
Unprecedented thiocarbamidation of nitroarenes: a facile one-pot route to unsymmetrical thioureas†
Soumya
Dutta
a,
Manas
Mondal
a,
Tubai
Ghosh
b and
Amit
Saha
 *a
*a
aDepartment of Chemistry, Jadavpur University, Kolkata 700032, India. E-mail: amit.saha@jadavpuruniversity.in
bDepartment of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India
First published on 19th November 2018
Abstract
Aromatic nitro compounds undergo one-pot thiocarbamide functionalization upon reacting with in situ generated dithiocarbamate anions in the presence of K2CO3 and DMF solvent to produce unsymmetrical thiourea compounds in good yields. Various cyclic and acyclic 2° amines, 1° amines, and aromatic amines reacted with CS2 to form dithiocarbamate anions which combine with different nitroarenes resulting in the formation of thiourea compounds. The reaction is assumed to proceed through a nitrosobenzene intermediate.
Introduction
Thiocarbamide or thiourea derivatives are of immense importance due to their potential uses in drug development. Thiocarbamide moieties are found in many biologically active compounds which show anticancer,1 analgesic,2 anti-inflammatory,3 antimicrobial,4 and insect repellent5 properties. In addition, they are extensively valuable as organocatalysts,6 ligands,7 chemosensors,8 textile-treating agents9 and synthetic building blocks.10 Thiourea also serves as a protecting group for various amines.11 The traditional method to synthesize thiourea is the direct or catalyzed addition of primary/secondary amines to isothiocyanate substrates.12 In many cases, isothiocyanates are generated in situ13 during the course of thiourea synthesis. Generally, isothiocyanate compounds and their precursors require tedious synthetic procedures and suffer from long term stability. Therefore, many non-isothiocyanate routes14 have been developed by various research groups. One of the popular methods includes the nucleophilic addition–elimination reaction of amines with the thiocarbonyl group of thiuram disulfide,14e dithiocarbamate anions,14b,c and dithiocarbamic acids14d and its esters14a (Scheme 1). However, the