
Pillar[5]arene pseudo[1]rotaxane-based redox-responsive supramolecular vesicles for controlled drug release†
Ya-Han
Cui
a,
Rong
Deng
 a,
Zheng
Li
a,
Xu-Sheng
Du
a,
Qiong
Jia
a,
Xing-Huo
Wang
a,
Chun-Yu
Wang
b,
Kamel
Meguellati
*a and
Ying-Wei
Yang
*ac
a,
Zheng
Li
a,
Xu-Sheng
Du
a,
Qiong
Jia
a,
Xing-Huo
Wang
a,
Chun-Yu
Wang
b,
Kamel
Meguellati
*a and
Ying-Wei
Yang
*ac
aInternational Joint Research Laboratory of Nano-Micro Architecture Chemistry (NMAC), College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: kamel_m@jlu.edu.cn; ywyang@jlu.edu.cn
bState Key Lab of Supramolecular Structure and Materials, Institute of Theoretical Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China
cThe State Key Laboratory of Refractories and Metallurgy, School of Chemistry & Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China
First published on 7th May 2019
Abstract
Supramolecular chemotherapy represents an emerging research field, where the construction of new supramolecular amphiphilic assemblies for the targeted release of anticancer drugs in a controlled manner is of great importance although still very challenging. In this work, we construct a unique amphiphilic pillar[5]arene-based pseudo[1]rotaxane (PPR) with a dynamic redox-responsive disulfide bond in the self-included linker and employ it as the hydrophobic core entity followed by the covalent attachment of a biocompatible poly(ethylene glycol) (PEG) polymer as a hydrophilic long arm via a DCC/DMAP-mediated coupling reaction. A number of amphiphilic PPR molecules can self-assemble to form uniform supramolecular vesicles with good colloidal stability in an aqueous solution, which can be disassembled in the presence of a high concentration of glutathione (GSH) resulting from the breakage and exchange of the disulfide bonds in the system. Interestingly, the vesicle system is capable of encapsulating anticancer drug doxorubicin (DOX) with a high loading efficiency to realize controlled drug delivery. Significantly, in vitro experiments reveal that the free vesicles possess non-cytotoxicity while the DOX-loaded vesicles can not only achieve a precise release of DOX but also enhance the efficiency of killing cancer cells such as human lung cancer cell line A549. To the best of our knowledge, this is the first use of pseudo[1]rotaxane-based materials to protect a vulnerable responsive bond in drug delivery systems, which specifically realizes zero premature release of drugs, leaving the healthy cells unharmed, and offers a new possibility for the application of supramolecular amphiphiles and molecular machinery in precise cancer therapy.
Introduction
Supramolecular amphiphiles1,2 have attracted growing interest due to the fascinating characteristics of dynamic and reversible nature of noncovalent interactions that endow them with a wide range of potential applications in many research areas including smart drug delivery3–6 and advanced functional nanomaterials.7–10 Representative amphiphiles with good responsiveness to various stimuli, such as pH changes,11–13 enzymes,14,15 and light irradiation,16,17 have been designed and utilized to control drug action and enhance drug internalization efficiency in a tumour region based on the inherent attributes of the target sites in the intracellular microenvironment. Among them, glutathione (GSH)-responsive systems are of special interest, since it has been reported that certain tumorous intracellular compartments are characterized by reductive GSH at concentrations of ca. 2–10 mM, which are 100–1000 times higher than those in extracellular milieu (ca. 2–20 μM).18,19 Therefore, GSH-responsive nanoparticles have been recognized as a promising intracellular drug delivery nanosystem for precise cancer therapy. Although substantial aggregates employing GSH as a trigger for controlled drug release have been constructed,20–23 these nanocarriers could not protect their responsive bonds, typically disulfide bonds, from non-specific damage due to their direct exposure in the reducing environment. As far as we know, the redox substances of cells consist of not only GSH but also thioredoxin reductase (TrxR),24 lysosomal mercaptan reductase (GILT),25 sulfo-aminolactic acid, etc.,26,27 which could also destroy the disulfide bonds, thus causing disassembly of nanoparticles and the premature release or leakage of loaded drugs, resulting in severe side effects toward normal cells, tissues, and organs. Therefore, reasonable protection of the responsive bonds of the drug carriers is essential for precision medicine.As a relatively new class of supramolecular macrocyclic hosts, pillar[n]arenes, have aroused significant attention during the past years on account of their unique structural features and related physicochemical properties.28–33 The inherent nature of rigid structures and the combination of several easily modifiable repeating units in one macrocycle make them suitable for easy preparation and further modification to obtain new functional systems for a broad range of applications in various areas.34–45 Recently, many amphiphilic pillar[5]arene-based functional systems46–54 have been reported since the first amphiphilic pillar[5]arene was reported by Huang and coworkers in 2012.55 Taking advantage of the above-mentioned unique features, we envision that a functional amphiphilic pillar[5]arene system can be designed and synthesized so that one could make full use of the pillarene cavity to protect responsive bonds from unwanted breakage due to unnecessary and unexpected factors. Recently, we have fabricated a functional [1]rotaxane and a typical pseudo[1]rotaxane and successfully applied it to the catalysis of the Knoevenagel reaction and dynamic covalent chemistry.56,57 Therefore, it is highly possible to construct supramolecular amphiphiles based on a pillar[5]arene-pseudo[1]rotaxane with the responsive bonds residing inside the cavity for protection to help further understand the control and mechanism of supramolecular chemotherapy based on functional nanostructured materials. On the other hand, to realize positive treatment outcomes against the intricate physiological conditions, poly(ethylene glycol) (PEG) polymer has been extensively studied in constructing amphiphiles to protect hydrophobic cores, which is favourable for a longer retention time in the blood circulation,7,51,58 thus accordingly, PEG polymer becomes an excellent candidate to be incorporated in our design.
