DOI:
10.1039/C9QO00493A
(Research Article)
Org. Chem. Front., 2019,
6, 2877-2883
Visible light-mediated photo-oxygenation of arylcyclohexenes†
Received
8th April 2019
, Accepted 30th June 2019
First published on 4th July 2019
Abstract
Substituted styrenes constitute important molecular building blocks for various synthetic manipulations, including selective core and peripheral oxidations. The photo-oxidation of such substrates by a singlet oxygen ene reaction (Schenck ene reaction) is especially attractive as it relies upon the combination of three abundant and cheap components: air, visible light, and an organic dye. The resultant allyl hydroperoxides enable various functionalizations to alcohols, carbonyls, epoxides, triols etc. The synthetic potential and mechanistic minutiae of photo-oxidations of styrene derivatives are not fully understood and were hitherto explored only for a very limited set of substrates. The operation of multiple oxidation events, low selectivities and yields have limited further applications of this method. Now this study reports a concise investigation of such photo-oxidations of diverse cycloalkenylbenzenes under continuous flow conditions. The combination of synthetic, kinetic, spectroscopic, and thereotical data enabled us to provide a detailed mechanistic rationalization of the observed reactivities and chemoselectivities. We propose a rarely discussed zwitterionic key intermediate of the observed allylic and [4 + 2]-dioxygenations based on a detailed Hammett study and DFT calculations. The reaction conditions of such photo-oxidations have been optimized to allow short reaction times (<10 min), high reproducibilities, and high chemoselectivities at 0 °C. Various isolation strategies of the sensitive products and their conversion to stable compounds. A set of synthetically versatile 2-aryl-2,3-epoxy-1-cyclohexanols and 1-aryl-6-hydroxy-1-cyclohexenes has been isolated and fully characterized.
Introduction
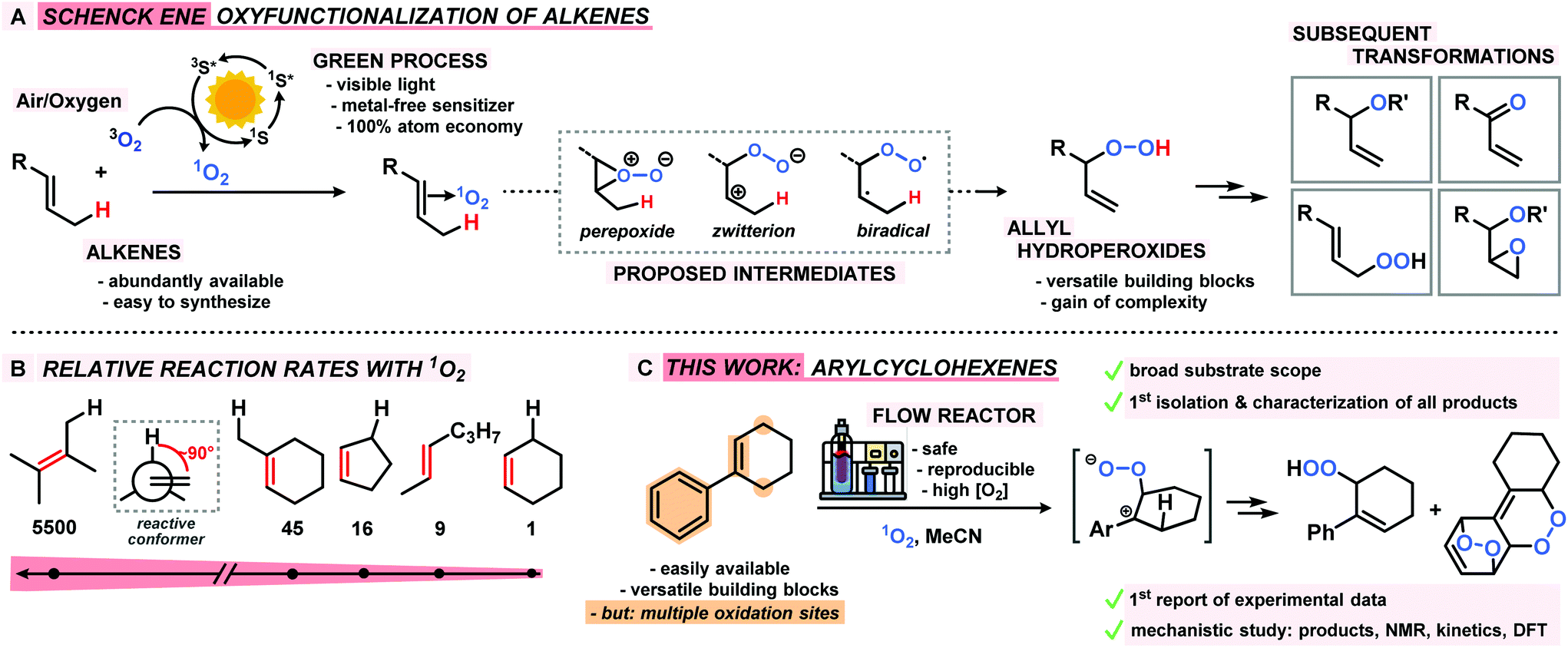
Benzylic and allylic oxidations are key steps of various molecular functionalizations, valorizations, and degradations in industrial and biological processes.1–3 The majority of available synthetic methods involve metal complexes and/or special stoichiometric oxidants (peracids, H2O2).4–6 In contrast, the use of the most abundant oxidant air (or dioxygen O2) in combination with a metal-free sensitizer (S) and visible light constitutes an especially desirable inexpensive and sustainable way of performing hydrocarbon oxidations (Scheme 1).7 Unlike biological oxidations with triplet oxygen 3O2, such photo-oxidations involve the generation of singlet oxygen 1O2, operate via closed-shell intermediates, and often exhibit high levels of regiocontrol. The Schenck-ene reaction is a prime example of a sustainable CH-oxidation method that relies on the combination of highly abundant components: simple unbiased alkenes, air (or O2), visible light, and a cheap organic dye as catalyst. This allylic oxygenation generally exhibits great versatility and operational simplicity and produces allyl hydroperoxides as highly reactive building block molecules (Scheme 1A). Beyond the technical multi-ton scale productions of the fragrance rose oxide and the antimalaria drug Artemisinin,8–101O2-ene reactions are still under-utilized in the context of oxyfunctionalizations. Reaction rates and selectivities (chemo, regio, stereo) are highly dependent on substitution patterns and conformations of the hydrocarbon substrates. In the past decades, the majority of research activities has been devoted to photo-oxidations of electron-rich acyclic alkenes that often bear hetero-atomic functional groups or directing groups. Interestingly, the