lack of general applicability, time consuming procedures for the preparation of many thiocarbonyl donors, and incidences of side reactions made it necessary to invoke an alternative clean method to prepare thiourea compounds from easily available starting materials. In our present study, we have developed a facile one-pot route to prepare unsymmetrical thiourea derivatives by a unique C–N coupling of nitroarenes with dithiocarbamate anions in DMF solution (Scheme 1).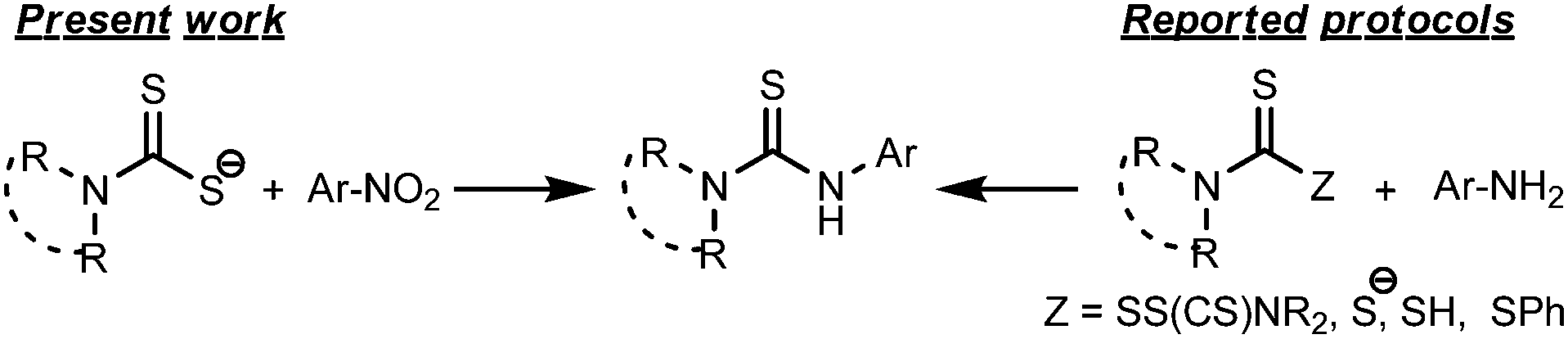 | ||
| Scheme 1 Synthesis of unsymmetrical thioureas. | ||
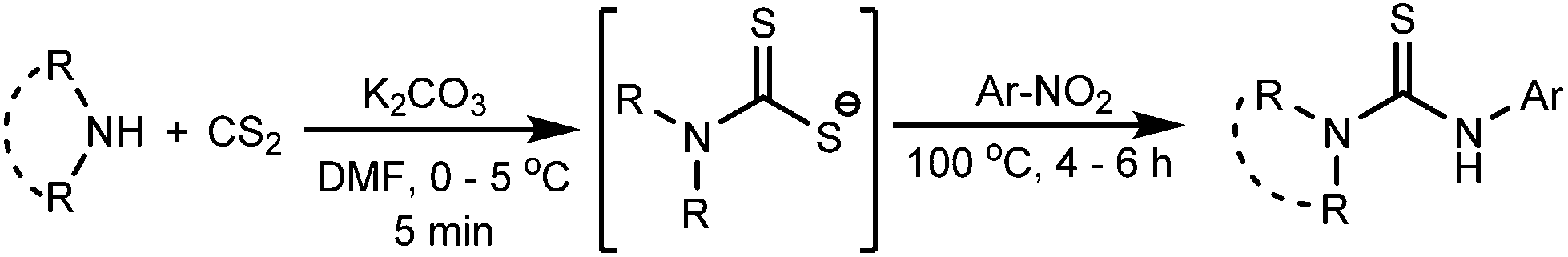
The dithiocarbamate anions, generated in situ at low temperature by the reaction of an amine and carbon disulfide, react with the aromatic nitro compounds in the presence of K2CO3 and DMF solvent at 100 °C to afford the unsymmetrically substituted thiourea compounds in good yields (Scheme 2).
 | ||
| Scheme 2 Thiocarbamidation of aromatic nitro compounds to obtain thiourea derivatives. | ||
Results and discussion
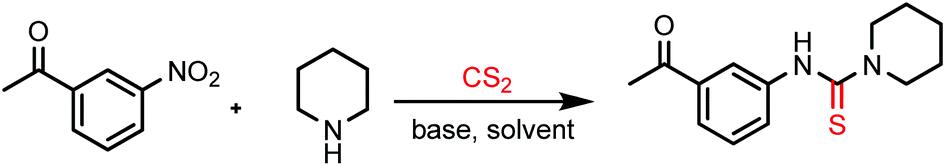
The experimental procedure is very simple and easy to perform. A nitroarene was added to a cooled (0–5 °C) mixture of an amine, carbon disulfide and K2CO3 in DMF solvent and the reaction mixture was heated for a certain time period (monitored by TLC). The crude product was extracted with EtOAc by the usual work-up procedure and was purified by column chromatography.In our initial attempt, piperidine and 3-nitroacetophenone were chosen as the model substrates for the reaction. Dithiocarbamate anions, formed by the reaction of piperidine and carbon disulfide, were allowed to react with 3-nitroacetophenone in the presence of the acetonitrile solvent and K2CO3 at 80 °C, but most of the starting materials remained unreacted with the formation of the desired thiourea product only in a trace amount (5%) (entry 1, Table 1). The reaction also failed in a 1,4-dioxane solvent medium (entry 2, Table 1). Interestingly, when the reaction was performed in the DMF solvent (entry 3, Table 1), the thiourea compound was obtained in a remarkably good yield (80%). Possibly, the