Herein, we report on the fabrication of a facile and efficient GSH-responsive delivery system from a pseudo[1]rotaxane macrocyclic amphiphile based on a PEG-modified pillar[5]arene (Fig. 1). A fragile disulfide bond can be self-included into the cavity of pillar[5]arene, completely shielded, and protected as well, by forming a stable pillarene-based pseudo[1]rotaxane that can act as a hydrophobic core component with the additionally attached PEG chain as a potential hydrophilic shell entity for further self-assembly. Interestingly, these amphiphiles would undergo a typical self-assembly process to produce supramolecular vesicles under aqueous conditions and also encapsulate hydrophobic cargo with a high loading capability to form cargo-loaded nanoparticles. When entering into certain cancer cells such as the lung cancer cell line A549, these nanoparticles will dissociate due to the presence of high level GSH that cleaves the –S–S– bonds. Accompanied by the disassembly of these vesicles, the drug molecules loaded inside the vesicles can be released followed by effective killing of the cancer cells, which makes it a versatile platform for both targeted and efficient intracellular pharmaceutical cargo release in a controlled manner. This represents a good example emphasizing the importance of protecting responsive bonds in dealing with the unfavourable premature drug release and achieving controlled delivery and accurate release of anticancer drugs.
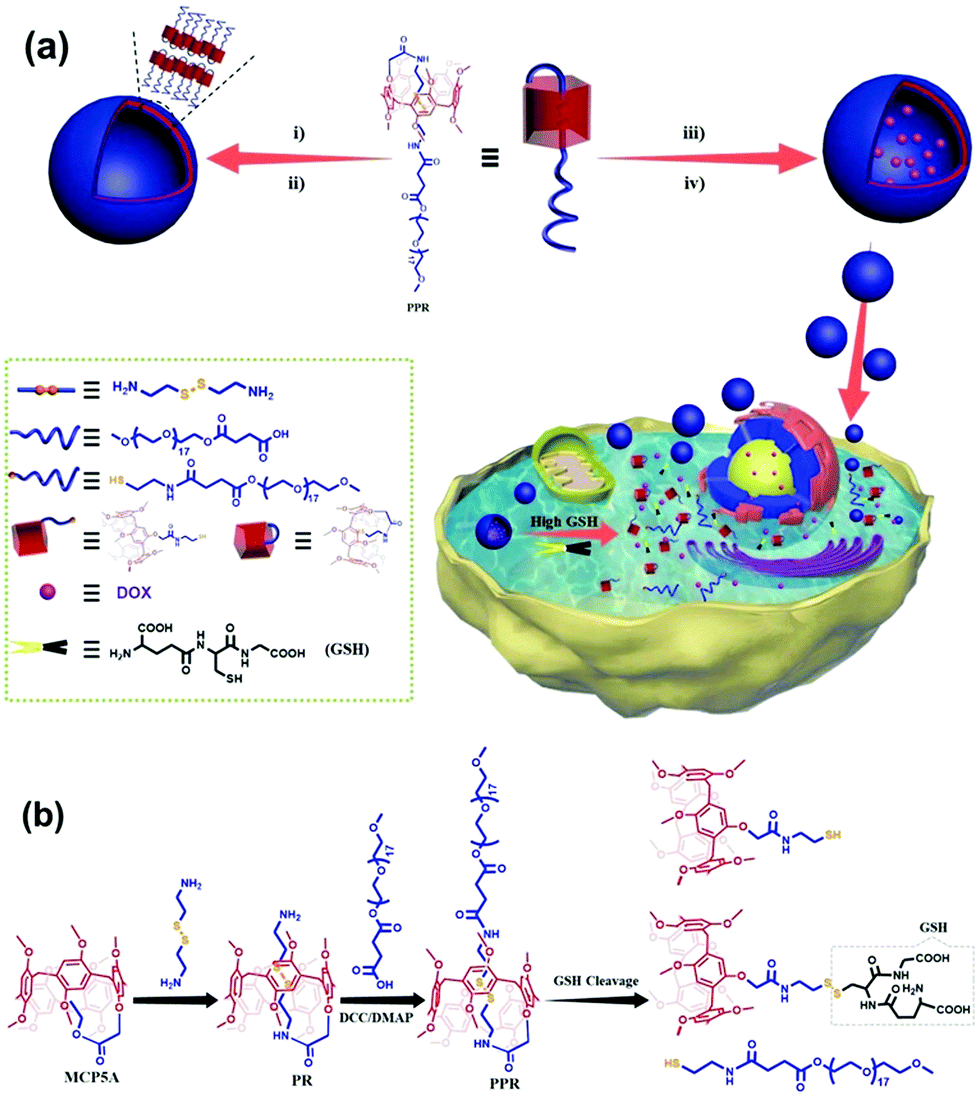 | ||
| Fig. 1 (a) Illustration of the construction of a redox-responsive smart vesicular nanocarrier, and its loading and controlled release of a hydrophobic drug in response to GSH. (i) Self-assembly of PPR in water, (ii) dialysis, (iii) self-assembly of PPR in water in the presence of doxorubicin (DOX), realizing drug loading in one pot, (iv) dialysis; note: 17 in the structure of PPR is the average number, and for details, see the Experimental section; (b) synthetic route to the amine-terminated pillar[5]arene-based pseudo[1]rotaxane (PR) and the amphiphilic pillar[5]arene-based pseudo[1]rotaxane (PPR), and the GSH-responsive process, showing the cleavage of the disulfide bond of PPR that is responsible for aggregate disassembly and drug release. | ||
Results and discussion
Synthesis and characterization