abundantly available cyclohexene derivatives are an especially unreactive class of substrates for CH-oxidations, which is mostly a direct consequence of unfavourable chair-like conformations in the transition states (Scheme 1B).11 Here, we report the first concise study of the photo-oxidation of arylcyclohexenes which presents tangible advances over the state-of-the-art: a series of 14 functionalized 1-aryl-1-cyclohexenes were oxidized under optimized Schenck-ene conditions; continuous flow reactors were applied to enable high selectivities; the products from competing hydrocarbon, alkene, and arene oxidations were isolated and characterized; a concise mechanistic study (by-products, kinetics, NMR, DFT) was performed; the mechanistic postulate of a zwitterionic intermediate was corroborated (Scheme 1C).12
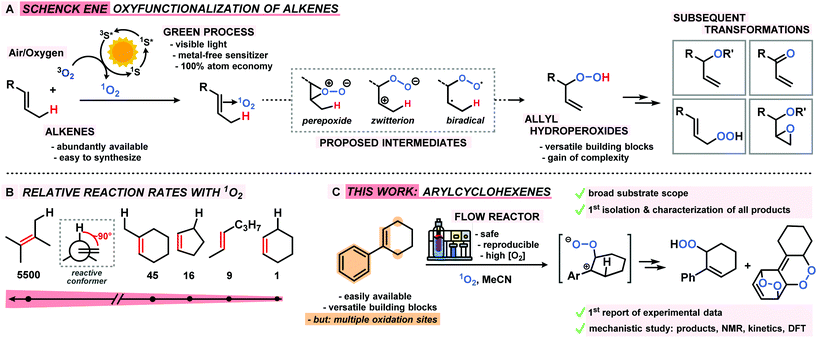 |
| | Scheme 1 The Schenck ene reaction: (A) general reaction, intermediates, primary oxidation product and follow-up manipulations; (B) relative rates of alkene substrate classes; (C) this work: arylcyclohexenes as new substrate class, reaction set-up, proposed intermediate, and main oxidation products. | |
Results and discussion
Photooxygenation of 1-phenyl-1-cyclohexene
The mechanism of the Schenck ene reaction is strongly depending on the stereoelectronic properties of the substrate, directing group effects, solvent polarity and proticity. For most reactions, a per-epoxide intermediate has been postulated as key intermediate which converts to a transition state that adopts an orthogonal orientation of the abstracting allylic H-atom and the evolving alkene plane.13–16
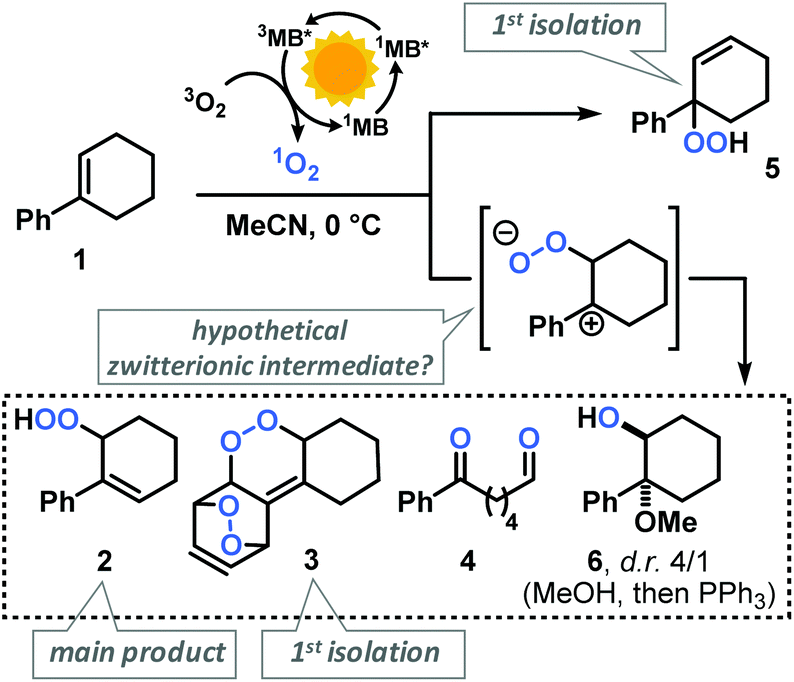
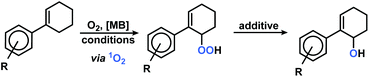
Our group has recently investigated the use of strained bicyclic alkenes for enhanced Schenck ene reactions,17 others have exploited confinement effects to access reactive conformations.18 We surmised that introduction of an aryl substituent in 1-position of cyclohexenes would result in a considerable stereoelectronic modulation of the ground state and transition state conformations which ultimately affect the rate of photo-oxidation. On the other hand, the presence of aryl substituents in cyclohexenes may enable competitive arene, alkene, and CH-oxidations under similar conditions and thereby result in the formation of complex reaction mixtures.19 We initially probed the reactivity of the model substrate 1-phenyl-1-cyclohexene (1) under the photo-oxidation conditions of the Schenck ene reaction in acetonitrile with 1 mol% methylene blue as sensitizer and pressurized O2.20 We have applied and optimized a self-made continuous-flow microreactor.17 Such reactor setups exhibit several advantages over batch reactions which enabled highly reproducible reactions,7 such as effective temperature control, excellent absorbance characteristics and energy efficiency using high power LEDs,21 a thin capillary for short reaction times, and high operational safety with regard to the formation of hazardous peroxides.22 We observed a strong rate enhancement of the reaction of 1-phenyl-1-cyclohexene (1) vs. cyclohexene which is in full accord with our hypothesis that introduction of the phenyl moiety (in conjugated position, resulting in (slight) steric repulsion from the ortho-CH) directly affects the transition state conformations.23 However, arylcyclohexenes such as 1 are a highly challenging class of substrates due to the presence of multiple oxidation-sensitive sites which can lead to various oxidation products in reactions with 1O2. Besides allylic oxidation to allyl hydroperoxides, the same conditions could induce [4 + 2]-cycloaddition to endoperoxides or [2 + 2]-cycloaddition with following 