poor solubility of the in situ generated dithiocarbamate salt in acetonitrile and 1,4-dioxane media inhibits the smooth progress of the reaction. The yield of the product was further improved by the elevation of the reaction temperature to 100 °C (entry 4, Table 1). However, the best reaction conditions were optimized to obtain the desired product in 92% yield by heating the reaction mixture at 100 °C for a 5 h time period (entry 5, Table 1). The reaction does not initiate at room temperature (entry 7, Table 1). The reaction in the NMP solvent (entry 12, Table 1) provided much lower yield (55%). In water medium, the nitroarene was completely insoluble and the reaction was greatly hampered (entry 13, Table 1). The necessity of the base was realized from the reaction in the absence of K2CO3 (entry 6, Table 1) where the product was formed in much lower yield (30%). K2CO3 was found to be superior compared to other bases like NaOAc, Na2CO3, NaHCO3, and NaOH (entries 8–11, Table 1) in terms of the yield of the product. Increasing the reaction temperature beyond 100 °C does not improve the yield of the desired thiourea compound.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Temp. (°C) | Time (h) | Yield (%) |
| a Reaction conditions: A mixture of piperidine (1.2 mmol), CS2 (2 mmol), and a base (1.5 mmol) in 3 ml of the solvent was stirred at 0–5 °C followed by the addition of 3-nitroacetophenone (1 mmol) and heating the resulting reaction mixture for a certain time period. | |||||
| 1 | MeCN | K2CO3 | 80 | 4 | 5 |
| 2 | 1,4-Dioxane | K2CO3 | 80 | 4 | — |
| 3 | DMF | K2CO3 | 80 | 4 | 78 |
| 4 | DMF | K2CO3 | 100 | 4 | 85 |
| 5 | DMF | K 2 CO 3 | 100 | 5 | 92 |
| 6 | DMF | — | 100 | 5 | 30 |
| 7 | DMF | K2CO3 | rt | 10 | — |
| 8 | DMF | NaOAc | 100 | 5 | 43 |
| 9 | DMF | Na2CO3 | 100 | 5 | 82 |
| 10 | DMF | NaHCO3 | 100 | 5 | 80 |
| 11 | DMF | NaOH | 100 | 5 | 63 |
| 12 | NMP | K2CO3 | 100 | 5 | 55 |
| 13 | H2O | K2CO3 | 100 | 5 | — |
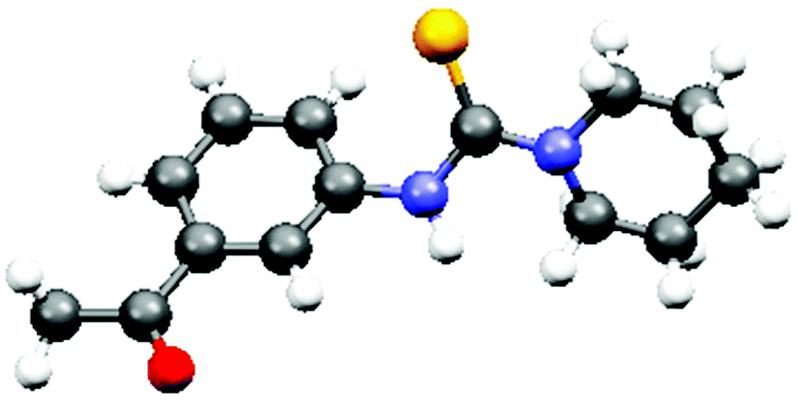
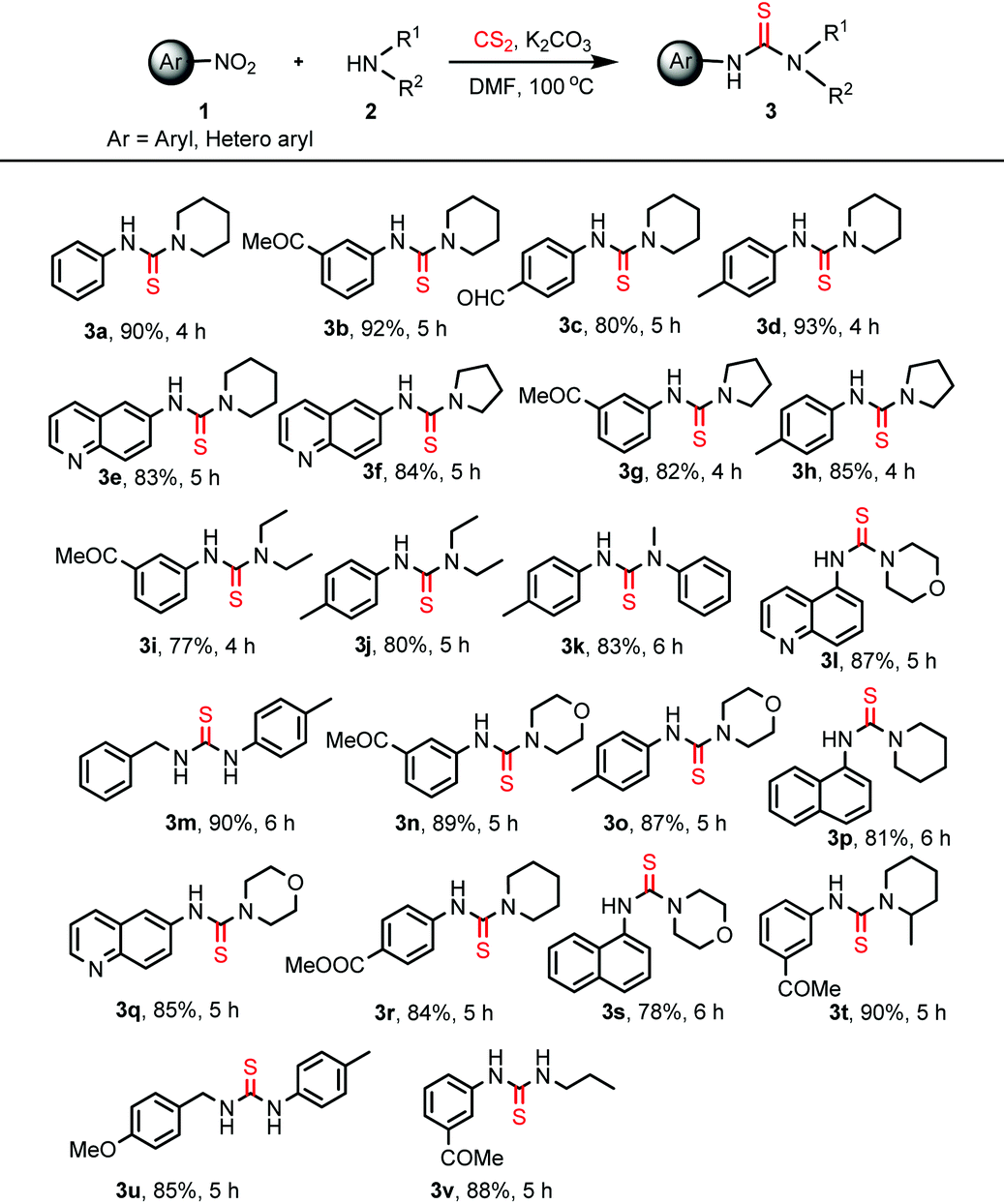