The amine-terminated pillar[5]arene-based pseudo-[1]rotaxane (PR) was first synthesized via the aminolysis of a pillar[5]arene precursor (MCP5A) by cystamine (Fig. 1b). For comparison, ethylmethoxy phenoxy acetate was also exposed to aminolysis by cystamine to give the corresponding monomer. As shown in Fig. 2a, the resonance signals of the protons on PR exhibited upfield shifts in comparison with the free monomer, which can be attributed to the self-inclusion of the chain of cystamine attached onto the pillar[5]arene macrocycle, forming a pseudo[1]rotaxane. 2D NMR experiment was conducted to further verify the configuration of pseudo[1]rotaxane. With the aid of analysis on the 1H–1H COSY and 1H–13C HSQC spectra of PR (Fig. S9 and S10, ESI†) besides the 1H NMR spectrum in Fig. 2a, we can easily assign the side arm peaks of PR and further identify that the methylene peaks of Hb–c shifted upfield obviously, so the Hb–c must be in a highly shielded environment, that is, in the cavity of pillar[5]arene. The partial 2D NOESY spectrum in Fig. 1b clearly shows that the protons of the side chain of PR are close to pillarene itself because of the strong correlations of Ha–d with –OCH3 and ArH. The –NH– from the amide group is close to Ha–b and –OCH3 and ArH as well. Therefore, we can conclude that PR is in a conformation of pseudo[1]rotaxane, that is, the disulfide bond between Hb and Hc has been fully sheltered by the pillar[5]arene cavity. Along this line, the chemical shift values of the 1H NMR spectra of PR at different concentrations also confirmed the self-included structure of PR in CDCl3 which exhibited high stability in the solution state (Fig. S18, ESI†).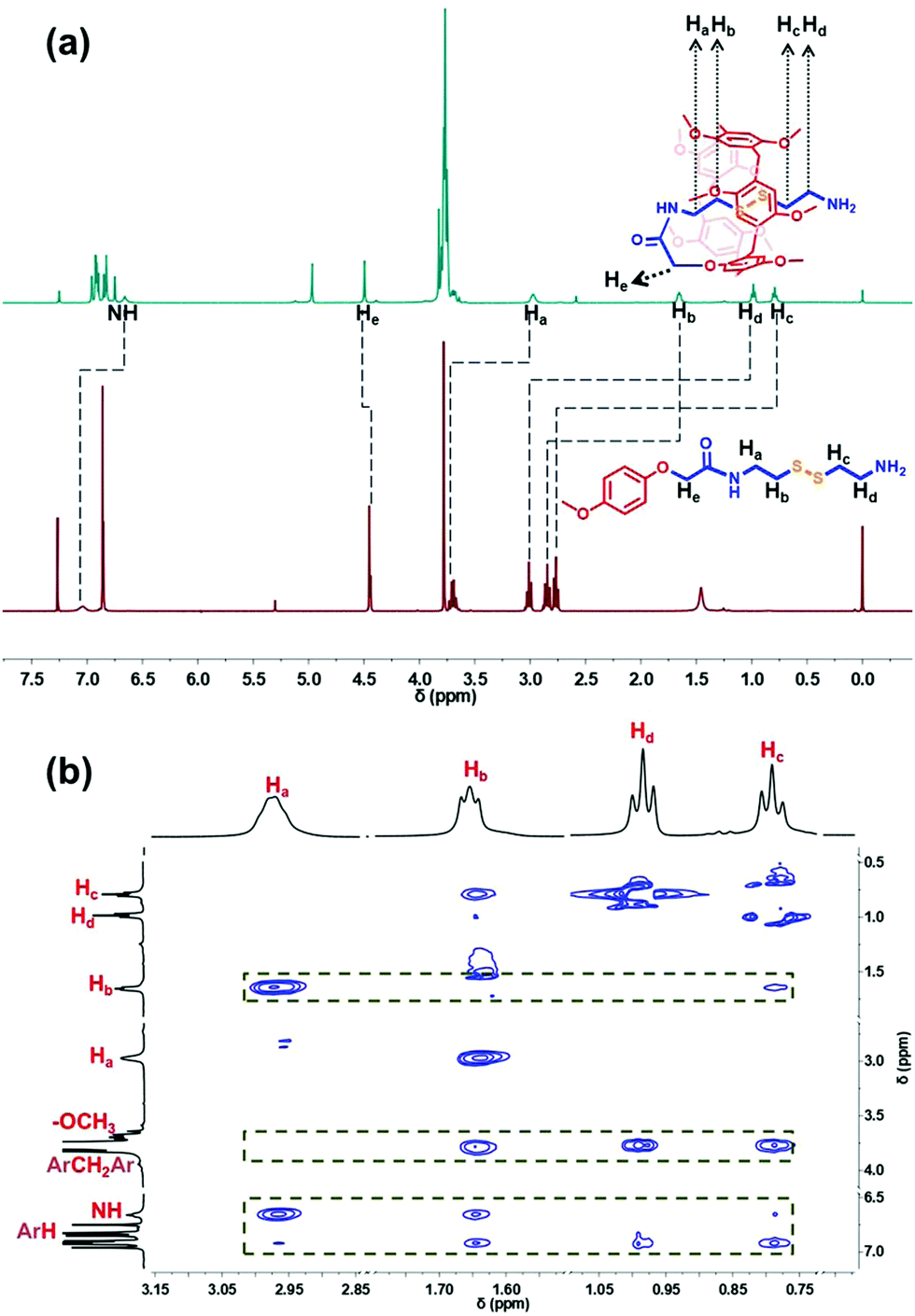 | ||
| Fig. 2 (a) 1H NMR spectra (400 MHz, CDCl3, 298 K) of the monomer and PR; (b) partial NOESY spectrum of PR. | ||
This interesting observation promotes us to develop a potential new application of pseudo[1]rotaxane in protecting responsive bonds. To clarify the protective capability of pillarene rings, further functionalization of PR with a PEG polymer chain by covalent bonds induced the formation of an amphiphilic pillar[5]arene-based pseudo[1]rotaxane (PPR) as a novel nanocarrier upon self-assembly for controlled drug release. By virtue of the comparison of the 1H NMR spectra of PR and PPR, a negligible change could be found in the chemical shifts of Ha–b, which further suggests that the inner part of the side arms within the cavity of pillar[5]arene can keep a stable self-included conformation. In addition, self-inclusion of Ha–b in the cavity of pillar[5]arene was also verified by IR spectroscopy, which was also used by others to check the conformation