1,6-dicarbonyl formation.24,25 With our standard conditions (50 s irradiation in flow reactor, red LEDs, 30 bar O2, 0 °C, 0.1 M 1 in MeCN, 1 mol% methylene blue (MB)), we observed the formation of two main products: the major O2-ene product 6-hydroperoxy-1-phenyl-1-cyclohexene (2) and the double [4 + 2]-cycloadduct (3). Three minor products were isolated: 6-oxo-6-phenylhexanal (4), the other 1O2-ene regioisomer 5, and after reductive workup with triphenylphosphine (PPh3) the 1,2-diol derivative 6 (anti/syn = 4/1). In contrast to the few reported photo-oxygenations of open-chain styrenes,25–27 reactions of arylcycloalkenes in this study did not form isolable mono-[4 + 2]-cycloadducts (Scheme 2).28 Our observations are remarkable in that they significantly expand the knowledge of photo-oxygenation pathways and product distributions of this family of easily available and synthetically versatile arylcyclohexenes. The desired allyl hydroperoxide 2 indeed was the major product under our photo-oxidation conditions. This is in stark contrast to earlier work by Foote et al. who observed only trace amounts of O2-ene products from the related styrene derivative trans-anethole using tetra-phenylporphyrin (TPP) as sensitizer and a high-power Xe lamp.25 Furthermore, the formation of the second O2-ene regioisomer, the tertiary allyl hydroperoxide 5, has yet not been reported.29 The postulated intermediacy of a peroxy zwitterion would suggest a strong dependence on the proticity of the solvent and the interception of the carbocation by competent nucleophiles.25,30,31 Both hypotheses were experimentally substantiated: the change from acetonitrile to methanol resulted in the increased formation of desired allyl hydroperoxide 2 at total conversion (MeCN: 2/3 = 2.1; MeOH: 2/3 = 5.0; vide infra). The formation of the 1,2-dioxy derivative 6 in MeOH is indicative of a carbocation intermediate which underwent nucleophilic attack by MeOH followed by reduction with PPh3.30–32
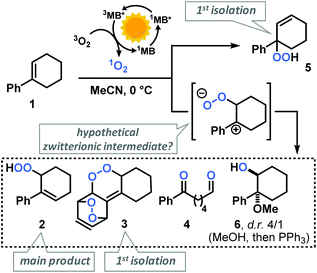 |
| | Scheme 2 Isolated products of the photo-oxygenation of phenylcyclohexene 1. | |
We then set out to explore the substrate scope of this photo-oxygenation and establish structure–activity relationships. The variation of the aryl substitution pattern in a Hammett study would directly address the nature of the reactive intermediate of the photo-oxygenation reaction. The postulated zwitterion bearing a carbocation in benzylic position would be subject to effective charge stabilization by donor substituents at the arene.33
Mechanistic studies
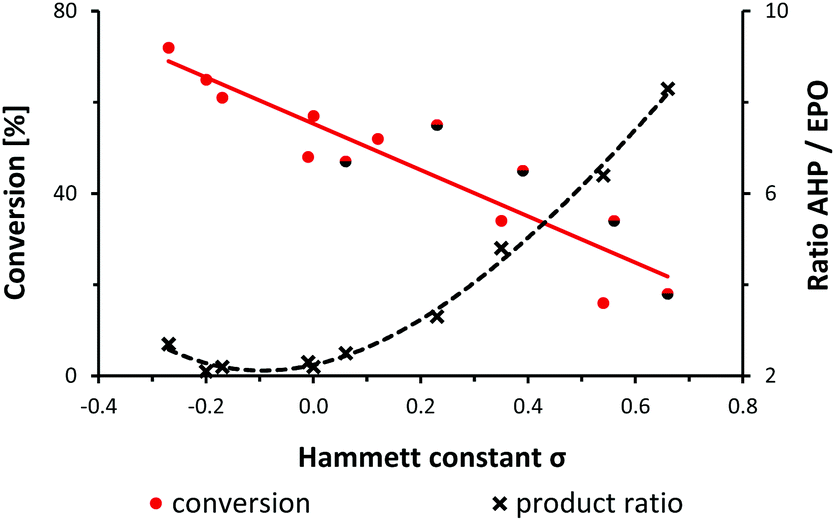
A preliminary mechanistic discussion of the photo-oxygenation of trans-anethole by Foote and coworkers involved the potential intermediacy of a zwitterion containing a benzyl cation. Peroxy-anion addition to the electrophilic ortho-arene position could account for the observed endoperoxide formation which was largely suppressed in the presence of protic solvents or acidic additives.25 In an effort to explore the mechanistic minutiae of the photo-oxygenation of 1-aryl-1-cyclohexenes, we performed a concise Hammett study that generally considers electric field effects, polarizabilities, inductive and resonance contributions within the characteristic substituent constants σ.34–37 We prepared a set of 14 meta- and para-substituted 1-aryl-1-cyclohexenes and subjected them to photo-oxygenation under standard conditions (Table 1, Fig. 1). Consistent with our hypotheses, the overall reaction rates exhibited a strong linear correlation with the electronic properties of the aryl substituents (σmeta, σpara). Electron donors gave faster conversions, with the 4-methoxy derivative reaching 72% conversion within 50 s irradiation in the flow reactor (entry 13). 