The optimized reaction conditions were then explored to check the general applicability of the protocol (Table 2). Various aliphatic secondary amines such as piperidine, pyrrolidine, morpholine, 2-methyl piperidine, and diethylamine participated in the reactions smoothly to give the unsymmetrical thiourea products in good yields. N-Methylaniline was employed as an aromatic secondary amine to perform the thiocarbamidation of 4-nitrotoluene giving rise to the product 3k. Primary amines like benzylamine, 4-methoxybenzylamine and propylamine also responded to identical reactions with nitroarenes providing unsymmetrical disubstituted thioureas, 3m, 3u and 3v, which are difficult to obtain by many reported protocols. Substituents like –COMe, –CHO, –CO2Me, and –Me present in the aromatic rings of nitroarenes remained unaffected under the present reaction conditions. Heteroaryl nitro compounds like 5-nitroquinolene and 6-nitroquinolene worked equally well producing the desired thiourea products 3l and 3e, 3f, and 3q, respectively. 1-Nitronaphthalene has been used for the preparation of unsymmetrical thiourea compounds, 3p and 3s, in satisfactory yields. However, the reaction with the aliphatic nitro compound gives a complex reaction mixture without the formation of the desired product. The structure of the product, 3b, was confirmed by X-ray crystallographic analysis as shown in Fig. 1 (CCDC 1856683†).
In general, all the reactions are clean and high yielding. A detailed mechanistic study is still under way in our laboratory. The aromatic nitro compound is known to be reduced to aniline by sulfur nucleophiles15 like thiol where the thiol gets converted to the corresponding disulfide. Similarly, in our protocol, the in situ generated dithiocarbamate anion is postulated to be transformed into thiuram disulfide by reducing the nitroarene to the corresponding aniline which is believed to further react with the thiuram disulfide intermediate14e to produce the desired thiourea compound. However, we could not identify any aniline or thiuram disulfide intermediate out of any reactions even if the reactions are arrested in between. Thus, we ruled out the possibility of the thiuram disulfide pathway.