of mono-amide derivatives of pillar[5]arene (Fig. S20, ESI†).59 The narrow band at 3400 cm−1 resulted from the formation of a strong intramolecular hydrogen bond between the NH proton of the amide fragment and the oxygen atom of the oxymethylene group. The existing intramolecular hydrogen bond stabilized the self-inclusion complexes formed by a variety of monoamide derivatives of pillar[5]arene. The above results are in good agreement with literature reports on the synthesis of monosubstituted pillar[5]arenes incorporating an –OCH2C(O)NH entity.50,59–61 The formation of self-included complexes occurred regardless of the dielectric constant and dipole moment of the solvent.59
Characterization of vesicles
With the above-mentioned stable amphiphilic PPR in hand, we further investigated whether PPR could self-assemble into high-ordered aggregates in an aqueous environment. The self-assembly of PPR was prepared using the nanoprecipitation method. Interestingly, the 1H NMR spectrum of PPR in D2O only shows the proton resonances of the PEG segment, meaning that the pillarene part is shielded (Fig. S19, ESI†). Moreover, the existence of abundant nanoparticles could be strongly confirmed as evidenced by the remarkable Tyndall effect in Fig. 3a. And the critical micelle concentration (CMC) value of the amphiphiles was identified to be 4.4 × 10−2 mg mL−1 (Fig. 3a) in aqueous solution using pyrene as a fluorescent probe.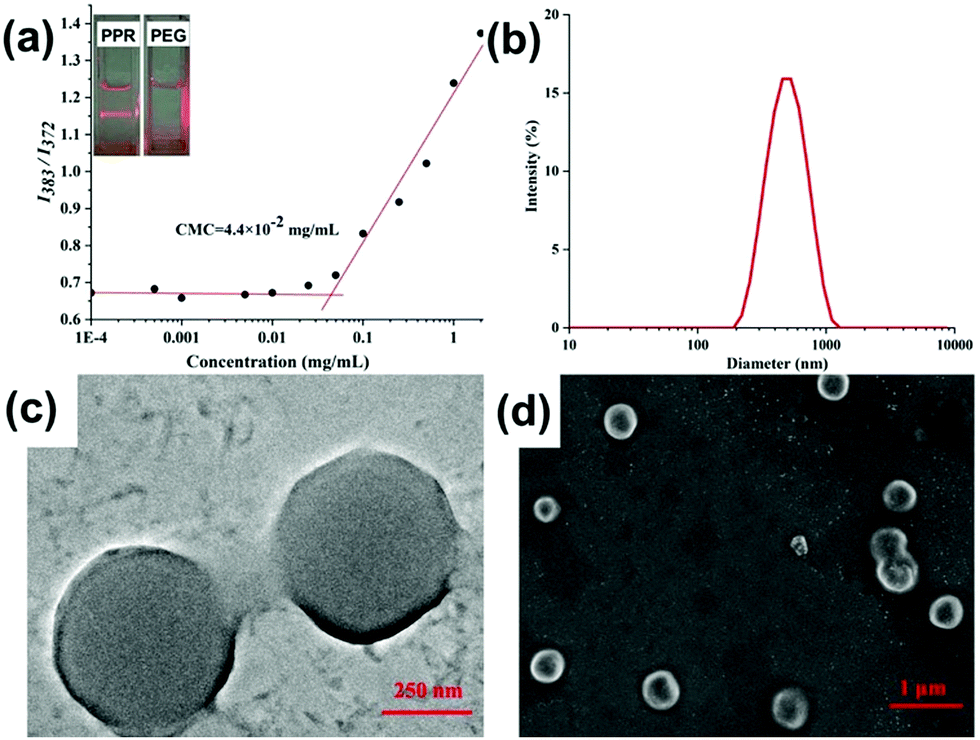 | ||
| Fig. 3 (a) Plot of the I383/I372 ratio with different concentrations of PPR vesicles. The inset images show the Tyndall effect (right: the aqueous solution of pure PEG. Left: The solution of PPR aggregates). (b) DLS result of PPR nanoparticles. (c) The TEM image of PPR vesicles. (d) The SEM image of PPR vesicles. | ||
Subsequently, dynamic light scattering (DLS) and transmission electron microscopy (TEM) were carried out to characterize the size and morphology of the nanoparticles. The DLS result (Fig. 3b) suggests that the formed aggregate has a narrow size distribution located at ca. 490 nm, and the TEM image (Fig. 3c) clearly shows the spherical structure of the amphiphile. Moreover, the periphery and central parts of the spherical structures could be distinguished by an obvious contrast, suggesting the formation of hollow vesicles. Additionally, the formation of vesicles self-assembled from PPR was identified from SEM images (Fig. 3d), showing the spherical structures with an average diameter of ca. 480 nm.