1-(4-Trifluoromethylphenyl)-1-cyclohexene was the least reactive substrate (16% conversion, entry 3). These observations argue in favour of the formation of an intermediate benzyl cation as rate-determining step. The experimental results were complemented with theoretical studies (PCM solvation model, MeCN; B3LYP 6-311++G(2d,p)) which clearly documented the stabilization of the postulated zwitterion by 3.3 kcal mol−1 for the 4-methoxy derivative (vs. the parent substrate 1) and destabilization by 2.2 kcal mol−1 for the 4-trifluoromethyl derivative (vs.1). Photo-oxygenation of the 4-(N,N-dimethylamino) derivative resulted in complete inhibition, most likely as a consequence of effective physical quenching of 1O2 by the amine (entry 14).38 The trend line of conversion vs. σ inhabited the (pseudo)halogen substituents F, Cl, Br, CN as outliers (see half-black dots in Fig. 1) which are known to often deviate from standard Hammett trends.39–41 The R2 value of trend line accuracy improves from 0.84 to 0.96 when eliminating the data points of m-Br, p-Cl, p-F, and m-CN. The chemoselectivity of the reaction was also strongly depending on the aryl substituents (Table 1, Fig. 1). The ratios of the two main primary oxidation products, the allyl hydroperoxide (AHP) and the endoperoxide (EPO), AHP/EPO increased with strongly electron-withdrawing para-substituents (large σp values, e.g. CN, CF3, OCF3). The formation of different regioisomers and stereoisomers of the endoperoxides from meta-substituted aryl-cyclohexenes prohibited a reliable product analysis so that no product ratios are given. The Hammett plot minimum for weakly electron-donating substituents without mesomeric stabilization (H, Me, tBu; entries 9, 11, 12 in Table 1) might indicate a change of reaction mechanism (Fig. 1).37
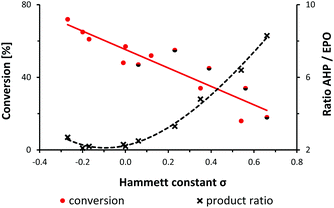 |
| | Fig. 1 Relation between alkene conversion, product ratios (allyl hydroperoxide, AHP; endoperoxide, EPO), and Hammett substituent constants σ. See Table 1. | |
Table 1 Hammett data of photo-oxidations of m- and p-substituted arylcyclohexenes
| Entry |
Substituent |
σ
m
|
σ
p
|
Conversiona [%] |
AHP/EPOb |
Hammett constants are given for meta-(σm) and para-substituents (σp).35 Conditions: 50 s irradiation in the flow reactor with red LEDs, O2 (∼30 bar), 0 °C, c(MB) = 1 mM, c(substrate) = 0.1 M. Conversion was determined by GC-FID. Ratio of allyl hydroperoxide (AHP)/endoperoxide (EPO) was determined by 1H-NMR of the crude reactions. Product ratios could not be determined (n.d.) for meta-substituted substrates. MeCN/toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used for solubility issues. :1) was used for solubility issues. |
| 1 |
–CN |
|
0.66 |
18 |
8.3 |
| 2 |
–CN |
0.56 |
|
34 |
n.d. |
| 3 |
–CF3 |
|
0.54 |
16 |
6.4 |
| 4 |
–Br |
0.39 |
|
45 |
n.d. |
| 5 |
–OCF3 |
|
0.35 |
34 |
4.8 |
| 6 |
–Cl |
|
0.23 |
55 |
3.3 |
| 7 |
–OMe |
0.12 |
|
52 |
n.d. |
| 8 |
–F |
|
0.06 |
47 |
2.5 |
| 9 |
–H |
|
0.00 |
57 |
2.2 |
| 10 |
–Phc |
|
−0.01 |
48 |
2.3 |
| 11 |
–Me |
|
−0.17 |
61 |
2.2 |
| 12 |
–tBu |
|
−0.20 |
65 |
2.1 |
| 13 |
–OMe |
|
−0.27 |
72 |
2.7 |
| 14 |
–NMe2 |
|
−0.83 |
0
|
n.d. |
