, S. W. Chung and C. A. Mirkin, Science, 2004, 303, 348–351 CrossRef CAS PubMed.
- J. He, B. Yu, M. J. Hourwitz, Y. Liu, M. T. Perez, J. Yang and Z. Nie, Angew. Chem., Int. Ed., 2012, 51, 3628–3633 CrossRef CAS PubMed.
- P. A. Rupar, L. Chabanne, M. A. Winnik and I. Manners, Science, 2012, 337, 559–562 CrossRef CAS PubMed.
- J. Zhang, X. Wang, D. Wu, L. Liu and H. Zhao, Chem. Mater., 2009, 21, 4012–4018 CrossRef CAS.
- J. Zhang, J. Jin and H. Zhao, Langmuir, 2009, 25, 6431–6437 CrossRef CAS PubMed.
- B. Liu, W. Wei, X. Qu and Z. Yang, Angew. Chem., Int. Ed., 2008, 120, 4037–4039 CrossRef.
- J. Yan, C. B. Sung and S. Granick, Adv. Mater., 2015, 27, 874–879 CrossRef CAS PubMed.
- G. Liu, J. Tian, X. Zhang and H. Zhao, Chem. – Asian J., 2014, 9, 2597–2603 CrossRef CAS PubMed.
- B. Wang, B. Li, B. Zhao and C. Y. Li, J. Am. Chem. Soc., 2008, 130, 11594–11595 CrossRef CAS PubMed.
- B. Li, C. Ni and C. Y. Li, Macromolecules, 2008, 41, 149–155 CrossRef CAS.
- Y. K. Takahara, S. Ikeda, S. Ishion, K. Tachi, K. Ikeue, T. Sakata, T. Hasegawa, H. Mori, M. Matsumura and B. Ohtani, J. Am. Chem. Soc., 2005, 127, 6271–6275 CrossRef CAS PubMed.
- Z. Nie, W. Li, M. Seo, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2006, 128, 9408–9412 CrossRef CAS PubMed.
- J. Liu, G. Liu, M. Zhang, P. Sun and H. Zhao, Macromolecules, 2013, 46, 5974–5984 CrossRef CAS.
- X. Ji, Y. Zhang and H. Zhao, Chem. – Eur. J., 2018, 24, 3005–3012 CrossRef CAS PubMed.
- Y. Jiang, T. I. Löbling, C. Huang, Z. Sun, A. H. E. Müller and T. P. Russell, ACS Appl. Mater. Interfaces, 2017, 9, 33327–33332 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51–56 CrossRef CAS PubMed.
- S. Z. Zhu, Z. W. Li and H. Zhao, Langmuir, 2015, 31, 4129–4136 CrossRef CAS PubMed.
- Z. Nie, D. Fava, E. Kumacheva, S. Zou, G. C. Walker and M. Rubinstein, Nat. Mater., 2007, 6, 609–614 CrossRef CAS PubMed.
- T. L. Nghiem, T. I. Löbling and A. H. Gröschel, Polym. Chem., 2018, 9, 1583–1592 RSC.
- T. Wang, J. Zhuang, J. Lynch, O. Chen, Z. Wang, X. Wang, D. LaMontagne, H. Wu, Z. Wang and Y. C. Cao, Science, 2012, 348, 358–363 CrossRef PubMed.
- Z. Zhang and S. C. Glotzer, Nano Lett., 2004, 4, 1407–1413 CrossRef CAS PubMed.
- Z. W. Li, Y. L. Zhu, Z. Y. Lu and Z. Y. Sun, Soft Mater., 2016, 12, 741–749 RSC.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381–384 CrossRef CAS PubMed.
- F. Romano and F. Sciortino, Nat. Mater., 2011, 10, 171–173 CrossRef CAS PubMed.
- Q. Chen, E. Diesel, J. K. Whitmer, S. C. Bae and E. Luijten, J. Am. Chem. Soc., 2011, 133, 7725–7727 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2018, 89, 61–75 CrossRef.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- Y. Li, K. Xiao, W. Zhu, W. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef CAS PubMed.
- C. I. C. Crucho, ChemMedChem, 2015, 10, 24–38 CrossRef CAS PubMed.
- H. Y. Lin and J. L. Thomas, Langmuir, 2004, 20, 6100–6106 CrossRef CAS PubMed.
- C. G. Pappas, T. Mutasa, P. W. J. M. Frederix, S. Fleming, S. Bai, S. Debnath, S. M. Kelly, A. Gachagan and R. V. Ulijn, Mater. Horiz., 2015, 2, 198–202 RSC.
- F. Zhou, M. Xie and D. Chen, Macromolecules, 2014, 47, 365–372 CrossRef CAS.
- W. Fan, L. Liu and H. Zhao, Angew. Chem., Int. Ed., 2017, 56, 8844–8848 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- W. Hou, Y. Feng, B. Li and H. Zhao, Macromolecules, 2018, 51, 1894–1904 CrossRef CAS.
- B. Zhao, W. J. Brittain, W. Zhou and S. Z. D. Cheng, J. Am. Chem. Soc., 2000, 122, 2407–2408 CrossRef CAS.
- E. B. Zhulina, C. Singh and A. C. Balazs, Macromolecules, 1996, 29, 8254–8259 CrossRef CAS.
- H. Zhao, B. P. Farrell and D. A. Shipp, Polymer, 2004, 45, 4473–4481 CrossRef CAS.
- L. Sun and H. Zhao, Langmuir, 2015, 31, 1867–1973 CrossRef CAS.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429–4440 CrossRef CAS.
- R. Bleul, R. Thiermann and M. Maskos, Macromolecules, 2015, 48, 7396–7409 CrossRef CAS.
- B. Bednár, K. Edwards, M. Almgren, S. Tormod and Z. Tuzar, Makromol. Chem., Rapid Commun., 1988, 9, 785–790 CrossRef.
- Z. Zhu, S. P. Arms and S. Liu, Macromolecules, 2005, 38, 9803–9812 CrossRef CAS.
- J. Yin, S. Shi, J. Hu and S. Liu, Langmuir, 2014, 30, 9551–9559 CrossRef CAS.
- Y. Ju, C. Xing, D. Wu, Y. Wu, L. Wang and H. Zhao, Chem. – Eur. J., 2017, 23, 3366–3374 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: SEC, UV-vis, TGA, SEM and TEM results. See DOI: 10.1039/c8qm00627j |
| ‡ These authors contributed equally to this research. |
| This journal is © the Partner Organisations 2019 |