Doxorubicin (DOX) loading and release of the vesicles
To assess the feasibility of using supramolecular vesicles as a drug nanocarrier, we initially encapsulated DOX (we use DOX to represent doxorubicin hydrochloride throughout the paper), a chemotherapeutic drug as a drug model, into the vesicle. The solution changed from colorless to orange (inset image in Fig. 4a), proving the successful loading of DOX. Meanwhile, different from the DOX-free vesicular solution, the absorption intensity of the DOX-containing vesicular solution distributed from 470 to 500 nm becomes significantly stronger, in accordance with the characteristic absorption of DOX (Fig. 4a). In addition, the loading capacity of the DOX-loaded PPR nanocarrier (DPPR) is ca. 21.3%, as determined by further drug loading experiments, confirming a good drug loading capability.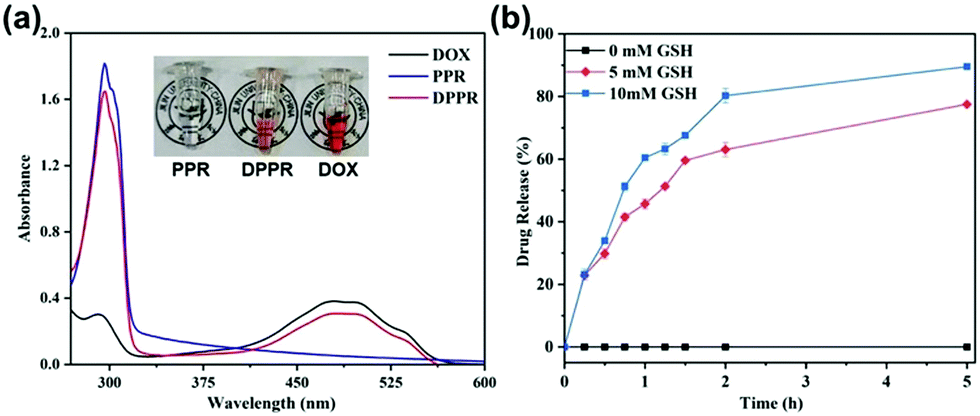 | ||
| Fig. 4 (a) UV-Vis absorption spectra of the solutions of DOX, DOX-free vesicle PPR and DOX-loaded vesicle DPPR at 25 °C in aqueous solutions. Inset images: PPR solution (left), DPPR solution (middle), and DOX solution (right). (b) Drug release profiles of DPPR assemblies in the presence of different concentrations of GSH. | ||
Furthermore, to demonstrate the protective ability of this novel pseudo[1]rotaxane toward disulfide bonds, that is, to explore the stimuli responsiveness of this vesicle, we treated them with different concentrations of GSH (0 mM, 5 mM and 10 mM) to mimic the microenvironment of cancer cells. To our great satisfaction, drug release from DPPR was severely limited in the absence of GSH (Fig. 4b), proving that these particles are very stable under physiological conditions. Coupling with the elevated concentration of GSH, a faster drug release could be found due to the reductive cleavage of disulfide bonds threaded in the pillarene cavity; in other words, higher concentration of GSH tends to cut off the disulfide bonds more easily, thus resulting in the disassembly of PPR vesicles. All of the experimental results illustrated that the DOX-loaded material, DPPR, enabled sensitive drug release in a GSH-responsive manner in the cancerous microenvironment without any premature release due to the excellent protection of pillarene rings, achieving a controlled release capability, therefore paving a promising avenue for the application of pseudo[1]rotaxane-based materials in precision medicine.