Based on few literature precedents and our experimental and theoretical studies,22–24 we propose a reaction mechanism that involves a charge-separated zwitterion with benzyl cation character as key intermediate (Scheme 3). The Mulliken charges and stabilities of three zwitterions with different para-aryl substituents (OMe, H, CF3) were calculated by DFT (B3LYP, 6-311+G(d,p), Scheme 3, top). These theoretical data support the notion of competing allyl hydroperoxide (AHP) and endoperoxide (EPO) formation. The introduction of a 4-methoxy substituent results in strong stabilization of the zwitterion (−14 kJ mol−1vs. 4-H-substitution), lower acidity of the β-proton, higher nucleophilicity of the peroxy anion, and higher electrophilicity of the ortho-aryl position. These conditions should enhance the formation of the endoperoxide which is in full accord with the experiments (AHP/EPO 2.7, Table 1). With the 4-trifluoro-methyl group, allyl hydroperoxide formation is much more favoured due to the destabilization of the intermediate benzyl cation (+9 kJ mol−1vs. 4-H), higher acidity of the β-H, lower nucleophilicity of the peroxy anion, and lower electrophilicity at the ortho-aryl position (experiment: AHP/EPO 6.4, see Table 1). As the zwitterion formation is the rate-determining step, the conversions of electron-deficient arylcyclohexenes are low (Table 1, Fig. 1).
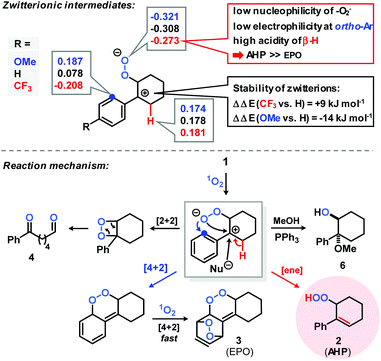 |
| | Scheme 3 Top: Proposed zwitterionic intermediate with calculated Mulliken charges at reactive O, C, and H-sites and thermodynamic stabilities. Bottom: Postulated mechanism of the photo-oxygenation of arylcyclohexenes. | |
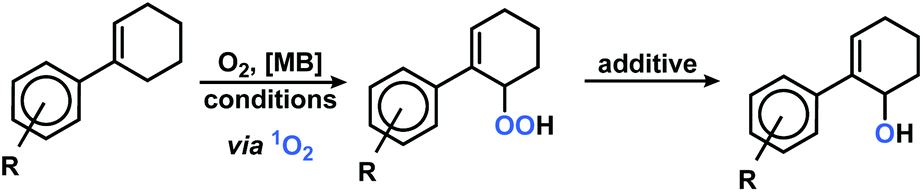
Synthesis, isolation and conversion of primary oxidation products
The optimization of the continuous-flow photo-oxidation revealed that reactions at 0 °C provided the best compromise between reaction time and selectivity toward the allyl hydroperoxide (AHP), whereas the temperature did not have a significant impact on the ratio AHP/EPO. However, the sensitizer methylene blue could undergo oxidative decomposition if reaction times are long.42 The ratio of the two 1O2-ene regioisomers (from phenylcyclohexene: 2 and 5, see Scheme 2) was ∼7/1 and nearly constant over the whole set of different meta- and para-substituted substrates and different conditions. Importantly, the regioselectivity of the 1O2-ene reactions was much higher with ortho-substituted arylcyclo-hexenes as <3% of the tert-hydro-peroxide isomers formed. This cannot be explained by electronic effects whereas steric hindrance around the benzylic carbon prevents 1O2 attack. On preparative scales, the formation of further oxidation products in minor quantities (such as 4 and 5) hamper the isolation of pure compounds. NMR monitoring studies with (1-(4-chlorophenyl)-1-cyclohexene documented the generally low reaction selectivity by formation of further oxidation products beside the major products AHP (and regioisomer) and EPO (Fig. 2). 1H resonances at 10–12 ppm might stem from further double bond oxidation, hydroperoxide degradation or polymerization. It is important to note that – unlike earlier reports – the endoperoxide appeared to be stable under the reaction conditions.43
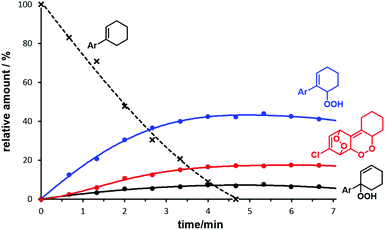 |
| | Fig. 2 Conversion and major products of the photo-oxygenation of 4-chloro-1-phenyl-1-cyclohexene from 1H NMR monitoring (Ar = 4-chlorophenyl). | |
We probed various isolation and derivatization strategies for preparative scales reactions. The rather non-polar starting materials could be easily separated from the products by SiO2 gel flash chromatography. However, the several oxidation products were difficult to separate from each other. SiO2 chromatography resulted in significant losses of materials to give only moderate to low isolated yields of reactions that exhibited good NMR yields. The bulkier ortho-substituted arylcyclohexenes underwent more selective photo-oxygenations and resulted in more stable products so that chromatographic isolations were effective. Most isolated allyl hydroperoxides and endoperoxides could be stored for days to months in a freezer and only slowly decomposed due to the low dissociation energy of the O–O bond.44,45 An alternative strategy