We believe that the dithiocarbamate anion, A, formed by the reaction of the amine and CS2, attacks the oxygen atom of the nitro group in the nitroarene substrate. The nucleophilic attack on the oxygen atom of the nitro group is not unprecedented and the same is described in the Bartoli indole synthesis.16 Thus, the nitroarene is reduced to the corresponding nitroso compound C by the dithiocarbamate anion which gets oxidized to Dvia the intermediate B (Scheme 3). Another nucleophilic attack of the dithiocarbamate anion (A) on the oxygen atom of the nitroso group of C produces E which rearranges to give the desired thiourea product and sulfur monoxide17 as transient species. Sulfur monoxide immediately reacts with D to produce the intermediate F. The expulsion of sulfur dioxide18 from F regenerates the dithiocarbamate anion, A, which initiates the next reaction cycle upon the reaction with a new molecule of the nitroarene. The nucleophilic attack on the oxygen atom of nitrosobenzene is also explained in the mechanism of the Bartoli indole synthesis.16 Probably, the base K2CO3 consumes SO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and helps the respective equilibrium to push towards the forward direction. To prove the involvement of the nitroso intermediate in our protocol, the nitrosobenzene prepared separately (detailed experimental procedures are given in the ESI†) was allowed to react with the dithiocarbamate anion generated in situ by the reaction of piperidine and CS2, under identical reaction conditions (heating the reaction mixture in the DMF solvent at 100 °C for a 4 h time period) (Scheme 4) and we found the formation of the desired thiourea compound along with some other unidentified side products. This experiment proves the intermediacy of nitrosobenzene in the reaction pathway as described in Scheme 3. The generation of the nitrosoarene (C) intermediate was also detected by the FT-IR and HRMS analyses of a crude reaction mixture obtained from an incomplete reaction of piperidine, CS2 and 3-nitroacetophenone (heating at 100 °C in DMF for a 1 h time period) in the presence of K2CO3, where the appearance of the characteristic IR signal of the N
19 and helps the respective equilibrium to push towards the forward direction. To prove the involvement of the nitroso intermediate in our protocol, the nitrosobenzene prepared separately (detailed experimental procedures are given in the ESI†) was allowed to react with the dithiocarbamate anion generated in situ by the reaction of piperidine and CS2, under identical reaction conditions (heating the reaction mixture in the DMF solvent at 100 °C for a 4 h time period) (Scheme 4) and we found the formation of the desired thiourea compound along with some other unidentified side products. This experiment proves the intermediacy of nitrosobenzene in the reaction pathway as described in Scheme 3. The generation of the nitrosoarene (C) intermediate was also detected by the FT-IR and HRMS analyses of a crude reaction mixture obtained from an incomplete reaction of piperidine, CS2 and 3-nitroacetophenone (heating at 100 °C in DMF for a 1 h time period) in the presence of K2CO3, where the appearance of the characteristic IR signal of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency at 1456 cm−1 and the HRMS signal of [M + H]+ corresponding to the respective nitrosoarene (m-NOC6H4COCH3) at m/z = 150.0462 (calcd: 150.0477) supports the formation of the nitroso intermediate.
O stretching frequency at 1456 cm−1 and the HRMS signal of [M + H]+ corresponding to the respective nitrosoarene (m-NOC6H4COCH3) at m/z = 150.0462 (calcd: 150.0477) supports the formation of the nitroso intermediate.
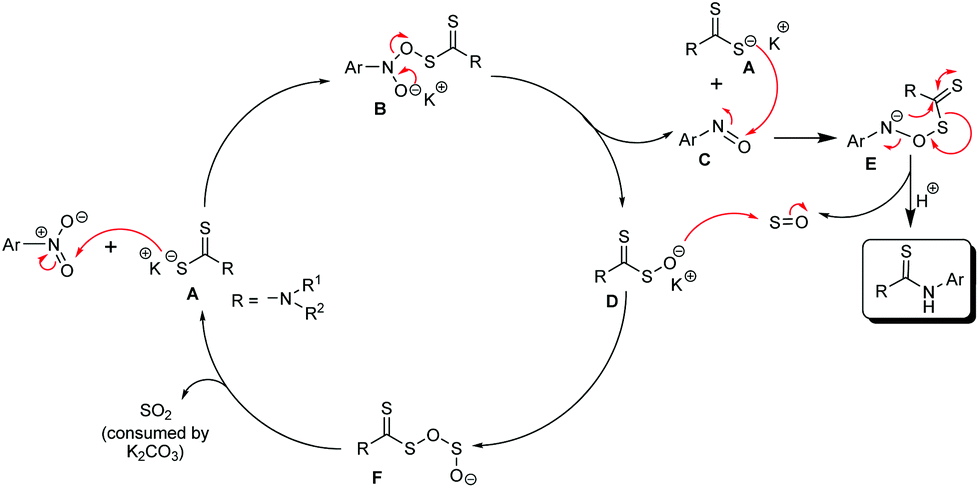 | ||
| Scheme 3 Plausible mechanistic pathway. | ||
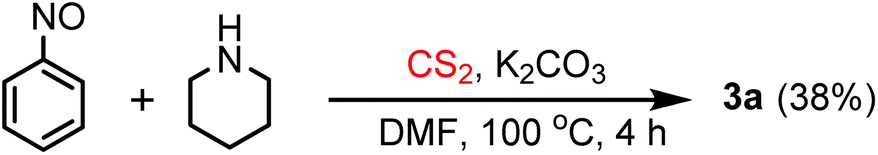 | ||
| Scheme 4 Control experiment with nitrosobenzene. | ||
The DMF solvent, having a higher dielectric constant compared to NMP, possibly stabilizes the ionic intermediates (intermediates A, B, D, E, and F) more effectively for easy progress of the reaction.