In vitro cell cytotoxicity and cellular uptake of nanoparticles
The internalization of DOX-loaded supramolecular vesicles DPPR was further studied by fluorescence microscopy using the A549 cell line. From the fluorescence microscopy images (Fig. 5a), relatively stronger fluorescence was witnessed at the cytoplasmic matrix after 4 h of incubation of DPPR. With an extended incubation time, the fluorescence was observed distinctly in cell nuclei. These results imply that DOX was efficiently released in the microenvironment of cancer cells (cytoplasmic with high GSH concentration), reflecting that the endocytosis of DPPR by A549 cells occurs in a time-dependent manner. Considering the DPPR size of ca. 550 nm (Fig. S26, ESI†) and the soft material nature, we consider they enter the cells mainly through the macropinocytosis endocytosis pathway.62,63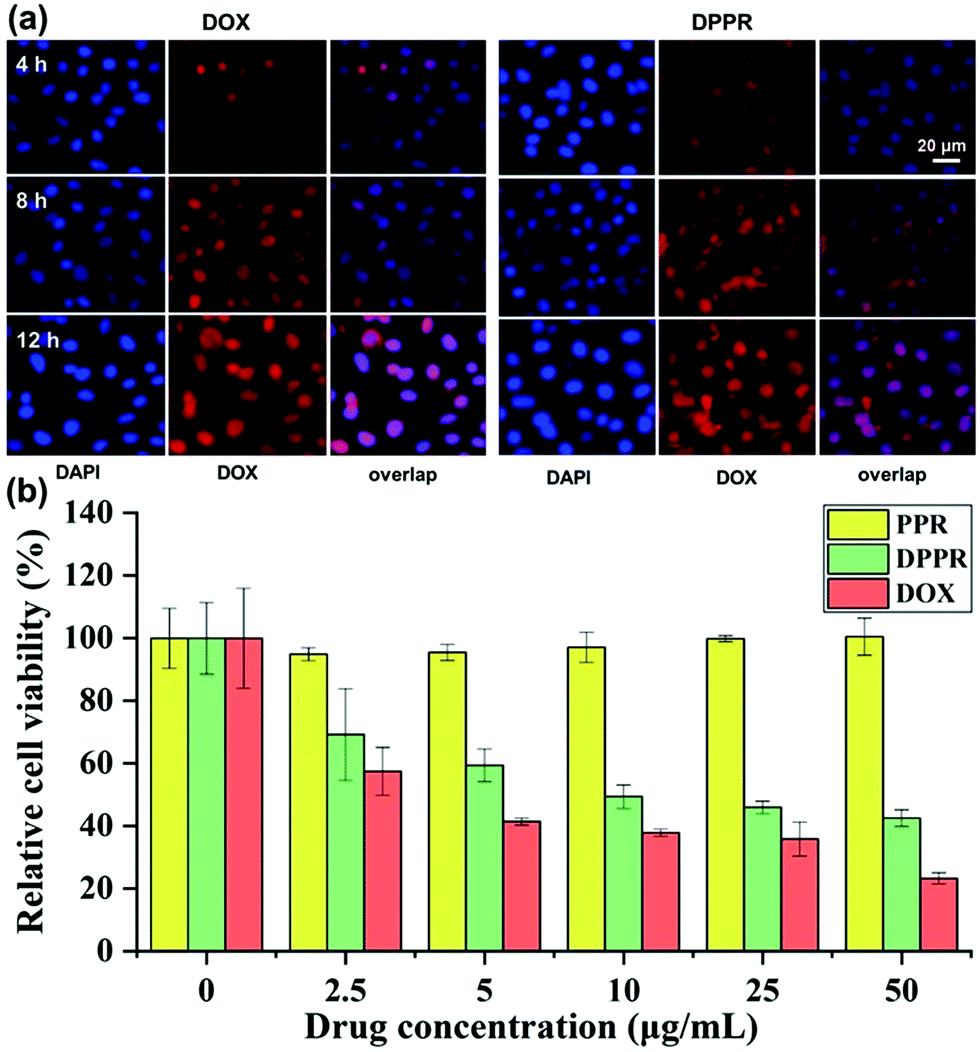 | ||
| Fig. 5 (a) Fluorescence microscope recording the cellular uptake of free DOX and the DPPR platform by A549 cells (blue represents nuclei and red represents DOX). (b) Viability of A549 cells assessed by the CCK-8 assay in the treatment of different concentrations of free DOX, PPR vesicles, and DPPR platform. | ||
To further evaluate the therapeutic effect of the newly developed nanomaterials against cancer cells, we assess the cytotoxicity of PPR and DPPR (the vesicles without and with DOX loaded) on A549 cells using CCK-8 assay. As shown in Fig. 5b, free PPR nanoparticles did not exhibit obvious cytotoxicity to A549 cells even with a higher concentration of 50 μg mL−1, showing the excellent biocompatibility of the assembled vehicles. However, the DPPR nanomaterial loaded with DOX could effectively kill A549 cells with the increase of drug dosage. All of the above-mentioned experimental results verified that these new supramolecular vesicles could serve as rational materials for precise drug delivery.
Conclusions
In summary, we successfully constructed a new PEG-modified pillar[5]arene-based pseudo[1]rotaxane macrocyclic amphiphile, which could self-assemble into spherical supramolecular vesicles in aqueous solution with a low CMC, exhibiting effective and sustained drug release in a reducing environment. Furthermore, considerable drug loading capacity and in vitro release experiments supported that DOX could be successfully encapsulated by such vesicles, and the resulting DOX-loaded vesicles possess not only satisfactory stability but also remarkable GSH-responsiveness, and can release the encapsulated drug DOX in a high-GSH-concentration environment similar to that of tumor cells, making them a suitable material for cancer therapy. Cytotoxicity experiments based on the CCK-8 assay as a proof-of-concept study revealed that drug-free vesicles are biocompatible with a negligible toxic effect on the A549 cell while DOX-loaded vesicles exhibited significant inhibition of cancer cell proliferation. This new strategy will open a new door for the application of supramolecular amphiphile-based molecular machinery in supramolecular chemotherapy and modern nanomedicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (51673084, 21871108), Jilin Province-University Cooperative Construction Project-Special Funds for New Materials (SXGJSF2017-3), Jilin University Talents Cultivation Program, and the Fundamental Research Funds for the Central Universities for financial support.Notes and references
- Z. Xu, S. Jia, W. Wang, Z. Yuan, B. Jan Ravoo and D.-S. Guo, Nat. Chem., 2019, 11, 86–93 CrossRef CAS PubMed.