to the isolation of the labile peroxo products is their derivatization to more stable secondary products. We have mostly applied reductions of the allyl hydroperoxides with by triphenylphosphine to give the corresponding allylic alcohols.46 PPh3 is also known to reduce endoperoxides to highly polar polyols47 which can easily be separated. Table 2 displays a selection of isolated allyl hydro-peroxides and their reduced allyl alcohol derivatives. We have also applied the general photo-oxygenation conditions to the ring size homologues 1-phenyl-1-cyclopentene (7) and 1-phenyl-1-cyclo-heptene (8). Both substrates showed much higher conversions than 1 and near-perfect AHP selectivities (Fig. 3). After 50 s in the flow reactor, quantitative conversions were observed and the exclusive formation of the desired Schenck-ene products (with isolated yields of 43% and 65%, respectively). These results manifest the facile thermal accessibility of reactive conformations of cyclopentenes and cycloheptenes bearing orthogonal orientation of the allylic CH bond to the plane of the alkene.48
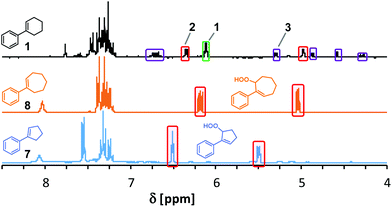 |
| | Fig. 3 Comparison of crude 1H-NMR spectra after photo-oxygenation of 1-aryl-1-cycloalkenes (see Table 1 for conditions). Signals of starting materials (green box), O2-ene products (red boxes), and [4 + 2]-cycloadducts (violet boxes). | |
Table 2 Photo-oxygenation products and their subsequent reduction with PPh3
|
|
| Entry |
R |
Conversion [%] |
Yield [%] |
Additivea |
Yield [%] |
| Conditions: 8 min irradiation in the flow reactor with red LEDs, O2 (30 bar), MeCN, 0 °C, 1 mol% methylene blue (MB). Addition of 1 equiv. PPh3 without prior work-up. Determined by 1H-NMR. 12 min of irradiation; 85% conversion, 48% yield. 5% of the regioisomeric O2-ene-derived allyl alcohol was formed. |
| 1 |
— |
100 |
39b |
PPh3 |
33d |
| 2 |
2-Ph |
100 |
63c |
— |
|
| 3 |
4-Cl |
100 |
n.d. |
PPh3 |
31d |
| 4 |
2-Me |
90 |
31 |
— |
|
| 5 |
4-CN |
90 |
n.d. |
PPh3 |
39 |
We explored the scope of the photo-oxygenation with further substrates (Scheme 4): 6-hydroxy-1-phenyl-1-cyclohexene, the PPh3-reduced product of the photo-oxygenation of 1, itself is a good substrate for another photo-oxygenation.49,50 Doubled reaction time (compared to the standard substrate 1) led to full conversion.51 Two sterically congested arylcyclohexenes with ortho,ortho-aryl substitution were also tested. 1-(2′,6′-Dimethyl-phenyl)-1-cyclohexene was unreactive towards oxidation even under prolonged irradiation times. DFT calculations showed that the alkene and arene planes are twisted out of conjugation so that the 1O2 attack is sterically hindered. 1-(Pentamethylphenyl)-1-cyclohexene gave full conversion but formed complex mixtures of arene oxidation products gave full conversion (Scheme 4).51
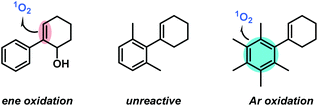 |
| | Scheme 4 Chemoselective conversions of substituted arylcyclohexenes. | |
The photo-oxygenation products derived from reactions of aryl-cyclohexenes with 1O2 are versatile building blocks of great synthetic utility. Reduction of the peroxy functions (by PPh3 or Na2SO3) gives the corresponding alcohols. Base-mediated acetylation with pyridine/Ac2O has been used to prepare α,β-unsaturated carbonyls.11 Another interesting follow-up reaction is the titanium-catalyzed “self”-epoxidation. 2,3-Epoxy alcohols can be prepared from allyl hydroperoxides in high diastereoselectivity by a Sharpless epoxidation protocol in the absence of any further oxidant.52,53 The presence of the alkene and hydroperoxide groups within the same molecule enables an intramolecular expoxidation at the Ti(IV) catalyst to give the syn-epoxy alcohol (dr 99%) in good yields over two steps (Scheme 5).