Conclusions
In conclusion, a new, convenient, and one-pot synthetic route for unsymmetrical thiourea has been developed by using easily available starting materials. All the reactions proceeded smoothly giving rise to high yields of the desired products. To the best of our knowledge, this is the first report of the C–N coupling between aromatic nitro compounds and dithiocarbamate anions which results in the formation of thiourea compounds. The reaction mechanism is proposed to involve a nitrosobenzene intermediate.Experimental
IR spectra were recorded on a JASCO FT/IR-4600 under neat conditions. NMR spectra were recorded using a Bruker Spectrometer at 300, 400, 500, and 600 MHz for 1H spectra and at 75, 100, 125, and 150 MHz for 13C spectra in CDCl3 and DMSO-d6 solution. High-resolution mass spectra (HRMS) were obtained in a methanol solvent on a Waters Micromass Q-tof Micromass spectrometer by an electrospray ionization method. Melting points were determined using a LabX India digital melting point apparatus.General experimental procedure for the one-pot synthesis of unsymmetrical thiourea
2 mmol of CS2 was added dropwise to a cooled solution of the amine (1.2 mmol) in DMF (3 ml) at 0–5 °C. K2CO3 (1.5 mmol) was added to the resulting mixture and it was stirred for 5 min at 0–5 °C. Nitroarene (1 mmol) was added to the reaction mixture followed by heating at 100 °C for the required time period (monitored by TLC) as mentioned in Table 2. After the completion of the reaction, the reaction mixture was diluted with EtOAc. Then the EtOAc layer was washed with water (three times) for the complete removal of DMF and other inorganic residues. The EtOAc solution was dried over anhydrous Na2SO4 and concentrated under vacuum to obtain the crude product. The crude product was purified by column chromatography using an EtOAc–hexane solvent mixture.All the products listed in Table 2 were characterized by their melting points, and by FTIR and 1H and 13C NMR spectroscopy. HRMS were recorded for all unknown compounds. The known compounds were identified by comparing the literature data of melting points, and FTIR, 1H NMR and 13C NMR spectra.