- C. Wang, Q. Chen, H. Xu, Z. Wang and X. Zhang, Adv. Mater., 2010, 22, 2553–2555 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- H. Zhang, X. Ma, K. T. Nguyen and Y. Zhao, ACS Nano, 2013, 7, 7853–7863 CrossRef CAS PubMed.
- L. Gao, B. Zheng, W. Chen and C. A. Schalley, Chem. Commun., 2015, 51, 14901–14904 RSC.
- Y. Chen, L. Rui, L. Liu and W. Zhang, Polym. Chem., 2016, 7, 3268–3276 RSC.
- L. Rui, L. Liu, Y. Wang, Y. Gao and W. Zhang, ACS Macro Lett., 2016, 5, 112–117 CrossRef CAS.
- J. Sun, Y. Liu, Y. Chen, W. Zhao, Q. Zhai, S. Rathod, Y. Huang, S. Tang, Y. T. Kwon, C. Fernandez, R. Venkataramanan and S. Li, J. Controlled Release, 2017, 258, 43–55 CrossRef CAS PubMed.
- L.-L. Tan, H. Li, Y. Tao, S. X.-A. Zhang, B. Wang and Y.-W. Yang, Adv. Mater., 2014, 26, 7027–7031 CrossRef CAS PubMed.
- W. Si, L. Chen, X.-B. Hu, G. Tang, Z. Chen, J.-L. Hou and Z.-T. Li, Angew. Chem., Int. Ed., 2011, 50, 12564–12568 CrossRef CAS PubMed.
- D.-S. Guo, K. Wang, Y.-X. Wang and Y. Liu, J. Am. Chem. Soc., 2012, 134, 10244–10250 CrossRef CAS PubMed.
- G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489–19497 CrossRef CAS PubMed.
- Q. Duan, Y. Cao, Y. Li, X. Hu, T. Xiao, C. Lin, Y. Pan and L. Wang, J. Am. Chem. Soc., 2013, 135, 10542–10549 CrossRef CAS PubMed.
- Y. Kang, Z. Cai, X. Tang, K. Liu, G. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 4927–4933 CrossRef CAS PubMed.
- J. Li, K. Liu, Y. Han, B. Z. Tang, J. Huang and Y. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 27987–27995 CrossRef CAS PubMed.
- K. M. Park, K. Baek, Y. H. Ko, A. Shrinidhi, J. Murray, W. H. Jang, K. H. Kim, J.-S. Lee, J. Yoo, S. Kim and K. Kim, Angew. Chem., Int. Ed., 2018, 57, 3132–3136 CrossRef CAS PubMed.
- Z. Sun, G. Liu, J. Hu and S. Liu, Biomacromolecules, 2018, 19, 2071–2081 CrossRef CAS PubMed.
- H. F. Gilbert, Methods Enzymol., 1995, 251, 8–28 CAS.
- Y. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- X.-D. Xu, L. Zhao, Q. Qu, J.-G. Wang, H. Shi and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 17371–17380 CrossRef CAS PubMed.
- L. Jiang, X. Huang, D. Chen, H. Yan, X. Li and X. Du, Angew. Chem., Int. Ed., 2017, 56, 2655–2659 CrossRef CAS PubMed.
- Y. Kang, X. Ju, L.-S. Ding, S. Zhang and B.-J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 4475–4484 CrossRef CAS PubMed.
- X. Liu, K. Jia, Y. Wang, W. Shao, C. Yao, L. Peng, D. Zhang, X.-Y. Hu and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 4843–4850 CrossRef CAS PubMed.
- C. V. Smith, D. P. Jones, T. M. Guenthner, L. H. Lash and B. H. Lauterburg, Toxicol. Appl. Pharmacol., 1996, 140, 1–12 CrossRef CAS PubMed.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- Z. Deng, S. Yuan, R. X. Xu, H. Liang and S. Liu, Angew. Chem., Int. Ed., 2018, 130, 9034–9038 CrossRef.
- J. Wu, L. Zhao, X. Xu, N. Bertrand, W. Choi, B. Yameen, J. Shi, V. Shah, M. Mulvale, J. L. MacLean and O. C. Farokhzad, Angew. Chem., Int. Ed., 2015, 54, 9218–9223 CrossRef CAS PubMed.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- W. Cheng, H. Tang, R. Wang, L. Wang, H. Meier and D. Cao, Chem. Commun., 2016, 52, 8075–8078 RSC.
- B. Shi, K. Jie, Y. Zhou, J. Zhou, D. Xia and F. Huang, J. Am. Chem. Soc., 2016, 138, 80–83 CrossRef CAS PubMed.
- S. Wang, Z. Xu, T. Wang, T. Xiao, X.-Y. Hu, Y.-Z. Shen and L. Wang, Toxicol. Appl. Pharmacol., 2018, 9, 1737 Search PubMed.