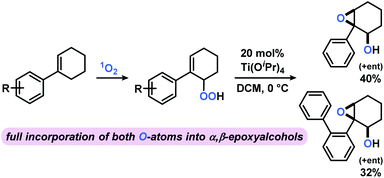 |
| | Scheme 5 Sequential photo-oxygenation and Ti-catalyzed “self”-epoxidation results in full incorporation of both O-atoms from O2 into α,β-epoxy alcohols. Flash chromatographic separation of hydroperoxide prior to Ti(OiPr)4 addition (20 mol%) at 0 °C in dichloromethane (DCM). Photo-oxidation conditions in Table 2. | |
Conclusion
The development of oxidations of unbiased hydrocarbons with oxygen/air as the stoichiometric oxidant is a prime objective in the quest for sustainable chemical reactions. Cyclohexenes are notoriously unreactive substrates for photo-oxygenations with singlet oxygen. This concise synthetic and mechanistic study has established that easily available 1-aryl-1-cyclohexenes are a highly competent class of substrates that display enhanced reactivity. For the first time, optimized conditions in a custom-made photo flow reactor have enabled photo-oxygenations of a diverse set of arylcycloalkenes bearing various substitution patterns and ring sizes. The major products are the desired allyl hydroperoxides (1O2-ene products). Minor oxidation products include endoperoxides which appeared to be rather stable and could be isolated and characterized for the first time. Detailed mechanistic investigations including a Hammett study, reaction profile analyses, the isolation of various by-products, and DFT calculations have been performed. We postulate a reaction mechanism that involves a zwitterionic intermediate that undergoes rapid allyl hydroperoxide formation under protic conditions, with electron-deficient aryl substituents, or with (slight) steric hindrance by ortho-aryl substituents. The major allyl hydro-peroxides were isolated, fully characterized, and converted to stable allyl alcohol derivatives (by reduction) or to α,β-epoxy alcohols (by intramolecular oxygen-transfer). The photo-oxygenation tolerated various functional groups (F, Cl, Br, CN. CF3, OCF3, OMe, OH).
Conflicts of interest
The authors declare no conflict of interest.
Notes and references
-
R. A. Sheldon and H. van Bekkum, in Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2000, pp. 473–551 Search PubMed.
- G. Zheng, H. Liu and M. Wang, Chin. J. Chem., 2016, 34, 519–523 CrossRef CAS.
- J. L. Jeffrey, E. S. Bartlett and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 2194–2197 CrossRef CAS PubMed.
- R. D. Bach, C. Canepa, J. E. Winter and P. E. Blanchette, J. Org. Chem., 1997, 62, 5191–5197 CrossRef CAS.
-
A. Goti and F. Cardona, in Green Chemical Reactions, Springer, Dordrecht, 2006, pp. 191–212 Search PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- D. Kopetzki, F. Lévesque and P. H. Seeberger, Chem. – Eur. J., 2013, 19, 5450–5456 CrossRef CAS PubMed.
-
T. Noël, M. Escriba Gelonch and K. Huvaere, in Photochemical Processes in Continuous-Flow Reactors, World Scientific Publishing Europe Ltd., London, 2017, pp. 245–267 Search PubMed.
- X. Liu, H. Chen, Z. Xu, Y. Wu and B. Liu, Chin. J. Chem., 2017, 35, 465–476 CrossRef CAS.
- M. Prein and W. Adam, Angew. Chem., Int. Ed. Engl., 1996, 35, 477–494 CrossRef CAS.
- An early report of Jefford et al.29 already mentioned the formation of a hydroperoxide and an endoperoxide from the 1O2-oxidation of 1-phenyl-1-cyclohexene. However, insufficient experimental and analytical data were reported (no information on yield, mechanism, substituent effects, product characterization, experimental details, purity, side products).
- A. A. Frimer, Chem. Rev., 1979, 79, 359–387 CrossRef CAS.
- M. Orfanopoulos, I. Smonou and C. S. Foote, J. Am. Chem. Soc., 1990, 112, 3607–3614 CrossRef CAS.