The spectral data of N-(3-acetylphenyl)piperidine-1-carbothioamide, 3b (Table 2), are given below:
White solid (241 mg, 92% yield); Rf – 0.2 (EtOAc/hexane = 20/80, v/v); melting point: 140–142 °C; IR (neat) ν 3553, 3462, 2687, 1669, 1482, 1397, 1266, 895, 727 cm−1. 1H NMR (CDCl3, 500 MHz) δ 7.74 (s, 1H), 7.69 (s, 1H), 7.62 (d, J = 10 Hz, 1H), 7.43 (d, J = 5 Hz, 1H), 7.32 (m, 1H), 3.80–3.79 (m, 4H), 2.5 (s, 3H), 1.62 (s, 6H) ppm. 13C NMR (CDCl3, 125 MHz) δ 197.86, 181.69, 140.85, 137.44, 128.79, 128.65, 124.59, 123.22, 50.23, 26.61, 25.48, 24.02 ppm. HRMS: calcd for C14H18N2OS [M]+ 262.1140; found 262.7686.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Soumya Dutta is thankful to the CSIR, India for his fellowship. The authors are also thankful to Mr Srikanta Jana of Dept. of Chemistry, Jadavpur University for his assistance in crystal structure analysis. We are pleased to acknowledge the financial and infrastructural support from the UGC-CAS program in Chemistry, Jadavpur University. The DST-PURSE Program and the DST-FIST Program at Jadavpur University are also acknowledged.References
-
(a) A. Ullrich and J. Schlessinger, Cell, 1990, 61, 203 CrossRef CAS PubMed
; (b) A. M. M. E. Omar, A. M. Farghaly, A. A. B. Hazzai, N. H. Eshba, F. M. Sharabi and T. T. Daabees, J. Pharm. Sci., 1981, 70, 1075 CrossRef CAS PubMed
- J. Lee, J. Lee, M. Kang, M. Shin, J. M. Kim, S. U. Kang, J. O. Lim, H. K. Choi, Y. G. Suh, H. G. Park, U. Oh, H. D. Kim, Y. H. Park, H. J. Ha, Y. H. Kim, A. Toth, Y. Wang, R. Tran, L. V. Pearce, D. J. Lundberg and P. M. Blumberg, J. Med. Chem., 2003, 46, 3116 CrossRef CAS PubMed
- A. Ranise, A. Spallarossa, O. Bruno, S. Schenone, P. Fossa, G. Menozzi, F. Bondavalli, L. Mosti, A. Capuano, F. Mazzeo, G. Falcone and W. Filippelli, Farmaco, 2003, 58, 765 CrossRef CAS PubMed
-
(a) S. Cunha, F. C. Macedo, G. A. N. Costa, M. T. Rodrigues, R. B. V. Verde, L. C. de Souza Neta, I. Vencato, C. Lariucci and F. P. Sá, Monatsh. Chem., 2007, 138, 511 CrossRef CAS
-
(a) F. J. Ruder, W. Guyer, J. A. Benson and H. Kayser, Pestic. Biochem. Physiol., 1991, 41, 207 CrossRef CAS
-
(a) Y. Hoashi, T. Okino and Y. Takemoto, Angew. Chem., Int. Ed., 2005, 44, 4032 CrossRef CAS PubMed
-
(a) Y. Tang, L. Deng, Y. Zhang, G. Dong, J. Chen and Z. Yang, Org. Lett., 2005, 7, 1657 CrossRef CAS
-
(a) E. B. Veale, G. M. Tocci, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Org. Biomol. Chem., 2009, 7, 3447 RSC
-
B. Mertschenk, F. Beck and W. Bauer, Thiourea and Thiourea Derivatives, in Ullmann's Encyclopedia of Industrial Chemistry, 2002, Wiley-VCH Verlag GmbH & Co. KGaA Search PubMed
-
(a) K. Biswas and M. F. Greaney, Org. Lett., 2011, 13, 4946 CrossRef CAS
-
F. Duus, in Comprehensive Organic Chemistry, ed. N. J. Jones, Pergamon Press, Oxford, 1979, vol. 3, pp. 465 Search PubMed
-
(a) J. Bhattacharjee, S. Das, R. K. Kottalanka and T. K. Panda, Dalton Trans., 2016, 45, 17824 RSC
-
(a) K. Singh and S. Sharma, Tetrahedron Lett., 2017, 58, 197 CrossRef CAS
-
(a) Q. Cao, F. Liu, M. Wang, W. Xu, M.-T. Zeng, M. Liu, Y.-S. Li and Z.-B. Dong, J. Chem. Res., 2017, 41, 301 CrossRef CAS
-
(a) Z. Duan, S. Ranjit, P. Zhang and X. Liu, Chem. – Eur. J., 2009, 15, 3666 CrossRef CAS
- R. Dalpozzo and G. Bartoli, Curr. Org. Chem., 2005, 9, 163 CrossRef CAS
-
(a) D. M. Lemal and P. Chao, J. Am. Chem. Soc., 1973, 95, 922 CrossRef CAS
-
(a) H. Ouast and F. Kees, Chem. Ber., 1981, 114, 787 CrossRef
-
(a) H. Fortier, C. Zelenietz, T. R. Dahn, P. Westreich, D. A. Stevens and J. R. Dahn, Appl. Surf. Sci., 2007, 253, 3201 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data and 1H and 13C NMR spectra of all compounds listed in Table 2. Data of X-ray crystallographic studies. CCDC 1856683. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qo00752g |
| This journal is © the Partner Organisations 2019 |