- J.-F. Chen, Q. Lin, H. Yao, Y.-M. Zhang and T.-B. Wei, Mater. Chem. Front., 2018, 2, 999–1003 RSC.
- H. Zhang and Y. Zhao, Chem. – Eur. J., 2013, 19, 16862–16879 CrossRef CAS PubMed.
- T. Ogoshi, T.-A. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- D. Cao and H. Meier, Asian J. Org. Chem., 2014, 3, 244–262 CrossRef CAS.
- M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- C. Li, Chem. Commun., 2014, 50, 12420–12433 RSC.
- S. Pan, M. Ni, B. Mu, Q. Li, X.-Y. Hu, C. Lin, D. Chen and L. Wang, Adv. Funct. Mater., 2015, 25, 3571–3580 CrossRef CAS.
- Q. Lin, K.-P. Zhong, J.-H. Zhu, L. Ding, J.-X. Su, H. Yao, T.-B. Wei and Y.-M. Zhang, Macromolecules, 2017, 50, 7863–7871 CrossRef CAS.
- N. Song and Y.-W. Yang, Sci. China: Chem., 2014, 57, 1185–1198 CrossRef CAS.
- N. Song, T. Kakuta, T.-A. Yamagishi, Y.-W. Yang and T. Ogoshi, Chem, 2018, 4, 2029–2053 CAS.
- Y. Chen, F. Huang, Z.-T. Li and Y. Liu, Sci. China: Chem., 2018, 61, 979–992 CrossRef CAS.
- G. Yu, J. Zhou, J. Shen, G. Tang and F. Huang, Chem. Sci., 2016, 7, 4073–4078 RSC.
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
- H. Zhang, Z. Liu and Y. Zhao, Chem. Soc. Rev., 2018, 47, 5491–5528 RSC.
- X.-Y. Hu, L. Gao, S. Mosel, M. Ehlers, E. Zellermann, H. Jiang, S. K. Knauer, L. Wang and C. Schmuck, Small, 2018, 14, 1803952 CrossRef PubMed.
- S. Sun, M. Geng, L. Huang, Y. Chen, M. Cen, D. Lu, A. Wang, Y. Wang, Y. Shi and Y. Yao, Chem. Commun., 2018, 54, 13006–13009 RSC.
- J. Zhou, M. Chen and G. Diao, ACS Appl. Mater. Interfaces, 2014, 6, 18538–18542 CrossRef CAS PubMed.
- Y. Han, G.-F. Huo, J. Sun, J. Xie, C.-G. Yan, Y. Zhao, X. Wu, C. Lin and L. Wang, Sci. Rep., 2016, 6, 28748 CrossRef PubMed.
- Y. Zhou, K. Jie and F. Huang, Org. Chem. Front., 2017, 4, 2387–2391 RSC.
- C. Ke, N. L. Strutt, H. Li, X. Hou, K. J. Hartlieb, P. R. McGonigal, Z. Ma, J. Iehl, C. L. Stern, C. Cheng, Z. Zhu, N. A. Vermeulen, T. J. Meade, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17019–17030 CrossRef CAS PubMed.
- Y. Chang, K. Yang, P. Wei, S. Huang, Y. Pei, W. Zhao and Z. Pei, Angew. Chem., Int. Ed., 2014, 53, 13126–13130 CrossRef CAS PubMed.
- K. Yang, Y. Pei, J. Wen and Z. Pei, Chem. Commun., 2016, 52, 9316–9326 RSC.
- Y. Yao, M. Xue, J. Chen, M. Zhang and F. Huang, J. Am. Chem. Soc., 2012, 134, 15712–15715 CrossRef CAS PubMed.
- X.-S. Du, C.-Y. Wang, Q. Jia, R. Deng, H.-S. Tian, H.-Y. Zhang, K. Meguellati and Y.-W. Yang, Chem. Commun., 2017, 53, 5326–5329 RSC.
- X.-S. Du, Q. Jia, C.-Y. Wang, K. Meguellati and Y.-W. Yang, Chem. Commun., 2019, 55, 5736–5739 RSC.
- J. Yao, W. Wu, W. Liang, Y. Feng, D. Zhou, J. J. Chruma, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, Angew. Chem., Int. Ed., 2017, 129, 6973–6977 CrossRef.
- A. A. Nazarova, P. L. Padnya, A. I. Gilyazeva, A. A. Khannanov, V. G. Evtugyn, M. P. Kutyreva, V. V. Klochkov and I. I. Stoikov, New J. Chem., 2018, 42, 19853–19863 RSC.
- Y. Han, Y. Sun, J. Sun and C.-G. Yan, Tetrahedron, 2012, 68, 8256–8260 CrossRef CAS.
- B. Xia and M. Xue, Chem. Commun., 2014, 50, 1021–1023 RSC.
- S. Bhattacharya, D. Roxbury, X. Gong, D. Mukhopadhyay and A. Jagota, Nano Lett., 2012, 12, 1826–1830 CrossRef CAS PubMed.
- L. Chen, H. Wang, X. Li, C. Nie, T. Liang, F. Xie, K. Liu, X. Peng and J. Xie, RSC Adv., 2018, 8, 35246–35256 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization data including 1H NMR spectra, 13C NMR spectra, 2D NMR spectra, and mass spectra of related compounds. See DOI: 10.1039/c9qm00237e |
| This journal is © the Partner Organisations 2019 |