- A. A. Gorman, I. R. Gould and I. Hamblett, J. Am. Chem. Soc., 1982, 104, 7098–7104 CrossRef CAS.
- J. R. Hurst, S. L. Wilson and G. B. Schuster, Tetrahedron, 1985, 41, 2191–2197 CrossRef CAS.
- J. Schachtner, P. Bayer and A. Jacobi von Wangelin, Beilstein J. Org. Chem., 2016, 12, 1798–1811 CrossRef CAS PubMed.
- M. Stratakis and M. Orfanopoulos, Tetrahedron, 2000, 56, 1595–1615 CrossRef CAS.
- K. R. Kopecky and H. J. Reich, Can. J. Chem., 1965, 43, 2265–2270 CrossRef CAS.
- The organic dye and common 1O2 sensitizer methylene blue (MB) in MeCN was chosen over porphyrin systems in DCM for reasons of substrate solubilities and environmental impact.
- See the ESI† for microreactor productivity and performance.
-
H. Klenk, P. H. Götz, R. Siegmeier and W. Mayr, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- J. P. Finley and J. R. Cable, J. Phys. Chem., 1993, 97, 4595–4600 CrossRef CAS.
- M. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- A. Greer, G. Vassilikogiannakis, K.-C. Lee, T. S. Koffas, K. Nahm and C. S. Foote, J. Org. Chem., 2000, 65, 6876–6878 CrossRef CAS PubMed.
- D. Lerdal and C. S. Foote, Tetrahedron Lett., 1978, 19, 3227–3230 CrossRef.
- C. S. Foote, S. Mazur, P. A. Burns and D. Lerdal, J. Am. Chem. Soc., 1973, 95, 586–588 CrossRef CAS.
- Different products and product distributions were observed with (radical) decatungstate-sensitized or charge-transfer induced oxidations:
(a) I. N. Lykakis and M. Orfanopoulos, Synlett, 2004, 2131–2134 CAS;
(b) K. Masanobu, S. Hirochika and T. Katsumi, Bull. Chem. Soc. Jpn., 1987, 60, 3331–3336 CrossRef.
- C. W. Jefford and C. G. Rimbault, Tetrahedron Lett., 1976, 17, 2479–2482 CrossRef.
- C. W. Jefford, A. F. Boschung and C. G. Rimbault, Helv. Chim. Acta, 1976, 59, 2542–2550 CrossRef CAS.
- C. W. Jefford, Chem. Soc. Rev., 1993, 22, 59 RSC.
- The diastereomers and disatereomeric ratios were identified with the help of authentic samples on GC-MS and 1H-NMR.
- I.-H. Um, H.-J. Han, J.-A. Ahn, S. Kang and E. Buncel, J. Org. Chem., 2002, 67, 8475–8480 CrossRef CAS PubMed.
- I. Fernández and G. Frenking, J. Org. Chem., 2006, 71, 2251–2256 CrossRef PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
-
E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2006 Search PubMed.
- A. Farmilo and F. Wilkinson, Photochem. Photobiol., 1973, 18, 447–450 CrossRef CAS PubMed.
- J. E. Heffner, C. T. Wigal and O. A. Moe, Electroanalysis, 1997, 9, 629–632 CrossRef CAS.
- A. Hedström, U. Bollmann, J. Bravidor and P.-O. Norrby, Chem. – Eur. J., 2011, 17, 11991–11993 CrossRef PubMed.
- D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398–3416 CrossRef CAS.
- A. Xu, X. Li, S. Ye, G. Yin and Q. Zeng, Appl. Catal., B, 2011, 102, 37–43 CrossRef CAS.
- 4-Chloro-1-phenyl-1-cyclohexene was used for kinetic studies because it reacts slower than phenylcyclohexene.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- The allyl hydroperoxides are very stable in metal-free and solvent-free environments at low temperatures (−30 °C) and can even be stored for several months without detectable (NMR) decomposition. The endoperoxides were stable for several days under the same conditions.
- R. Hiatt, R. J. Smythe and C. McColeman, Can. J. Chem., 1971, 49, 1707–1711 CrossRef CAS.
- W. Adam and S. Weinkötz, Tetrahedron Lett., 1995, 36, 7431–7432 CrossRef CAS.
- T. Matsuura, A. Horinaka and R. Nakashima, Chem. Lett., 1973, 2, 887–890 CrossRef.
- A. G. Griesbeck, W. Adam, A. Bartoschek and T. T. El-Idreesy, Photochem. Photobiol. Sci., 2003, 2, 877–881 RSC.
- E. L. Clennan, D. Zhang and J. Singleton, Photochem. Photobiol., 2006, 82, 1226 CrossRef CAS PubMed.
- The collected spectroscopical data document the formation of various products. Several attempts to isolate, purify, and characterize pure compounds failed.
- W. Adam, A. Griesbeck and E. Staab, Angew. Chem., Int. Ed. Engl., 1986, 25, 269–270 CrossRef.
- W. Adam, M. Braun, A. Griesbeck, V. Lucchini, E. Staab and B. Will, J. Am. Chem. Soc., 1989, 111, 203–212 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00493a |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab